

Abstract
A short, asymmetric synthesis of the 1,2,9,9a-tetrahydrocyclopropa[c]benzo[e]indol-4-one (CBI) analogue of the CC-1065 and duocarmycin DNA alkylation subunits is described. Treatment of iodo-epoxide 5, prepared by late-stage alkylation of 4 with (S)-glycidal-3-nosylate, with EtMgBr at room temperature directly provides the optically pure alcohol 6 in 87% yield (99% ee) derived from selective metal–halogen exchange and subsequent regioselective intramolecular 6-endo-tet cyclization. The use of MeMgBr or i-PrMgBr also provides the product in high yields (82–87%), but requires larger amounts of the Grignard reagent to effect metal–halogen exchange and cyclization. Direct transannular spirocyclization of 7 following O-debenzylation of 6 provides N-Boc-CBI. This approach represents the most efficient (9-steps, 31% overall) and effective (99% ee) route to the optically pure CBI alkylation subunit yet described.
Introduction
CC-1065 (1)1 and duocarmycin SA (2)2 represent the key members of a class of naturally occurring antitumor agents that also includes duocarmycin A3 and yatakemycin4 and that derive their biological activity from their ability to selectively alkylate duplex DNA (Figure 1).5,6 The study of the natural products, their synthetic unnatural enantiomers,7 derivatives, and key analogues has defined the fundamental features that control the DNA alkylation selectivity, efficiency, and catalysis, providing a detailed understanding of the relationships between structure, reactivity, and biological properties.6
Figure 1.

Natural products
Among the earliest and most studied of the natural product alkylation subunit analogues is CBI8 (Figure 2), which is not only synthetically more accessible, but it was found to possess a number of features that have made its use more attractive than the natural alkylation subunits themselves. Most significantly, its derivative analogues were found to be 4-fold more stable and 4-fold more potent than the corresponding analogues bearing the 7-MeCPI subunit found in CC-1065 and to display a substantially enhanced intrinsic reaction regioselectivity (>20:1 vs 4–6:1).9 Additionally, its derivative analogues were found to alkylate DNA with an unaltered sequence selectivity at enhanced rates and with a greater efficiency than the corresponding 7-MeCPI analogues.10 Although not quite as potent as the corresponding duocarmycin SA analogues, the ability to easily prepare and evaluate CBI analogues designed to probe fundamental questions on structure, reactivity, and activity have resulted in its extensive use.6 Thus, it is on this alkylation subunit analogue that new design concepts are often explored, developed, and evaluated.
Figure 2.

Approaches to the asymmetric synthesis of CBI
Its subsequent exploration by others and our continued examination of its properties have provided alternative11 and iteratively improved syntheses12–15 of CBI. Nonetheless, most studies detailing its preparation and use rely on a Chiralcel OD chromatographic resolution16 of a racemic precursor. Although several approaches to the asymmetric synthesis of CBI have been reported, none have replaced this or related chromatographic resolutions.17 The asymmetric approaches (Figure 2) include our own strategies relying on an asymmetric hydroboration (73%, 80% ee),18 a Jacobsen epoxidation (60%, 92% ee),18 and a lipase-catalyzed resolution of a prochiral diol (88%, 99% ee).19 Complementary to these efforts are Lown’s lipase-catalyzed resolution of 3,20 the use of a Sharpless AD reaction (30–60%, 70% ee),21 and Mohamadi’s application of an asymmetric hydroboration reaction (58%, 40% ee).22 The unusual and structure-dependent activity of the unnatural as well as natural enantiomers with this class of natural products requires the evaluation of enantiomers of unusually high optical purities.23 Thus, even small amounts of natural enantiomer contaminant (<1%) in the unnatural enantiomer samples can provide skewed biological results, requiring the use of a route or technique that delivers the agents in ≥99% ee. In the course of our recent development of a unique class of reductively activated CBI-based prodrugs,24,25 we have explored and herein report an effective approach to the asymmetric synthesis of CBI.
Recently, we described the asymmetric total synthesis of (+)- and ent-(−)-yatakemycin and (+)- and ent-(−)-duocarmycin SA through use of a selective metal–halogen exchange and subsequent intramolecular regioselective 6-endo-tet epoxide opening for the late-stage introduction of the chirality, simplifying access to either enantiomer.26 Thus, treatment of the aryl iodide with i-PrMgCl (1.1 equiv of i-PrMgCl, −42 °C, 1 h) followed by transmetalation to the cuprate (0.2 equiv of CuI–PBu3, −78 °C, 2 h) provided the desired alcohol in 69% yield and 99% ee (eq. 1). Herein we report the extension of this approach to the asymmetric synthesis of the CBI alkylation subunit, representing the most efficient (9-steps, 31% overall) and effective (99% ee) synthesis to date.
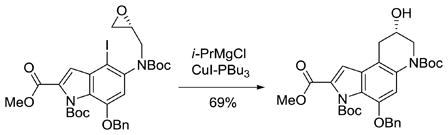 |
(1) |
Results and Discussion
Synthesis
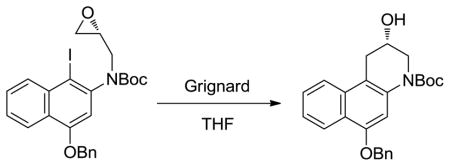
Alkylation of 4, prepared in 5-steps from 1,3-dihydroxynaphthalene,13 with recrystallized (S)-glycidal-3-nosylate27 (1.3 equiv, 1.5 equiv of NaH, DMF, 0 °C to 23 °C, 3 h, 90%) provided 5 with clean SN2 displacement of the nosylate versus epoxide opening and subsequent displacement of the nosylate (Scheme 1). Competitive epoxide opening would provide the enantiomeric epoxide, degrading the enantiomeric purity of the product. However, the product 5 is isolated in nearly perfect optical purity, confirming and establishing the regioselectivity of the alkylation. Initial attempts to afford the aryl Grignard by metal–halogen exchange with i-PrMgCl analogous to our efforts on yatakemycin only provided recovered starting material, even at elevated reaction temperatures (23 °C) and extended reaction times (72 h), presumably due to the use of a more electron-rich aryl iodide inherent in the naphthalene vs electron-deficient indole substrate. Thus, the more electron-rich naphthalene core slows the rate of metal–halogen exchange even with use of an aryl iodide, rendering most Grignard reagents ineffective. Subsequent efforts to overcome this problem by enlisting lithium–halogen exchange followed by transmetalation onto either a Grignard reagent or a cuprate did provide the desired alcohol 6. However, the best attainable yields were still low even after optimization (20–40%). These observations led to the examination of a range of more reactive Grignard reagents.28 Whereas such reagents including i-PrMgCl·LiCl, i-Pr2Mg·LiCl, and s-Bu2Mg·LiCl provided substantial amounts of side products arising from alkyl or chloride addition to the epoxide, the treatment of 5 with MeMgBr, EtMgBr, or i-PrMgBr provided 6 directly in high yields (82–87%), precluding the need for transmetalation onto copper. Formation of the aryl Grignard by metal–halogen exchange (2.0 equiv of EtMgBr, 23 °C, 30 min) was followed by the rapid intramolecular ring opening to give exclusively 6, the result of intramolecular 6-endo versus 5-exo addition to the epoxide. MeMgBr and i-PrMgBr also provide 6 in high yields (82–87%), albeit requiring larger amounts of the Grignard reagent (10 equiv). Representative studies defining reaction conditions and the number of equivalents of each reagent necessary to optimize yields are summarized in Table 1. Hydrogenolysis removal of the benzyl ether (10% Pd/C, HCO2NH4, 9:1 THF:MeOH) provided 7, and direct transannular Ar-3′ spirocyclization upon Mitsunobu activation of the secondary alcohol (3.0 equiv of 1,1′-(azodicarbonyl)dipiperidine (ADDP), 3.0 equiv of Bu3P, toluene, 23 °C, 1 h, 71%) provided N-Boc-CBI (8), which proved identical in all respects to authentic material. Confirmation of the absolute configuration was made by comparison ([α]D, HPLC Chiralcel OD, tR) with authentic material for which the stereochemical assignment was previously established by X-ray of a heavy atom derivative.9b The optical purity of 8 was determined by chiral phase HPLC (Chiralcel OD column, 7 mL/min, 10% i-PrOH/hexane) and established to be 99% ee (see Figure 3, Supporting Information).
Scheme 1.

Table 1.
Optimization of Grignard formation and cyclization
 | |||
|---|---|---|---|
| equiv | time | result | |
| EtMgBr | 1.0 | 120 min | 33% |
| 2.0 | 30min | 87% | |
| 4.0 | 30 min | 84% | |
| 6.0 | 30 min | 80% | |
| 10.0 | 10 min | 83% | |
| MeMgBr | 1.0 | 24 h | <1% |
| 2.0 | 24 h | 7% | |
| 3.0 | 24 h | 30% | |
| 4.0 | 24 h | 38% | |
| 10.0 | 30 min | 87% | |
| i-PrMgBr | 1.0 | 120 min | <1% |
| 2.0 | 120 min | 26% | |
| 3.0 | 120 min | 41% | |
| 4.0 | 120 min | 67% | |
| 10.0 | 10 min | 82% | |
Additionally, 8 can be converted to N-Boc-seco-CBI (9), resulting from a stereoelectronically-controlled regioselective cyclopropane cleavage, in near quantitative yield by treatment with 4 N HCl in EtOAc at −78 °C for 30 min, followed by solvent removal under a stream of nitrogen while at −78 °C (Scheme 2). A procedure for the direct attachment of the DNA binding subunits onto 8 was also developed. This was accomplished by treatment of 8 with 4 N HCl in EtOAc at −78 °C for 30 min, followed by warming to room temperature and stirring for 30 min, resulting in regioselective cleavage of the cyclopropane and subsequent N-Boc deprotection. Following solvent removal under a stream of nitrogen, the residue was treated with indole2-CO2H and EDCI in DMF and stirred for 16 h, providing seco-CBI-indole2 (10) in good yield (68%).
Scheme 2.

Conclusions
An effective (9-step, 31% overall) asymmetric synthesis of N-Boc-CBI was developed that provides the optically pure (99% ee) material. The key late stage step introducing the chirality proceeds in excellent yields (82–87%). Thus, treatment of key intermediate 5 with EtMgBr, MeMgBr, or i-PrMgBr afforded the desired secondary alcohol 6 directly in superb chemical yield and regioselectivity. This route should prove useful in the future preparation and evaluation of CC-1065 and duocarmycin analogues.
Experimental Section
(R)-tert-Butyl (4-(Benzyloxy)-1-iodonaphthalen-2-yl)(oxiran-2-ylmethyl)carbamate (5)
A solution of 4 (750 mg, 1.58 mmol) in anhydrous DMF (10 mL) was cooled to 0 °C and treated with NaH (60% dispersion in mineral oil, 76.0 mg, 1.90 mmol). The solution was stirred for 5 min before solid recrystallized (S)-glycidal-3-nosylate (99% ee, 491 mg, 1.90 mmol) was added. The reaction mixture was stirred at 0 °C for 30 min, after which it was warmed to room temperature and stirred for 2 h. The solution was quenched with the addition of saturated aqueous NH4Cl, diluted with EtOAc, washed with H2O and saturated aqueous NaCl, and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (15% EtOAc/hexane) to provide 5 (735 mg, 88%) as a white solid and as a mixture of rotamers (1:1): 1H NMR (acetone-d6, 400 MHz) δ 8.34–8.31 (m, 1H), 8.23–8.20 (m, 1H), 7.68 (t, J = 7.2 Hz, 1H), 7.64–7.56 (m, 3H), 7.44 (t, J = 7.2 Hz, 2H), 7.37 (t, J = 7.2 Hz, 1H), 7.23 (s, 0.5H), 7.14 (s, 0.5H), 5.40 (s, 2H), 4.12–4.02 (m, 1 H), 3.39–3.25 (m, 2H), 2.69–2.62 (m, 1H), 2.41–2.35 (m, 1H), 1.30 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 157.13, 157.07, 155.23, 155.16, 145.8, 145.4, 138.70, 138.67, 137.04, 136.98, 134.30, 134.28, 130.50, 130.48, 130.40, 129.84, 129.82, 129.7, 129.6, 129.4, 129.3, 128.3, 128.2, 127.2, 124.22, 124.20, 109.99, 109.96, 96.1, 96.0, 81.6, 81.5, 72.1, 72.0, 54.3, 53.0, 51.3, 50.8, 47.4, 47.0, 29.34, 29.32; IR (film) νmax 2975, 1700, 1589 cm−1; ESI-TOF HRMS m/z 532.0975 (M+H+, C25H26INO4 requires 532.0979).
5:[α]23D +8 (c 2.0, THF).
ent-5:[α]23D −8 (c 2.0, THF).
(S)-tert-Butyl 6-(Benzyloxy)-2-hydroxy-2,3-dihydrobenzo[f]quinoline-4(1H)-carboxylate (6)
A solution of 5 (126.5 mg, 0.2381 mmol) in distilled anhydrous THF (1.3 mL) at room temperature was treated with EtMgBr (476 μL, 0.4761 mmol, 1.0 M in THF). The reaction mixture was stirred for 30 min, after which the reaction was quenched with the addition of saturated aqueous NH4Cl, diluted with EtOAc, washed with H2O and saturated aqueous NaCl, and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (20% EtOAc/hexane) to provide 6 as a clear oil (84.5 mg, 87%): 1H NMR (acetone-d6, 400 MHz) δ 8.26 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 7.2 Hz, 2H), 7.54 (t, J = 8.4 Hz, 1H), 7.44 (t, J = 7.2 Hz, 3H), 7.37 (t, J = 7.2 Hz, 2H), 5.28 (s, 2H), 4.33 (d, J = 4.4 Hz, 1H), 4.28 (m, 1H), 3.99 (dd, J = 13, 3.2 Hz, 1H), 3.56 (dd, J = 12, 8.0 Hz, 1H), 3.37 (dd, J = 13, 6.4 Hz, 1H), 2.92 (dd, J = 17, 6.0 Hz, 1H), 1.53 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 155.7, 153.8, 139.3, 138.2, 134.8, 130.4, 129.7, 129.4, 128.7, 126.0, 125.3, 124.4, 124.0, 115.7, 106.4, 82.2, 71.8, 66.1, 51.9, 35.0, 29.5; IR (film) νmax 3414, 2975, 2928, 1692, 1622, 1592 cm−1; ESI-TOF HRMS m/z 406.2014 (M+H+, C25H27NO4 requires 406.2013).
6:[α]D +35 (c 0.41, THF).
ent-6:[α]D −34 (c 0.41, THF).
Treatment with MeMgBr (10 equiv) or i-PrMgBr (10 equiv) using the above procedure provide 6 in 87% and 82%, respectively.
(S)-tert-Butyl 2,6-Dihydroxy-2,3-dihydrobenzo[f]quinoline-4(1H)-carboxylate (7)
A solution of 6 (84.7 mg, 0.209 mmol) and ammonium formate (130 mg, 2.09 mmol) in 9:1 distilled THF:MeOH (8 mL) at room temperature was treated with 10% Pd/C (40 mg). The reaction mixture was stirred for 30 min, at which point the solution was filtered through Celite. The solvent was removed under reduced pressure to provide 7 as a white foam (63.9 mg, 97%), identical in all respects to authentic material: 1H NMR (acetone-d6, 400 MHz) δ 8.85 (s, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.27 (s, 1H), 4.25 (m, 2H), 3.98 (dd, J = 9.2, 2.4 Hz, 1H), 3.51 (dd, J = 12, 7.6 Hz, 1H), 3.35 (dd, J = 16, 6.0 Hz, 1H), 2.88 (dd, J = 16, 6.0 Hz, 1H), 1.51 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 155.6, 152.4, 138.3, 135.0, 128.4, 125.3, 124.7, 124.4, 124.2, 114.3, 108.2, 81.9, 66.3, 51.9, 35.0, 29.4; IR (film) νmax 3306, 2975, 2929, 1670, 1626, 1594 cm−1; ESI-TOF HRMS m/z 316.1540 (M+H+, C18H21NO4 requires 316.1543).
7:[α]D +42 (c 0.94, THF).
ent-7:[α]D −41 (c 1.13, THF).
1,2,9,9a-Tetrahydrocyclopropa[c]benzo[e]indol-4-one (N-Boc-CBI, 8)
A solution of 7 (10.6 mg, 0.0336 mmol) in toluene (1 mL) at room temperature was treated with ADDP (42.4 mg, 0.168 mmol) and tributyl phosphine (42 μL, 0.168 mmol). The reaction mixture was stirred for 30 min, at which point the solvent was removed under reduced pressure and the residue was purified by flash chromatography (30% EtOAc/hexane) to provide 8 (7.1 mg, 71%) as a white solid, identical in all respects to authentic material: 1H NMR (acetone-d6, 400 MHz) δ 8.08 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.39 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 6.78 (s, 1H), 4.04 (m, 2H), 3.03 (m, 1H), 1.66 (dd, J = 7.6, 4.0 Hz, 1H), 1.54 (s, 9H), 1.49 (t, J = 4.4 Hz, 1H); 13C NMR (acetone-d6, 150 MHz) δ186.3, 162.0, 153.3, 142.5, 134.6, 133.4, 127.9, 127.8, 123.7, 109.5, 84.0, 54.8, 34.9, 30.4, 29.2, 25.4; IR (film) νmax 2975, 2929, 1723, 1626, 1600, 1564 cm−1; ESI-TOF HRMS m/z 298.1439 (M+H+, C18H19NO3 requires 298.1438).
8:[α]D +131 (c 0.53, THF).
ent-8:[α]D −129 (c 0.60, THF).
(S)-tert-Butyl 1-(Chloromethyl)-5-hydroxy-1H-benzo[e]indole-3(2H)-carboxylate (N-Boc-seco-CBI, 9)
A sample of 8 (2.5 mg, 0.0084 mmol) at −78 °C was treated with 4 N HCl in EtOAc (0.5 mL) and stirred at −78 °C for 30 min. While keeping the solution cooled, the solvent and HCl gas was removed under a stream of nitrogen, providing 9 as a light brown solid (2.7 mg, 96%), identical in all respects to authentic material: 1H NMR (acetone-d6, 400 MHz) δ 8.19 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.73 (br s, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.31 (t, J = 8.4 Hz, 1H), 4.20 (dd, J = 11, 2.4 Hz, 1H), 4.15 (t, J = 10 Hz, 1H), 4.07 (m, 1H), 3.99 (dd, J = 11, 3.2 Hz, 1H), 3.66 (dd, J = 11, 8.8 Hz, 1H), 1.57 (s, 9H); 13C NMR (acetone-d6, 150 MHz) δ 156.4, 153.9, 132.6, 129.2, 127.0, 125.2, 124.2, 124.0, 123.4, 115.6, 100.8, 82.5, 54.6, 48.9, 42.9, 29.6; ESI-TOF HRMS m/z 334.1218 (M+H+, C18H20ClNO3 requires 334.1204).
(S)-N-(2-(1-(Chloromethyl)-5-hydroxy-2,3-dihydro-1H-benzo[e]indole-3-carbonyl)-1H-indol-5-yl)-1H-indole-2-carboxamide (seco-CBI-indole2, 10)
A sample of 8 (13.5 mg, 0.0455 mmol) at −78 °C was treated with 4 N HCl in EtOAc (1.0 mL) and stirred at −78 °C for 30 min. The solution was warmed to 23 °C and stirred for 30 min, at which point the solvent and HCl gas was removed under a stream of nitrogen. The resulting residue was dissolved in anhydrous DMF (1.0 mL) and EDCI (26.1 mg, 0.136 mmol) and indole2-CO2H (16.0 mg, 0.0500 mmol) were added. The resulting reaction mixture was stirred for 18 h, at which point the solution was diluted with EtOAc, washed with 1 M HCl, saturated aqueous NaHCO3, saturated aqueous NaCl, and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (40% THF/hexane) to provide 10 as a pale yellow solid (16.5 mg, 68%), identical in all respects to authentic material: 1H NMR (DMSO-d6, 400 MHz) δ 11.74 (s, 1H), 11.72 (s, 1H), 10.44 (s, 1H), 10.17 (s, 1H), 8.23 (s, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.99 (s, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.59 (dd, J = 8.8, 1.6 Hz, 1H), 7.55–7.47 (m, 3H), 7.43 (s, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.24 (s, 1H), 7.20 (t, J = 7.2 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 5.28 (s, 2H), 4.83 (t, J = 10 Hz, 1H), 4.58 (d, J = 7.2 Hz, 1H), 4.24 (m, 1H), 4.04 (dd, J = 11, 3.2 Hz, 1H), 3.89 (dd, J = 11, 7.2 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 160.0, 159.5, 154.1, 142.2, 136.6, 133.3, 131.8, 131.6, 131.3, 129.8, 127.2, 127.05, 127.02, 123.4, 123.1, 123.0, 122.7, 122.1, 121.5, 119.7, 119.2, 114.9, 112.8, 112.3, 112.2, 105.6, 103.3, 100.3, 55.0, 47.6, 41.1; ESI-TOF HRMS m/z 535.1544 (M+H+, C31H23ClN4O3 requires 535.1531).
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA 041986) and the Skaggs Institute for Chemical Biology and thank R. Rodriguez for initial studies. JPL is a Skaggs Fellow.
Footnotes
Supporting Information Available: General experimental details, Figure 3, and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. J Antibiot. 1981;34:1119. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]
- 2.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. J Antibiot. 1990;43:1037. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. J Antibiot. 1988;41:1915. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 4.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. J Antibiot. 2003;56:107. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 5.For duocarmycin SA, see: Boger DL, Johnson DS, Yun W. J Am Chem Soc. 1994;116:1635.For yatakemycin, see: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. J Am Chem Soc. 2003;125:10971. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Nat Chem Biol. 2006;2:79. doi: 10.1038/nchembio761.Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2007;129:10858. doi: 10.1021/ja072777z.For CC-1065, see: Hurley LH, Lee CS, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Biochemistry. 1988;27:3886. doi: 10.1021/bi00410a054.Hurley LH, Warpehoski MA, Lee CS, McGovren JP, Scahill TA, Kelly RC, Mitchell MA, Wicnienski NA, Gebhard I, Johnson PD, Bradford VS. J Am Chem Soc. 1990;112:4633.Boger DL, Johnson DS, Yun W, Tarby CM. Bioorg Med Chem. 1994;2:115. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. J Am Chem Soc. 1990;112:4623.For duocarmycin A, see: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1990;112:8961.Boger DL, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. Bioorg Med Chem Lett. 1992;2:759.Boger DL, Yun W. J Am Chem Soc. 1993;115:9872.Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. Tetrahedron. 1991;47:2661.
- 6.Reviews: Boger DL, Johnson DS. Angew Chem Int Ed Engl. 1996;35:1438.Boger DL. Acc Chem Res. 1995;28:20.Boger DL, Johnson DS. Proc Natl Acad Sci USA. 1995;92:3642. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043.Searcey M. Curr Pharm Des. 2002;8:1375. doi: 10.2174/1381612023394539.Tichenor MS, Boger DL. Natural Prod Rep. 2008;25:220. doi: 10.1039/b705665f.MacMillan KS, Boger DL. J Med Chem. 2009;52:5771. doi: 10.1021/jm9006214.
- 7.(a) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:1321. [Google Scholar]; (b) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:4796. [Google Scholar]; (c) Boger DL, Machiya K. J Am Chem Soc. 1992;114:10056. [Google Scholar]; (d) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. J Am Chem Soc. 1993;115:9025. [Google Scholar]; (e) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1996;118:2301. [Google Scholar]; (f) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1997;119:311. [Google Scholar]; (g) Tichenor MS, Kastrinsky DB, Boger DL. J Am Chem Soc. 2004;126:8346. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]
- 8.(a) Boger DL, Ishizaki T, Wysocki RJ, Jr, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1989;111:6461. [Google Scholar]; (b) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823. [Google Scholar]; (c) Boger DL, Ishizaki T. Tetrahedron Lett. 1990;31:793. [Google Scholar]; (d) Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. Bioorg Med Chem Lett. 1991;1:55. [Google Scholar]; (e) Boger DL, Ishizaki T, Sakya SM, Munk SA, Kitos PA, Jin Q, Besterman JM. Bioorg Med Chem Lett. 1991;1:115. [Google Scholar]; (f) Boger DL, Munk SA, Ishizaki T. J Am Chem Soc. 1991;113:2779. [Google Scholar]; (g) Boger DL, Yun W. J Am Chem Soc. 1994;116:5523. [Google Scholar]; (h) Boger DL, Yun W, Han N, Johnson DS. Bioorg Med Chem. 1995;3:611. doi: 10.1016/0968-0896(95)00048-l. [DOI] [PubMed] [Google Scholar]; (i) Boger DL, Yun W, Cai H, Han N. Bioorg Med Chem. 1995;3:761. doi: 10.1016/0968-0896(95)00066-p. [DOI] [PubMed] [Google Scholar]; (j) Boger DL, Yun W, Han N. Bioorg Med Chem. 1995;3:1429. doi: 10.1016/0968-0896(95)00130-9. [DOI] [PubMed] [Google Scholar]
- 9.Parrish JP, Trzupek JD, Hughes TV, Hwang I, Boger DL. Bioorg Med Chem. 2004;12:5845. doi: 10.1016/j.bmc.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 10.Boger DL, Munk SA. J Am Chem Soc. 1992;114:5487. [Google Scholar]
- 11.Drost KJ, Cava MP. J Org Chem. 1991;56:2240. [Google Scholar]
- 12.Boger DL, Yun W, Teegarden BR. J Org Chem. 1992;57:2873. [Google Scholar]
- 13.Boger DL, McKie JA. J Org Chem. 1995;60:1271. [Google Scholar]
- 14.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Tetrahedron Lett. 1998;39:2227. [Google Scholar]
- 15.Review: Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem Rev. 1997;97:787. doi: 10.1021/cr960095g.
- 16.Boger DL, Yun W. J Am Chem Soc. 1994;116:7996. [Google Scholar]
- 17.Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823. [Google Scholar]
- 18.Boger DL, McKie JA, Boyce CW. Synlett. 1997:515. [Google Scholar]
- 19.Kastrinsky DB, Boger DL. J Org Chem. 2004;69:2284. doi: 10.1021/jo035465x. [DOI] [PubMed] [Google Scholar]
- 20.(a) Ling L, Xie Y, Lown JW. Heterocycl Commun. 1997;3:405. [Google Scholar]; (b) Lei L, Lown JW. Chem Commun. 1996:1559. [Google Scholar]
- 21.Aristoff PA, Johnson PD. J Org Chem. 1992;57:6234. [Google Scholar]
- 22.Mohamadi F, Spees MM, Staten GS, Marder P, Kipka JK, Johnson DA, Boger DL, Zarrinmayeh H. J Med Chem. 1994;37:232. doi: 10.1021/jm00028a005. [DOI] [PubMed] [Google Scholar]
- 23.Tse WC, Boger DL. Chem Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 24.(a) Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, Boger DL. J Med Chem. 2010;53:7731. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, Hwang I, Boger DL. J Am Chem Soc. 2007;129:15391. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 26.Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J Am Chem Soc. 2006;128:15683. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanson RM. Chem Rev. 1991;91:437. [Google Scholar]
- 28.Krasovskiy A, Straub BF, Knochel P. Angew Chem Int Ed. 2006;45:159. doi: 10.1002/anie.200502220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.