

Abstract
Fas ligand (FasL) mediated hepatocyte apoptosis occurs in the context of acute liver injury that can be accompanied by intravascular coagulation (IC). We tested the hypothesis that analysis of selected protein fractions from livers undergoing apoptosis will shed light on mechanisms that are involved in liver injury which might be amenable to intervention. Proteomic analysis of the major insoluble liver proteins after FasL exposure for 4 to 5 hours identified fibrinogen-γ (FIB-γ) dimers and FIB-γ-containing high molecular mass complexes among the major insoluble proteins that are visible by Coomassie blue staining. Presence of the FIB-γ-containing products was confirmed using FIB-γ-specific antibodies. The FIB-γ-containing products partition selectively and quantitatively into the liver parenchyma after inducing apoptosis. Similar formation of FIB-γ products occurs after acetaminophen administration. The observed intrahepatic IC raised the possibility that heparin therapy may ameliorate the FasL-mediated liver injury. Notably, heparin administration into mice 4-hours before or up to 2-hours after FasL-injection had a dramatic reduction of the liver injury including liver hemorrhage, serum alanine aminotransferase, caspase activation and liver apoptosis as compared to heparin-untreated mice. Heparin did not directly interfere with FasL-induced apoptosis in isolated hepatocytes and heparin-treated mice survived the FasL-induced liver injury longer compared to heparin-untreated animals. There was a sharp near-simultaneous rise in FasL-induced intrahepatic apoptosis and coagulation, with IC remaining stable while apoptosis continued to increase.
Conclusions
Formation of FIB-γ dimers and their high molecular mass products are readily detectable within the liver during mouse apoptotic liver injury. Heparin provides a potential therapeutic modality since it not only prevents the extensive FasL-related liver injury but also limits the extent of injury if given at early stages of injury exposure.
Keywords: Hepatitis, Acute liver failure, Intravascular coagulation
INTRODUCTION
Apoptosis occurs in the context of acute and chronic injury, which provides an important target for intervention and amelioration of the injury.1,2 A major mechanism that leads to hepatocyte apoptosis is the interaction of a cell surface death receptor such as Fas with its ligand, the Fas ligand (FasL). Similarly, tumor necrosis factor-α (TNF-α) can interact with its cell surface receptor to lead to apoptosis. Both TNF-α and FasL interactions with their respective receptors lead to down-stream activation of caspases which function as the executioners of cell death. The activated caspases, in the context of pathological conditions, cleave key cellular proteins thereby leading cells to undergo apoptosis. Numerous caspase substrates have been identified including cytoplasmic proteins such as keratins,3,4 and nuclear proteins such as the lamins.5 Mice that are exposed to the functional anti-Fas receptor antibody, Jo2, which serves as a FasL, develop fulminant liver failure and die within hours after administration of the antibody,6,7 thereby mimicking the cell death that occurs in the context of a variety of acute and chronic liver diseases.8
Acute liver injury is associated with several changes in the hemostatic system that may lead to intrahepatic or intravascular coagulation (IC) and changes that promote both bleeding and thrombosis.9 Fibrinogen, a major blood protein that consists of three pairs of polypeptide chains (fibrinogen Aα, Bβ and γ), is synthesized and secreted by liver parenchymal cells.10,11 Apart from its essential role in blood clotting, fibrinogen-γ (FIB-γ) contains binding sites for several proteins including clotting factors, growth factors and integrins.12,13 FIB-γ forms dimers upon various cellular conditions via transamidation and cross-linking of FIB-γ-chains between a lysine at position 406 of one γ-chain and a glutamine at position 398 or 399 of a second chain.14 High amounts of FIB-γ dimers have been detected in patients with tumors, but not in control patients suffering from acute infection or inflammation. These findings suggest that the amount of crosslinked FIB-γ dimer may correlate with tumor-associated fibrin deposition, and may be useful as a biomarker.15,16 However, characterization of FIB-γ dimers during liver damage has not been studied.
Depending upon the context, hemostatic imbalance in acute liver failure (ALF) may contribute to cell injury or may have a protective function.9 The therapeutic effect of the anti-coagulant antithrombin-III (AT-III), a protease inhibitor of thrombin, has been evaluated in dimethylnitrosamine (DMN) and CCl4-induced rat liver damage.17 Upon treatment with AT-III, DMN-intoxicated rats benefited while CCl4-treated rats showed no improvement, thereby suggesting that IC may complicate certain types of acute liver injury and contribute to its aggravation.17 In addition, pretreatment with heparin decreased acetaminophen-induced liver injury in mice.18 Anti-coagulant treatment of human ALF was also reported in a few patients. For example, nine patients with hemorrhagic diathesis due to acute hepatic necrosis were treated with heparin and no one survived,19 while three patients with ALF and one with severe relapse of viral hepatitis accompanied by IC were treated with heparin and fresh frozen plasma and survived.19 Therefore, the efficacy of treatment with heparin in the context of apoptotic liver injury remains unclear although the major concern is an increased risk of bleeding.9
In this study, we initially sought to identify unique proteins that are relatively insoluble and are associated with FasL-associated liver apoptosis in mice. This led us to identify FIB-γ dimers and other FIB-related high molecular species as among the major insoluble proteins in the liver after FasL administration. Based on this finding, we hypothesized that pretreatment of mice with heparin before administering FasL, or treatment of mice with heparin after FasL lead to a protective effect. Our findings provide support for this hypothesis and raise the possibility that targeted anticoagulation may have a beneficial effect in some forms of ALF.
EXPERIMENTAL PROCEDURES
Toxin and heparin administration, liver tissue and blood analysis
FasL (Jo2 clone, BD Pharmingen) was injected intraperitoneally into age/sex-matched (10–12 week/female) FVB mice (0.15μg/g) to induce liver apoptosis. Heparin (AAP Pharmaceuticals) was administered (dorsal midline, level of scapula) by subcutaneous injection (20USP units/mouse, with mice weighing ~25 grams). FVB/N female mice were sacrificed by CO2 inhalation at the indicated time points, or survival time was monitored for the lethality experiments. Livers were isolated and divided into pieces that were either stored in liquid nitrogen for biochemical analysis or fixed in 10% formalin for hematoxylin and eosin (H&E) staining and histological analysis. Blood samples were collected from the sacrificed mice by intracardiac puncture and stored (4 oC overnight) prior to analysis.
Serum alanine aminotransferase (ALT) levels were determined using Vetscan-vs2 (ABAXIS) employing the comprehensive diagnostic profile. Serum fibrinogen levels were determined employing a mouse fibrinogen ELISA kit (GenWay).
Sample preparation and Biochemical analysis
High salt extraction (HSE) was performed as previously described.20 Supernatants (TX100 and high salt fractions) were saved where necessary and used after mixing with 2x SDS sample buffer. Total liver lysates (TLL) were prepared from liver tissues by homogenization using 2x SDS sample buffer. Serum, plasma and clot fractions were also mixed (serum and plasma) or homogenized (clot) using 2x SDS sample buffer. Proteins were separated with SDS-PAGE, then stained with Coomassie blue or transferred to polyvinylidene fluoride membranes followed by blotting with antibodies to fibrinogen-γ (Abcam), tubulin and actin (Neomarkers), active caspases-3 or 7 (Cell Signaling), tissue factor and plasminogen activator inhibitor-1 (R&D systems), and an antibody to keratin polypeptide 18 (K18) p29 apoptotic fragment.21
Determination of percent apoptotic cells and extent of hemorrhage
Apoptotic cells were detected using a TUNEL assay kit (Roche). The percentage of apoptotic cells in the liver was determined by counting the total number of nucleated cells (DAPI-stained) and the number of apoptotic cells (TUNEL-stained) in five random high power fields. The extent of hemorrhage replacing normal architecture was scored qualitatively by a pathologist (D.S.M.) in a blinded fashion as follows: no hemorrhage (0), 1–10% hemorrhage (1=mild), 11–20% (2=mild-moderate), 20–30% (3=moderate), 30–40% (4=moderate-severe), and >40% (5=severe). The scores were then used for statistical analysis.
Statistical analysis
All statistical analyses were performed employing one-way ANOVA or Log-rank (Mantel-Cox) Test (for the lethality experiments) using GraphPad Prism 5 statistical software.
Mass spectrometry and N-terminal sequencing
Mass spectrometry was carried out using standard protocols by the Michigan Proteome Consortium (University of Michigan). N-terminal sequencing was done by the Molecular Structure Facility (University of California, Davis).
RESULTS
Comparison of the liver insoluble protein fractions before and after Fas-mediated apoptosis
We used intraperitoneal injection of the FasL (Jo2) which is known to induce significant liver injury manifested by apoptosis and intrahepatic hemorrhage (Fig. 1A).6,7 We compared the insoluble fractions of livers obtained from control and FasL-injected mice using HSE that removes nonionic-detergent-soluble and high-salt-buffer-soluble proteins. Notably, 3 major proteins become clearly prominent in the livers of the FasL-treated mice (Fig. 1B, bands 1–3). Proteolysis followed by mass spectrometry identified bands 1–3 as fibrinogen-α/γ, fibrinogen-γ and actin, respectively. For band 2, the peptides that were predicted by mass spectrometry are displayed in bold lettering in Fig. 2A. N-terminal sequencing of band 2 identified 5 amino acids (Fig. 2A) of FIB-γ which indicates that band 2 (100-kDa) is a cleaved dimer of FIB-γ. It is already known that FIB-γ undergoes cleavage and dimerization during the coagulation cascade.14,22
Figure 1. Characterization of the insoluble liver proteins after FasL-induced apoptosis.

(A) FasL was administered to mice. After 4.5h, mice were sacrificed followed by H&E staining. Note the dramatic hemorrhage (arrows) in FasL-treated compared to untreated mice. (B) Insoluble protein fractions of livers from FasL-treated and untreated mice were isolated using HSE followed by SDS-PAGE and Coomassie staining. Bands that showed increased intensity after FasL-treatment (circles 1,2,3) were subjected to mass spectrometric analysis which, in turn, predicted the identity of bands 1–3 as: fibrinogen α/γ(1), fibrinogen-γ(2) and actin (3).
Figure 2. FIB-γ is the major insoluble liver protein after FasL-mediated injury.

(A) Fragment sequences of FIB-γ obtained from N-terminal sequencing (underlined) and mass spectrometry (bold) confirm the identity of band #2 shown in Fig. 1B. (B) Prediction of mass spectrometric results was verified by immunoblotting using antibodies to FIB-γ and actin. The FIB-γ antibody recognized several bands in the FasL-treated liver (250-kDa and 100-kDa) that were not present in the untreated controls. These species correspond to bands 1 and 2 (Fig. 1B), respectively. The actin blot demonstrated elevated levels of actin protein in FasL-treated liver compared to untreated control. Formation of the K18 apoptotic fragment confirmed the hepatocyte FasL-induced apoptosis.
To confirm the findings predicted by mass spectrometry, we used immunoblotting with antibodies specific to FIB-γ and actin. Consistent with the mass spectrometric and N-terminal sequence analysis, the anti-FIB-γ antibody recognized several protein species [including ~250-kDa and 100-kDa (Fig. 2B)] exclusively in livers of FasL-treated mice. The 250-kDa and 100-kDa species (Fig. 2B) correspond to bands 1 and 2 in Fig. 1B, respectively. As expected based on the predicted identity of band 3 (Fig. 1B), the actin blot demonstrated elevated levels of insoluble actin in FasL-treated livers as compared to untreated control (Fig. 2B). Hepatocyte apoptosis after FasL was also confirmed biochemically by immunoblotting using an antibody that recognized cleaved K18 after caspase digestion (Fig. 2B).
The shift in solubility of FIB-γ upon induction of apoptosis was also tested in individual fractions of liver homogenates from mice with or without exposure to FasL. Barely detectable levels of FIB-γ monomer (~50-kDa) are present in the livers under basal conditions (Fig. 3). In contrast, the FIB-γ blot of livers undergoing hepatocyte apoptosis showed two major bands (100-kDa and 250-kDa) that are present only in the total liver homogenate and are markedly enriched in the pellet fraction but not in the soluble TX100 or high salt fractions (Fig. 3). Taken together, these findings indicate that FIB-γ dimerizes and becomes insoluble upon FasL-mediated liver injury.
Figure 3. Solubility dynamics of FIB-γ during FasL-induced liver injury.

Solubility dynamics of FIB-γ during FasL-induced liver injury were analyzed biochemically by comparing the soluble TX100 fraction, high salt wash fraction (Salt) and the insoluble fraction (Pellet) from FasL-treated and untreated livers. TLL (Total) were used as controls. None of the fractions showed detectable levels of FIB-γ in untreated liver samples. However, FIB-γ antibody faintly recognized the FIB-γ monomer (50-kDa) in the TLL and the soluble TX100 fraction of untreated liver. In contrast, FIB-γ blot showed two major bands (100-kDa, 250-kDa) in FasL-treated TLL, but not in the soluble TX100 and wash fractions. A duplicate gel to that used for immunoblotting was stained with Coomassie blue. Athough the protein amounts in some of the lanes (3,4,7,8) were not enough to be visible with Coomassie staining, they were readily detected by the FIB-γ antibody (lane 8).
FIB-γ partitions from plasma to the liver during FasL-induced liver injury
The above findings led us to hypothesize that FIB-γ shifts it location from plasma to the liver upon apoptotic injury. We tested this hypothesis by comparing serum, plasma and liver FIB-γ levels before and after exposure to FasL. The FIB-γ 100-kDa dimer was detected in FasL-treated mouse serum but not in plasma, while this dimer and other high molecular weight (HMW) products were readily observed in the liver lysates (Fig. 4A). A separate analysis that compares the FIB-γ dimer and higher complexes in the clot from whole blood versus the intact liver (boxed panels in Fig. 4B) shows a clear and marked shift from the clot to the liver. Therefore, the FIB-γ dimer and its HMW complexes accumulate in the liver after FasL-mediated liver injury, which is consistent with intrahepatic IC. The intrahepatic IC is also supported by the increased levels of liver plasminogen activator inhibitor-1 in plasma and liver upon FasL-mediated liver injury, with concurrent increase in tissue factor levels in plasma (Fig. S1).
Figure 4. Intrahepatic accumulation of FIB-γ during FasL-induced injury.

Mice were sacrificed 4.5h after FasL-injection and blood components (serum, plasma, clot) and liver were collected. (A) FIB-γ antibody recognized the FIB-γ dimer (100-kDa) in FasL-treated mouse serum but not in FasL-untreated serum. FIB-γ dimers and other HMW products (~250-kDa) were readily observed in FasL-treated liver lysates. (B) FIB-γ dimers and other HMW products were clearly observed by immunoblotting in the clot fraction of untreated mice but not in the liver, while the opposite occurs after FasL treatment. Coomassie staining of duplicate gels to those used for blotting is included as loading control.
Prophylactic treatment with heparin reduces the extent of FasL-induced apoptotic liver injury
The extensive intravascular coagulation within liver parenchyma after FasL-induced hepatocyte apoptosis raised the hypothesis that anticoagulation using heparin may provide a protective effect. For this, we first defined a time period whereby heparin is administered and maintains its anticoagulant effect prior to injecting FasL. Using a dose range of 10–100 Units/mouse, we found that 20U/mouse provided anticoagulation that is similar to the higher tested doses (based on elevation of plasma fibrinogen levels without leading to significant hematoma formation, not shown). Based on this dosing regimen, heparin was administered subcutaneously followed 4h later by FasL administration. The extent of injury in these mice was then compared to mice given FasL alone (Fig. 5). Histological analysis of the livers showed a dramatic decrease in the extent of hemorrhage in mice that were given heparin (Fig. 5A-top, Fig. S2). Heparin pretreatment also resulted in a dramatic decrease in TUNEL staining (Fig. 5A). These findings were supported by significantly reduced serum ALT levels (4.8-fold) and lower liver apoptotic cell number in the heparin pretreated mice (Fig. 5B,C). In addition, biochemical analysis showed that the activation of caspases-3 and 7, and formation of the K18 apoptotic fragment were markedly blunted in mice that received heparin (Fig. 5D). These biochemical changes also paralleled the detection of the FIB-γ dimer (Fig. 5D). As expected, mice that received heparin showed elevated levels of serum fibrinogen (Fig. S3), thereby confirming its anticoagulative effect. Importantly, heparin-treated mice survived the FasL-induced liver injury longer compared to heparin-untreated mice (Fig. 5E). Taken together, these data indicate that prophylactic pre-treatment with heparin reduces the extent of FasL-induced apoptotic liver injury in FVB/N mice.
Figure 5. Pretreatment with heparin reduces FasL-induced liver injury.
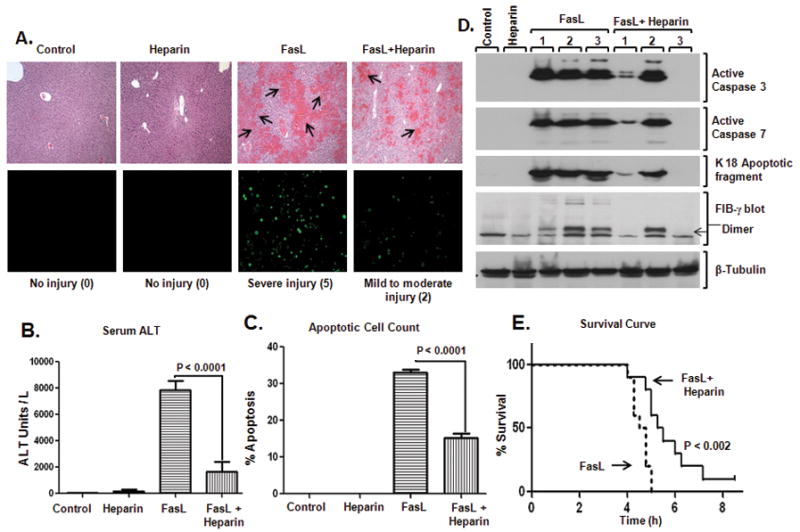
(A) Heparin was administered to age/sex-matched mice followed by FasL injection (4h after heparin), and the extent of liver damage was assessed. H&E sections show hemorrhage formation (top) and TUNEL staining shows apoptotic cells (bottom). Three mice were used for each of the control and heparin-alone arms, while 7 mice were used for each of the FasL and FasL+heparin arms. Numbers in parenthesis reflect the injury score (see Experimental Procedures and Fig. S2). (B) Serum ALT, (C) % Apoptotic cell count, and (D) Biochemical analysis of the livers from the indicated treatments and controls are shown. Note that all measured parameters are significantly less in mice that received heparin prior to FasL. In panel D, each lane represents an independent liver, and tubulin is shown as a loading control. (E) Heparin or saline were administered to mice (10–12 week-old; n=10/group) followed by FasL-injection (4h after heparin/saline) and survival time (hours) was recorded.
Therapeutic benefit of heparin in FasL-induced liver injury
Most cases of ALF occur in the context of an unanticipated exposure to an insult.23,24 Therefore, it is important to identify potential compounds that can be used as a treatment albeit prophylactic drugs do have a role. Given the significant benefit imparted by heparin when administered before the FasL insult, we examined its effect as a therapeutic. In a preliminary experiment, we verified the rapid onset heparin action to be as early as 15 minutes after subcutaneous injection (Fig. S4). For the treatment experiment, mice were first given FasL then given heparin 1h, 2h, 2.5h and 3h after FasL-injection. At 4.5h (i.e., the same time point used for the experiments in Fig. 1 and Fig. 5) after FasL injection, mice were sacrificed to evaluate the extent of injury using histological, serological and biochemical means. Notably, treatment with heparin 1h and even 2h following FasL administration significantly reduced hemorrhage as compared to heparin untreated mice (Fig. 6A, Fig. S5). Serum ALT levels were significantly lower (7.3-fold) in mice that received heparin treatment after 1h following FasL-injection but not at subsequent times (Fig. 6B). Quantification of the apoptotic cells showed a protective effect when heparin was given 1h or 2h after FasL (Fig. 6C), which is paralleled by findings using TUNEL staining (Fig. 6A). Similarly, the levels of activated caspases-3/7, and formation of the K18 apoptotic fragment were decreased, particularly at the 1h time point (Fig. 6D). Therefore, early treatment with heparin significantly reduces FasL-induced mouse liver injury.
Figure 6. Early treatment with heparin reduces FasL-induced liver injury.
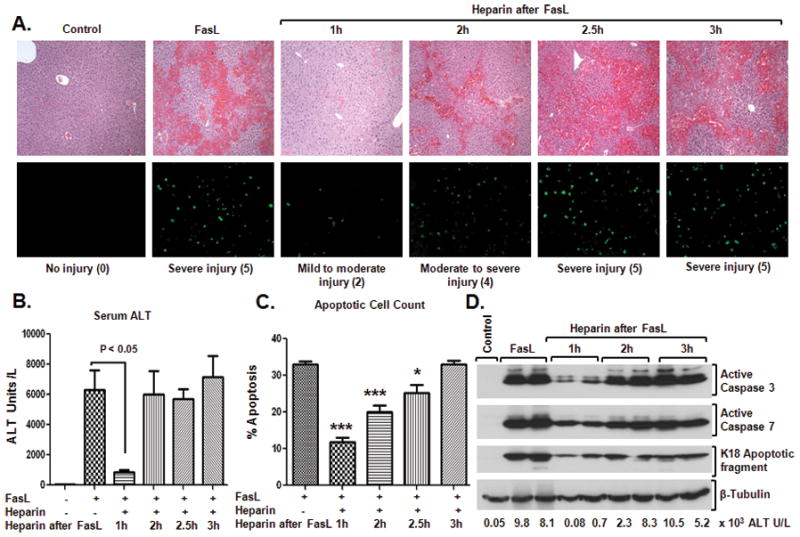
FasL was administered intraperitoneally followed by heparin injection 1h, 2h, 2.5h and 3h after FasL (5–7 mice/time point). After 4.5h of FasL injection, mice were sacrificed and liver damage was assessed. (A) H&E staining (top) and TUNEL staining (bottom) showed that treatment with heparin at 1h and 2h after FasL injection significantly reduced hemorrhage formation and apoptosis, indicative of only mild-to-moderate injury, as compared to heparin-untreated mice. Numbers in parenthesis reflect the injury score (see Experimental Procedures and Fig. S5). (B) and (C) show serum ALT levels and percentage of apoptotic cells, respectively. (D) Activation of caspase-3 and 7 is markedly reduced in mice that received heparin 1h following FasL-injection. Consistent with the decreased caspase activation, lower levels of the K18 apoptotic fragment were detected in livers of mice that received heparin treatment 1h following FasL injection as compared with mice that received FasL-alone. Serum ALT levels for each of the biochemically analyzed livers of each animal are shown. Tubulin blot is included as a loading control.
Relationship of apoptosis to intravascular coagulation within the liver
We addressed the time course of apoptosis progression versus IC within the liver. Administration of FasL followed by analysis of the livers at hourly intervals demonstrated that the readily detectable activation of caspases and keratin cleavage during apoptosis occur concurrently with FIB-γ dimer formation (Fig. 7). Notably, FIB-γ dimer formation shows a sharp rise then remains relatively constant as injury progresses while caspase activation and keratin fragmentation also display a sharp rise but continue to increase with time (compare lanes 6 and 7 with 8 and 9). An independent experiment using analysis at 0.5 hour intervals showed similar findings (Fig. S6).
Figure 7. Time course of caspase activation, K18 caspase-mediated digestion and FIB-γ dimer formation after FasL-induced injury.

Liver apoptosis was induced by FasL-injection. Mice were sacrificed at 1h, 2h, 3h and 4h after FasL-injection (2 mice/time point), followed by preparation of the total liver lysates and HSE then immunoblotting using antibodies to the indicated antigens. A tubulin blot is included as a loading control, and a Coomassie stain of a duplicate gel of the analyzed HSE samples that were analyzed by blotting is also included.
DISCUSSION
Our findings provide a model for FIB-γ dynamics during mouse liver injury (Fig. 8). Upon apoptotic liver injury, plasma fibrinogen moves from plasma and is deposited within liver parenchyma as part of an intrahepatic IC that is triggered by the apoptotic cell injury. This process is accompanied by FIB-γ cleavage, dimerization and formation of higher molecular weight species that are detected exclusively in the liver upon FasL-induced liver injury (Fig. 4). A similar behavior of FIB-γ proteolysis and solubility dynamics also occurs in acetaminophen-mediated acute liver injury (Fig. S7). In contrast, under basal conditions only small amounts of fibrinogen are present in the liver, where it is synthesized in both rodents and humans then secreted into the circulation.25 The significance of the FIB-γ dimer as a potential biomarker has been reported in cancer patients.15,16 However, to our knowledge, biochemical FIB-γ changes in the context of ALF have not been previously reported although fibrin deposition in mouse liver has been observed after acetaminophen-mediated acute injury.18 Notably, blood clots from mice undergoing apoptosis manifest a dramatic decrease in their 100-kDa FIB-γ levels (Fig. 4B; compare lanes 2,5). Further studies will be needed to determine whether loss of FIB-γ dimers (or other fibrinogen isoforms) in the clot of patients with ALF will serve as a potential useful marker of intrahepatic-IC and disease severity.
Figure 8. Model of intrahepatic vascular coagulation and epithelial cell apoptosis dynamics in response to FasL.
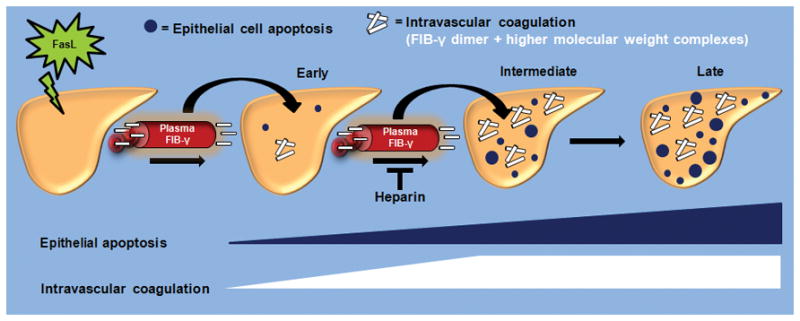
The early stages of FasL-induced liver injury are manifested by limited hepatocytes apoptosis and intrahepatic vascular coagulation. This is followed at the intermediate stages by a near-concurrent marked increase in liver caspase activation, epithelial apoptosis and intravascular coagulation. The IC is manifested by the formation of FIB-γ dimers and HMW products within the liver, in association with a dramatic shift of FIB-γ from plasma to liver. During the late stages that follow FasL exposure, IC remains relatively unchanged while hepatocytes apoptosis continues. Heparin provides a therapeutic modality that not only limits the extensive of FasL-related liver injury when given as prophylactic but also limits the extent of injury if given at the early stages of injury exposure.
The approach that we used to arrive at the importance of the hemostasis pathway during ALF utilized a limited proteomic analysis aimed at the characterization of insoluble proteins that accumulate as a consequence of FasL-induced liver injury. Given the relative short time from exposure to FasL to havesting of the livers (4–5h), we predicted that any new protein species that either appear or disappear after FasL exposure are likely related to posttranslational modification of resident proteins or to proteins derived from infiltrating cells. The observed increase in actin (Fig. 1) is likely due to actin that is derived from infiltrating erythrocytes that accompany the observed hemorrhage, although we cannot exclude the possibility of a posttranslational modification of actin that renders it insoluble.
As a result of IC, fibrin thrombi including the FIB-γ dimer and its cleaved higher mass complexes accumulate in the liver thereby altering normal blood flow. The consequent decreased blood flow to hepatocytes likely results in accumulation of reactive oxygen species and nitrogenous waste products in liver, thereby perpetuating the extent of liver injury. Therefore, heparin is predicted to act by disrupting the injury cycle as injury moves from an early to an intermediate stage (Fig. 8) and preventing the deposition of fibrin thrombi including the FIB-γ dimer and its complexes, and facilitating adequate blood supply to liver parenchymal cells. Heparin does not appear to directly inhibit FasL-mediated apoptosis since heparin pre-treatment of isolated mouse hepatocytes ex vivo did not alter the extent of FasL-induced caspase activation and K18 degradation (Fig. S8). We are not able to completely separate whether Fas and intrahepatic coagulation act simultaneously or sequentially, but it appears that once significant coagulation occurs it does not continue to increase while caspase activation and apoptosis do continue to increase (Fig. 7 and Fig. S6). This suggests that there may be a second wave of apoptosis/necrosis that is inhibited by heparin. In support of this notion, assessment of early time points after heparin pretreatment then FasL injection showed that heparin decreases caspase-3 activation, K18 caspase-mediated digestion and formation of FIB-γ dimers (Fig. S9). One important caveat is that heparin pretreatment delays, but does not prevent animal mortality (Fig. 5E). This indicates that Fas-FasL interaction continues to occur despite the presence of heparin.
Another important finding herein is the beneficial effect of heparin, not only to provide prophylaxis towards apoptosis-associated liver injury but also to treat the injury which is an important distinction in terms of potential therapeutic utility. Heparin use was described previously to be effective in providing protection when given before administering acetaminophen, but was not tested for its effect after induction of liver injury.18 In humans, therapy for ALF is primarily supportive unless liver transplantation is available or deemed required,24,26 though interventions such as the administration of N-acetylcysteine in non-acetaminophen-related ALF may be beneficial.27 Currently anticoagulation is not used in human ALF because of the risk of increased bleeding, especially in the context of invasive procedures.28,29 However, it appears that patients with ALF have complex hyper- and hypo-coagulable states,9,30 and patients undergoing liver transplantation with international normalized ratio values of >1.5 did well without plasma or red blood cell transfusions.31 The complex anti-fibrinolytic and pro-fibrinolytic milieu in patients with ALF suggests that a targeted anticoagulation approach that is patient and disease specific may be beneficial.
The dose of heparin that we used in the mice is predicted to be lower than what is currently used in patients who undergo treatment for deep venous thrombosis or other complications related to a hypercoagulable state. For example, the 20 USP units per 25 grams of mouse weight can be converted to a predicted human dose of ~5000 USP units, based on the recommended body surface area conversion,32 which is lower than the typical 10,000 or more USP bolus dosing that is used in adult humans before initiating continuous infusion.33 Our study provides a proof-of-principle approach that anticoagulation is effective in ameliorating FasL-induced liver injury. The use of the minimum effective dosing is of obvious importance in order to minimize bleeding complications. For example, when we used doses of 50–100 USP units/mouse, hematomas of variable sizes were frequently noted proximal to the site of injection (not shown). The use of other fibrinolytic approaches (e.g., low molecular weight heparin33) may also prove beneficial.
Supplementary Material
Intravascular coagulation observed in FasL-induced ALF was assessed by the expression of tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) in total liver lysates (A) and plasma (B). Elevated levels of TF and PAI-1 in the context of liver injury have been well documented18 [Mackman N. Blood Cells Molecules and Diseases 2006:36;104–107]. Nearly equal TF levels were observed in the livers of FasL-treated and untreated mice. However, plasma TF was detected only in FasL-treated liver. Higher PAI-1 total liver lysate and plasma levels were observed in FasL-treated mice as compared to untreated mice. The Coomassie stain of a duplicate gel is shown as a loading control.
The extent of hemorrhage was calculated as described in Experimental Procedures, with the hemorrhage score indicated in parentheses. The number of mice used for each experimental are is shown. Note that pretreatment with heparin significantly reduces hemorrhage formation after FasL-induced apoptotic liver injury.
To confirm that heparin is having a biological effect under the experimental conditions, total serum fibrinogen levels were measured for the indicated treatments as described in Experimental Procedures. The mean fold change in serum fibrinogen levels with standard error (error bars) for each group are displayed (Heparin, 3 mice; FasL, 6 mice; FasL+Heparin, 6 mice). The FasL-treated mice exhibit markedly lower levels of serum fibrinogen, while heparin-treated mice show high fibrinogen levels thereby confirming the anticoagulation effect of heparin.
Mice were injected with heparin then sacrificed after 15, 30 and 60 min. Total fibrinogen levels were measured as described in Experimental Procedures. The mean serum fibrinogen levels (ng/mL) with standard error (error bars) for each time point is shown (3 mice per experimental time point). Note that heparin is effective as an anticoagulant as early as 15 min after administration.
The extent of hemorrhage was calculated as described in Experimental Procedures. Significant reduction of the mean hemorrhage score (indicated in parentheses) was observed when heparin was administered 1h after FasL-injection.
This is an identical experiment to that shown in Fig. 7 except that the time course is more refined. Liver apoptosis in age/sex-matched FVB mice was induced by FasL-injection (2 mice/time point), then mice were euthanized after 1.5h, 2h, 2.5h and 3h. Liver tissues were analyzed by immunoblotting of total liver lysates and HSE as described in Fig. 7 legend. Note that in this experiment we are able to detect limited caspase activation, K18 fragmentation and formation of FIB-γ dimers at the early time points (as early as 1.5 h after FasL injection). However, as shown in Fig. 7, there is a dramatic increase in all the parameters at the 2.5 h time point with caspase activation and K18 caspase digestion continuing to increase at the 3 h time point while FIB-γ dimer formation remains relatively unchanged.
Age- and sex-matched FVB/N mice were fasted overnight followed by intraperitonial injection of APAP (700 mg/kg). Three mice were injected with APAP while one mouse was used as a control. After 6h, animals were sacrificed followed by biochemical and serologic analysis of high salt extracts that were prepared from the liver tissues. (A) The FIB-γ antibody recognized several protein species in the APAP-treated mice (in 2 of 3 mice that manifested elevated serum ALT levels; ALT levels are shown at the bottom of the panel) that were not present in the untreated controls. (B) The solubility dynamics of FIB-γ after APAP-induced liver injury were analyzed biochemically by comparing the soluble TX100 fraction and the insoluble fraction (Pellet) from APAP-treated and untreated livers. None of the fractions had detectable FIB-γ dimers in the untreated liver fractions (TX100 or pellet). In contrast, FIB-γ dimers and HMW complexes were observed in the APAP-treated insoluble pellet fractions but not in the soluble TX100 fractions of livers from APAP-treated mice. Duplicate gels to those used for immuniblotting in panels A and B were stained with Coomassie blue.
To test whether heparin directly interferes with Fas activity, primary hepatocytes were isolated from FVB mice then cultured in the absence or presence of heparin (10 units/ml or 50 units/ml) for 4h followed by the addition of FasL (0.5 μg/ml). After 8h in the presence of FasL, the hepatocytes were solubilized in sample buffer followed by analysis of the lysates by immunoblotting using antibodies to the indicated antigens. Notably, heparin treatment did not prevent the FasL-mediated damage as evidenced by the similar levels of activated caspase-3 and -7 and the similar formation of the K18 apoptotic fragment. As previously reported (W. Steven et al., Biochemical Pharmacology 1984;33:2223–2226) both doses of heparin-alone displayed some hepatotoxicity as evident by K18 degradation (lanes 3 and 5) which provides some evidence that heparin is effective under our culture conditions.
Heparin was administered to 10–12 weeks old female FVB mice followed by FasL-injection (4h after heparin), and the extent of liver damage was assessed biochemically at the indicated time points. Caspase-3 and 7 activations were detectable after 2.5 hours of FasL administration in one of 2 of both heparin-untreated and treated mice. Both caspase-3 activation and K18 apoptotic fragment formation were less in the heparin treated mice (2.5 h time point, compare lanes 10 with 7), thereby suggesting that even in the presence of heparin, Fas-FasL interaction still occurs and induces hepatocyte apoptosis but to a lesser extent. The less caspase-3 activation and less K18 fragmentation observed in heparin pre-treated mouse compared to heparin-untreated mouse at 2.5 h of FasL suggest that heparin prevents the second wave of injury by preventing Fibrinogen-γ accumulation in liver (no fibrinogen-γ accumulation seen in heparin treated mouse). Each lane represents an independent liver. The Coomassie stain of a duplicate gel is shown as a loading control.
Acknowledgments
This work was supported by NIH grant DK47918 and the Department of Veterans Affairs (M.B.O.). We thank Dr. Pavel Strnad for valuable comments.
Abbreviations
- ALF
acute liver failure
- ALT
alanine aminotransferase
- AT-III
anti-thrombin-III
- DMN
dimethylnitrosamine
- FasL
Fas ligand
- FIB-γ
fibrinogen-γ
- H&E
hematoxylin and eosin
- HMW
high molecular weight
- HSE
high salt extraction
- IC
intravascular coagulation
- K18
keratin polypeptide 18
- PAGE
polyacrylamide gel electrophoresis
- TLL
total liver lysates
- TNF-α
tumor necrosis factor-α
- TX-100
Triton X-100
References
- 1.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 2.Malhi H, Guicciardi ME, Gores JG. Hepatocyte Death: A Clear and Present Danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ku NO, Liao J, Omary MB. Apoptosis generates stable fragments of human type I keratins. J Biol Chem. 1997;272:33197–33203. doi: 10.1074/jbc.272.52.33197. [DOI] [PubMed] [Google Scholar]
- 4.Caulin C, Salvesen GS, Oshima RG. Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J Cell Biol. 1997;138:1379–1394. doi: 10.1083/jcb.138.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S. Essential roles of the Fas ligand in the development of hepatitis. Nat Med. 1997;3:409–413. doi: 10.1038/nm0497-409. [DOI] [PubMed] [Google Scholar]
- 7.Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–1230. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guicciardi ME. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lisman T, Porte RJ. Activation and regulation of hemostasis in acute liver failure and acute pancreatitis. Semin Thromb Hemost. 2010;36:437–443. doi: 10.1055/s-0030-1254052. [DOI] [PubMed] [Google Scholar]
- 10.Mosesson MW, Homandberg GA, Amrani DL. Human platelet fibrinogen gamma chain structure. Blood. 1984;63:990–995. [PubMed] [Google Scholar]
- 11.Drury DR, McMaster PD. The liver as a source of fibrinogen. J Exp Med. 1929;50:569. doi: 10.1084/jem.50.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosesson MW. Fibrinogen gamma chain functions. J Thromb Haemost. 2003;1:231– 238. doi: 10.1046/j.1538-7836.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 13.Farrel DH. Pathophysiologic roles of the fibrinogen gamma chain. Curr Opin Hematol. 2004;11:151–155. doi: 10.1097/01.moh.0000131440.02397.a4. [DOI] [PubMed] [Google Scholar]
- 14.Chen R, Doolittle RF. Cross-linking sites in human and bovine fibrin. Biochemistry. 1971;10:4487–4491. doi: 10.1021/bi00800a021. [DOI] [PubMed] [Google Scholar]
- 15.Vejda S, Posovszky C, Zelzer S, Peter B, Bayer E, Gelbmann D, Schulte-Hermann R, et al. Plasma from cancer patients featuring a characteristic protein composition mediates protection against apoptosis. Mol Cell Proteomics. 2002;1:387–393. doi: 10.1074/mcp.m200004-mcp200. [DOI] [PubMed] [Google Scholar]
- 16.Gerner C, Steinkellner W, Holzmann K, Gsur A, Grimm R, Ensinger C, Obrist P, et al. Elevated plasma levels of crosslinked fibrinogen gamma-chain dimer indicate cancer-related fibrin deposition and fibrinolysis. Thromb Haemost. 2001;85:494–501. [PubMed] [Google Scholar]
- 17.Fujiwara K, Ogata I, Ohta Y, Hirata K, Oka Y, Yamada S, Sato Y, et al. Intravascular coagulation in acute liver failure in rats and its treatment with antithrombin III. Gut. 1988;29:1103–1108. doi: 10.1136/gut.29.8.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganey PE, Luyendyk JP, Newport SW, Eagle TM, Maddox JF, Mackman N, Roth RA. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- 19.Rake MO, Shilkin KB, Winch J, Flute PT, Lewis ML, Williams R. Early and intensive therapy of intravascular coagulation in acute liver failure. The Lancet. 1971 doi: 10.1016/s0140-6736(71)90540-x. [DOI] [PubMed] [Google Scholar]
- 20.Ku NO, Toivola DM, Zhou Q, Tao GZ, Zhong B, Omary MB. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 2004;78:489–517. doi: 10.1016/s0091-679x(04)78017-6. [DOI] [PubMed] [Google Scholar]
- 21.Tao GZ, Li DH, Zhou Q, Toivola DM, Strnad P, Sandesara N, Cheung RC, et al. Monitoring of epithelial cell caspase activation via detection of durable keratin fragment formation. J Pathol. 2008;215:164–174. doi: 10.1002/path.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siebenlis KR, Mosesson MW. Progressive cross-linking of fibrin γ chains increases resistance to fibrinolysis. J Biol Chem. 1994;269:28414–28419. [PubMed] [Google Scholar]
- 23.Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142–152. doi: 10.1055/s-2008-1073114. [DOI] [PubMed] [Google Scholar]
- 24.Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190–201. doi: 10.1016/S0140-6736(10)60274-7. [DOI] [PubMed] [Google Scholar]
- 25.Tennent GA, Brennan SO, Stangou AJ, O’Grady J, Hawkins PN, Pepys MB. Human plasma fibrinogen is synthesized in the liver. Blood. 2007;109:1971–1974. doi: 10.1182/blood-2006-08-040956. [DOI] [PubMed] [Google Scholar]
- 26.Pathikonda M, Munoz SJ. Acute liver failure. Ann Hepatol. 2010;9:7–14. [PubMed] [Google Scholar]
- 27.Lee WM, Hynan LS, Rossaro L, Fontana RJ, Stravitz RT, Larson AM, Davern TJ, 2nd, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137:856–864. 864, 851. doi: 10.1053/j.gastro.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira SP, Langley PG, Williams R. The management of abnormalities of hemostasis in acute liver failure. Semin Liver Dis. 1996;16:403–414. doi: 10.1055/s-2007-1007253. [DOI] [PubMed] [Google Scholar]
- 29.Munoz SJ, Stravitz RT, Gabriel DA. Coagulopathy of acute liver failure. Clin Liver Dis. 2009;13:95–107. doi: 10.1016/j.cld.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Pernambuco JR, Langley PG, Hughes RD, Izumi S, Williams R. Activation of the fibrinolytic system in patients with fulminant liver failure. Hepatology. 1993;18:1350–1356. [PubMed] [Google Scholar]
- 31.Massicotte L, Beaulieu D, Thibeault L, Roy JD, Marleau D, Lapointe R, Roy A. Coagulation defects do not predict blood product requirements during liver transplantation. Transplantation. 2008;85:956–962. doi: 10.1097/TP.0b013e318168fcd4. [DOI] [PubMed] [Google Scholar]
- 32.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 33.Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest. 2004;126:188S–203S. doi: 10.1378/chest.126.3_suppl.188S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Intravascular coagulation observed in FasL-induced ALF was assessed by the expression of tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) in total liver lysates (A) and plasma (B). Elevated levels of TF and PAI-1 in the context of liver injury have been well documented18 [Mackman N. Blood Cells Molecules and Diseases 2006:36;104–107]. Nearly equal TF levels were observed in the livers of FasL-treated and untreated mice. However, plasma TF was detected only in FasL-treated liver. Higher PAI-1 total liver lysate and plasma levels were observed in FasL-treated mice as compared to untreated mice. The Coomassie stain of a duplicate gel is shown as a loading control.
The extent of hemorrhage was calculated as described in Experimental Procedures, with the hemorrhage score indicated in parentheses. The number of mice used for each experimental are is shown. Note that pretreatment with heparin significantly reduces hemorrhage formation after FasL-induced apoptotic liver injury.
To confirm that heparin is having a biological effect under the experimental conditions, total serum fibrinogen levels were measured for the indicated treatments as described in Experimental Procedures. The mean fold change in serum fibrinogen levels with standard error (error bars) for each group are displayed (Heparin, 3 mice; FasL, 6 mice; FasL+Heparin, 6 mice). The FasL-treated mice exhibit markedly lower levels of serum fibrinogen, while heparin-treated mice show high fibrinogen levels thereby confirming the anticoagulation effect of heparin.
Mice were injected with heparin then sacrificed after 15, 30 and 60 min. Total fibrinogen levels were measured as described in Experimental Procedures. The mean serum fibrinogen levels (ng/mL) with standard error (error bars) for each time point is shown (3 mice per experimental time point). Note that heparin is effective as an anticoagulant as early as 15 min after administration.
The extent of hemorrhage was calculated as described in Experimental Procedures. Significant reduction of the mean hemorrhage score (indicated in parentheses) was observed when heparin was administered 1h after FasL-injection.
This is an identical experiment to that shown in Fig. 7 except that the time course is more refined. Liver apoptosis in age/sex-matched FVB mice was induced by FasL-injection (2 mice/time point), then mice were euthanized after 1.5h, 2h, 2.5h and 3h. Liver tissues were analyzed by immunoblotting of total liver lysates and HSE as described in Fig. 7 legend. Note that in this experiment we are able to detect limited caspase activation, K18 fragmentation and formation of FIB-γ dimers at the early time points (as early as 1.5 h after FasL injection). However, as shown in Fig. 7, there is a dramatic increase in all the parameters at the 2.5 h time point with caspase activation and K18 caspase digestion continuing to increase at the 3 h time point while FIB-γ dimer formation remains relatively unchanged.
Age- and sex-matched FVB/N mice were fasted overnight followed by intraperitonial injection of APAP (700 mg/kg). Three mice were injected with APAP while one mouse was used as a control. After 6h, animals were sacrificed followed by biochemical and serologic analysis of high salt extracts that were prepared from the liver tissues. (A) The FIB-γ antibody recognized several protein species in the APAP-treated mice (in 2 of 3 mice that manifested elevated serum ALT levels; ALT levels are shown at the bottom of the panel) that were not present in the untreated controls. (B) The solubility dynamics of FIB-γ after APAP-induced liver injury were analyzed biochemically by comparing the soluble TX100 fraction and the insoluble fraction (Pellet) from APAP-treated and untreated livers. None of the fractions had detectable FIB-γ dimers in the untreated liver fractions (TX100 or pellet). In contrast, FIB-γ dimers and HMW complexes were observed in the APAP-treated insoluble pellet fractions but not in the soluble TX100 fractions of livers from APAP-treated mice. Duplicate gels to those used for immuniblotting in panels A and B were stained with Coomassie blue.
To test whether heparin directly interferes with Fas activity, primary hepatocytes were isolated from FVB mice then cultured in the absence or presence of heparin (10 units/ml or 50 units/ml) for 4h followed by the addition of FasL (0.5 μg/ml). After 8h in the presence of FasL, the hepatocytes were solubilized in sample buffer followed by analysis of the lysates by immunoblotting using antibodies to the indicated antigens. Notably, heparin treatment did not prevent the FasL-mediated damage as evidenced by the similar levels of activated caspase-3 and -7 and the similar formation of the K18 apoptotic fragment. As previously reported (W. Steven et al., Biochemical Pharmacology 1984;33:2223–2226) both doses of heparin-alone displayed some hepatotoxicity as evident by K18 degradation (lanes 3 and 5) which provides some evidence that heparin is effective under our culture conditions.
Heparin was administered to 10–12 weeks old female FVB mice followed by FasL-injection (4h after heparin), and the extent of liver damage was assessed biochemically at the indicated time points. Caspase-3 and 7 activations were detectable after 2.5 hours of FasL administration in one of 2 of both heparin-untreated and treated mice. Both caspase-3 activation and K18 apoptotic fragment formation were less in the heparin treated mice (2.5 h time point, compare lanes 10 with 7), thereby suggesting that even in the presence of heparin, Fas-FasL interaction still occurs and induces hepatocyte apoptosis but to a lesser extent. The less caspase-3 activation and less K18 fragmentation observed in heparin pre-treated mouse compared to heparin-untreated mouse at 2.5 h of FasL suggest that heparin prevents the second wave of injury by preventing Fibrinogen-γ accumulation in liver (no fibrinogen-γ accumulation seen in heparin treated mouse). Each lane represents an independent liver. The Coomassie stain of a duplicate gel is shown as a loading control.