

Abstract
Introduction
Chronic obstructive pulmonary disease (COPD) constitutes a worldwide health problem. There is currently an urgent and unmet need for the development of small molecule therapeutics capable of blocking and/or reversing the progression of the disorder. Recent studies have greatly illuminated our understanding of the multiple pathogenic processes associated with COPD. Of paramount importance is the key role played by proteases, oxidative stress, apoptosis, and inflammation. Insights gained from these studies have made possible the exploration of new therapeutic approaches.
Areas covered
An overview of major developments in COPD research with emphasis on low molecular weight neutrophil elastase inhibitors is described in this review.
Expert opinion
Great strides have been made toward our understanding of the biochemical and cellular events associated with COPD. However, our knowledge regarding the inter-relationships among the multiple pathogenic mechanisms and their mediators involved is till limited. The problem is further compounded by the unavailability of suitable validated biomarkers for assessing the efficacy of potential therapeutic interventions. The complexity of COPD suggests that effective therapeutic interventions may require the administration of more than one agent such as, for instance, an HNE or MMP-12 inhibitor with an anti-inflammatory agent such as a phosphodiesterase-4 inhibitor, or a dual function agent capable of disrupting the cycle of proteolysis, apoptosis, inflammation and oxidative stress
Keywords: chronic obstructive pulmonary disease, cigarette smoke, oxidative stress, neutrophils, macrophages, T cells, apoptosis, chemokines, cytokines, protease-antiprotease imbalance, inflammation, human neutrophil elastase, human neutrophil proteinase 3, macrophage metalloelastase, α1-proteinase inhibitor, secretory leukocyte proteinase inhibitor, tissue inhibitors of metalloproteinases, cystatins, small molecule therapeutics
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a multi-factorial, chronic inflammatory disorder characterized by enlargement of the airspaces and airflow obstruction (1). The disorder constitutes a major health problem that affects more than 16 million people in the U.S. and is currently the fourth most common cause of death (2–3). Furthermore, COPD is continuing to increase in both prevalence and mortality, accounting for 120,000 deaths per year in the U.S. and an annual financial burden in excess of $37 billion (4). Current therapy for COPD is symptomatic and there are no drugs on the market that arrest or slow the relentless progression of the disorder (5–6). This review is intended to serve as an updated sequel to earlier reviews on neutrophil elastase inhibitors and COPD (7–9) and highlights emerging concepts and paradigms related to the pathophysiology of the disorder and potential therapeutic modalities, with particular emphasis on low molecular weight neutrophil elastase inhibitors.
2. Pathogenesis of Chronic Obstructive Pulmonary Disease
The molecular mechanisms underlying the pathogenesis of COPD are complex and poorly understood, gravely hampering the development of novel therapeutics. However, although many fundamental questions related to COPD pathogenesis (for example, the precise pathogenic mechanisms responsible for the initiation and progression of the disorder, the role(s) and the relative importance of the multiple processes and mediators involved in COPD, etc) remain poorly-defined (4), recent seminal findings have greatly illuminated our understanding of the key biochemical and cellular events associated with the disorder. These could potentially be exploited, ultimately leading to the emergence of new and effective therapeutic interventions (10).
Polymorphonuclear leukocytes (neutrophils) ordinarily serve as an important host defense against bacterial and fungal infections. Invading pathogens are phagocytosed and then killed in phagolysosomes by the combined action of reactive oxygen species generated by the NADH oxidase and the intracellular release of antibacterial proteins and proteolytic enzymes (11–13). The latter are stored in the azurophilic and specific granules of neutrophils and include the serine proteases human neutrophil elastase (HNE), proteinase 3 (Pr 3) and cathepsin G (14). Several lines of evidence suggest that neutrophil-derived oxidative and proteolytic mediators (HNE, Pr 3) released extracellularly at sites of inflammation play an important role in the pathogenesis of a range of inflammatory diseases (15), including COPD, cystic fibrosis (16–17), acute lung injury (18) and others (19). Airflow obstruction arising from the hypersecretion of mucous into the airways, a characteristic feature of COPD and other chronic lung diseases, is due to the stimulation of goblet cells by HNE and Pr 3 (20–21).
The protease/antiprotease imbalance hypothesis, as originally framed, postulated that damage to lung connective tissue results from the massive migration of neutrophils to the lungs during smoke-induced inflammation and the subsequent release of proteolytic enzymes. Inadequate control of the activity of these enzymes due to depressed levels of their physiological protein inhibitors leads to a protease/antiprotease imbalance in the airways, ultimately allowing the degradation of elastin, the elastic component of lung connective tissue, and other components of the extracellular matrix (22–25). Thus, the hypothesis focused primarily on neutrophil and neutrophil elastase as the primary cell and enzyme responsible for COPD. However, in addition to HNE, other cell types and enzymes derived thereof, are of critical importance in COPD (26–28). Specifically, in addition to neutrophils, the inflammation associated with COPD involves the influx to the lungs of macrophages and CD8+ T lymphocytes. Indeed, alveolar macrophages are the most abundant defense cell in the lung under normal conditions and during chronic lung inflammation. They are a major source of metallo- (macrophage metalloelastase, MMP-12) and cysteine (cathepsin S, Cat S) proteases. The capacity of cathepsin S to degrade elastin is comparable to that of HNE and Pr 3. Studies have shown that cigarette smoke-induced emphysema is closely associated with the influx of macrophages to the lungs and their enhanced elastolytic activity (29). Indeed, the extent of lung destruction in emphysematous patients is directly related to the number of alveolar macrophages and CD8+ T-lymphocytes (30–31).
Cigarette smoke inhalation inactivates histone deacetylase and results in the release of nuclear factor-κB (NF-κB) leading to transcription of neutrophil chemokines and cytokines (TNF-α and IL-8) (32). The subsequent secretion of MMP-12 by macrophages leads to the release of tumor necrosis factor-α (TNF-α), an important pro-inflammatory cytokine which activates endothelial cells and leads to an influx of neutrophils to the lungs. Subsequent release of serine proteases by neutrophils and metalloproteases by macrophages is believed to account for most matrix destruction (33).
The influx of neutrophils and other phagocytic cells is associated with chronic airway inflammation, a cardinal pathophysiologic feature in patients with COPD (34–35), which is characterized by elevated levels of interleukin-8 (IL-8), a chemotactic cytokine produced by neutrophils, alveolar macrophages, lymphocytes and epithelial cells (36–38). Indeed, the sputum concentration of IL-8 is closely related to the degree of airway obstruction in COPD (39). Several lines of evidence indicate that neutrophil serine proteases play an important role in regulating inflammation by activating pro-inflammatory cytokines (40). For instance, Pr 3 is known to activate TNF-α, IL-1β, and IL-18, while HNE induces apoptosis of lung epithelial cells and activates epidermal growth factor receptor (EGFR) and toll-like receptor-4.
Oxidative stress, a hallmark of COPD, arises from the inhalation of cigarette smoke which contains a multitude of highly reactive species (41) and the production of reactive oxygen species by inflammatory cells recruited to the lungs (45). Oxidative stress leads to infiltration of neutrophils, macrophages, and cytotoxic T lymphocytes to the lungs which release an array of proteolytic enzymes (HNE, Pr 3, cathepsin S, MMP-12) (42–43). These mediate a multitude of signaling pathways (40, 44) and participate in the degradation of lung connective tissue and other components of the extracellular matrix due to the resultant protease/anti-protease imbalance (vide supra). Furthermore, oxidative stress leads to elevated airway inflammation, alveolar cell apoptosis, and impaired lung repair. Nuclear factor erythroid-2-related factor 2 (Nrf2), a transcription factor that plays an important role in protecting the lungs against cigarette smoke-induced inflammation, oxidative stress, and alveolar cell apoptosis by upregulating multiple antioxidant and detoxification genes (45), exhibits decreased activity in COPD and may play a critical role in the pathogenesis of the disorder (46).
In summary, the preponderance of evidence indicates that COPD involves the confluence and interplay of an oxidant/antioxidant imbalance (47–48), a protease/antiprotease imbalance (42–43), apoptosis (49–51), and chronic inflammation (34–35, 52), which are interlinked by elevated levels of an array of mediators, including ceramide, nuclear factor-κB (NF-κB), histone acetyltransferases, histone deacetylases, cytokines, and chemokines (40, 53–54) (Figure 1). Thus, it is clearly evident that the convergence of multiple pathogenic processes, in particular the pivotal role played by oxidative stress in alveolar cell apoptosis, inflammation, and proteolysis of lung elastin that results in lung maintenance and repair impairment, ultimately leads to the COPD phenotype (53–57).
Figure 1.
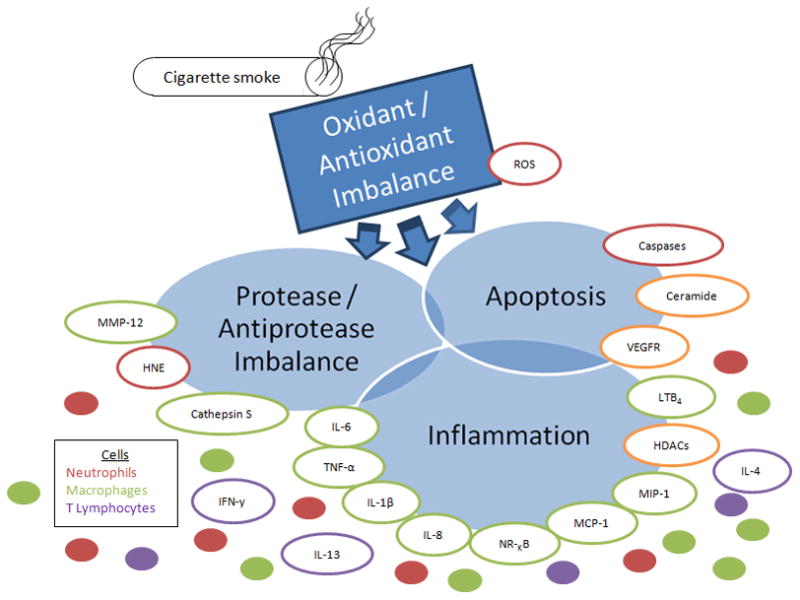
Confluence and interplay of major processes and mediators associated with COPD pathogenesis. Lung tissue damage resulting from the inhalation of free radicals in cigarette smoke results in oxidative stress and leads to the recruitment of neutrophils, macrophages and T lymphocytes to the lungs. Oxidative stress also leads to upregulation of ceramide, resulting in apoptosis of lung epithelial cells. Neutrophils release HNE while macrophages release MMP-12, and cathepsin S which collectively degrade lung elastin and exracellular matrix components. In addition, macrophages activate NF-κB and release various pro-inflammatory and chemotactic mediators which amplify inflammation while neutrophils release reactive oxygen species. MMP-12, oxidants in cigarette smoke, and reactive oxygen species inactivate alpha-1-proteinase inhibitor, the physiological inhibitor of HNE, resulting in a protease/anti-protease imbalance which allows the renegade proteases to degrade lung elastin. HNE inactivates TIMPs, the physiological inhibitor of MMP-12, augmenting elastin degradation. Elastin fragments are chemoattractants to macrophages and drive the progression of the disease. Alveolar wall homeostasis is eventually compromised, leading to enlargement of the airspaces and the COPD phenotype. Abbreviations: MMP-12, macrophage metalloelastase; HNE, human neutrophil elastase; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1beta; IL-8, interleukin 8; MCP-1, monocyte chemotactic protein; MIP-1, monocyte inflammatory protein; HDACs, histone deacetylases; LTB4, leukotriene B4; VEGFR, vascular endothelium growth fibroblast factor; NF-κB, nuclear factor kappa B; IFN-γ, interferon gamma; ROS, reactive oxygen species.
3. Neutrophil Elastase
HNE is a basic, 218 amino acid single polypeptide glycoprotein (Mr 29,500) whose primary structure shows considerable homology with proteinase 3 (54%), and cathepsin G (37%). HNE has an extended binding site and prefers hydrophobic substrates and inhibitors (58). The S1-S4 subsites of HNE have been mapped using peptidyl chromogenic substrates (59). The primary substrate specificity subsite S1 of HNE shows a preference for medium size P1 alkyl groups (isopropyl, n-propyl, isobutyl, n-butyl), however, P1 specificity is regulated by peptide length. Several X-ray crystal structures of HNE complexed to low molecular weight inhibitors (peptidyl chloromethyl ketones) and protein inhibitors (turkey ovomucoid inhibitor, secretory leukocyte protease inhibitor domain 2) have been reported. The X-ray crystal structure of HNE consists of two homologous β-barrels, each barrel composed of six antiparallel β-sheets, and a C-terminal α-helix. The catalytic triad residues (Ser195, His57, and Asp102) are located at the junction of the two β-barrels. As is true for all serine proteases, catalysis by HNE involves three steps, namely, substrate binding, acylation of Ser195, and deacylation (59, 60).
Endogenous protein inhibitors of proteases are divided into three main classes: serpins (serine protease inhibitors) (61–62), TIMPS (tissue inhibitors of metalloproteases) (63) and cystatins (cysteine protease inhibitors) (64). The activity of HNE is primarily regulated by the serpins α-1-proteinase inhibitor (α-1-PI) (65) and monocyte/neutrophil elastase inhibitor (MNEI, also called Serpin B1) (66), and the chelonianin family of canonical inhibitors that includes secretory leukocyte proteinase inhibitor (SLPI) (67), and elafin (68–69). The activity of Pr 3 is controlled by the same inhibitors, with the exception of SLPI which does not inhibit the enzyme. α-1-PI, a blood plasma glycoprotein (Mr 53 kDa), regulates the activity of HNE and Pr 3 in the lower respiratory tract, while SLPI protects the upper airways from proteolysis (70). MNEI (SerpinB1) is a potent inhibitor of HNE, as well as PR 3 and cathepsin G, while elafin, a 6 kDa protein isolated from bronchial secretions, is a potent inhibitor of HNE and Pr 3 (68–69). α-1-Macroglobulin serves as a general plasma inhibitor of serine, cysteine and metallo- proteases. There is increasing evidence that serine protease inhibitors (α-1-PI, SLPI, elafin), besides regulating inflammation by inhibiting the proteolytic activity of proteases, also directly affect leukocyte chemotaxis and pro-inflammatory mediator release, and may contribute to defense against invading pathogens. For instance, in addition to its anti-elastolytic activity, α-1-PI may alleviate emphysema by inhibiting smoke-induced inflammation via the suppression of tumor necrosis factor-α (71). Most importantly, because macrophage metalloelastase and neutrophil elastase degrade α-1-PI and TIMPS respectively (the two enzymes degrade each other’s inhibitor), the matrix degrading capacity of these enzymes is greatly augmented in emphysema. As stated earlier, oxidative processes play multiple roles in COPD (vide supra) (45–48). For instance, the reactive center of α-1-PI has a critical methionine residue (Met-358) that serves as the primary specificity residue P1 that is recognized and accommodated at the S1 subsite of HNE. The inhalation of oxidants during smoking and the presence of elevated levels of endogenous oxidants released by inflammatory cells, lead to the inactivation of α-1-PI via the oxidation of Met-358 and/or Met-351 to the corresponding sulfoxide (72). Oxidized α-1-PI is incapable of preventing the proteolysis of lung elastin by HNE (73) and, furthermore, interacts directly with lung epithelial cells to release chemokines (IL-8, MCP-1, TNF-α) that attract macrophages and neutrophils into the airways (74). Cigarette smoke also inactivates SLPI and histone deacetylases. Thus, oxidative processes influence the protease/antiprotease balance by decreasing the protease inhibitor screen and by increasing the protease burden in the lungs through the recruitment and degranulation of phagocytic cells.
4. HNE Inhibitors
The general principles related to the design of inhibitors of serine proteases in general, and HNE in particular, have been described in previous reviews (75–76). The design of HNE inhibitors has primarily focused on the attachment of a serine trap (CHO, CF3, CF2CF3, CF2COOR, CF2(C=O)NHR, CO(C=O)NHR, CO(C=O)OR, B(OH)2, and COHet, where Het = heterocycle) to a peptidyl recognition element that facilitates the binding of the inhibitor to the active site and the ensuing reversible formation of a tetrahedral adduct (Figure 2). Inhibitors of this type, termed transition state inhibitors, form a tetrahedral adduct with the active site serine and exploit favorable binding interactions with multiple subsites at the active site of the enzyme, consequently they are highly potent (inhibition constant, KI, is generally in the low nanomolar range).
Figure 2.

Interaction of a serine protease with a transition state inhibitor.
A second type of inhibitors included in this review are agents that acylate the active site serine and undergo slow deacylation (termed alternate substrate inhibitors). The likelihood of exhibiting side effects is greater with acylating agents because of their higher chemical reactivity. This review includes inhibitors of HNE reported in the patent and primary literature since 2005 (Table 1).
Table 1.
Low molecular weight inhibitors of human neutrophil elastase
| Entry | HNE inhibitors | IC50 (nM) |
|---|---|---|
| a) Transition state inhibitors | ||
| 1 | 1600a | |
| 2 | 29 | |
| b) Alternate substrate inhibitors | ||
| 3 | 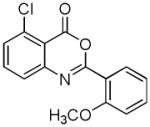 |
15.4 |
| 4 | 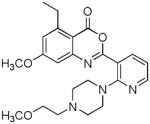 |
16 |
| 5 | 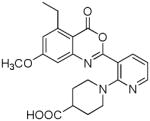 |
28 |
| 6 | 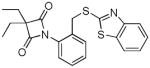 |
99.3a |
| 7 | 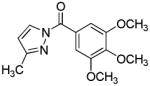 |
21a |
| c) Carbamylating agent | ||
| 8 | 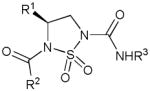 |
350 – 1260b |
| d) Mechanism-based inhibitor | ||
| 9 | 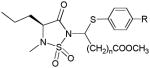 |
20 – 4580b |
| e) Reversible competitive inhibitors | ||
| 10 | 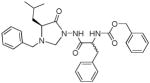 |
7340 |
| 11 | 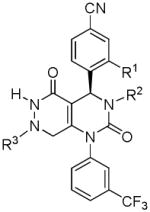 |
3 – 32 |
| 12 |  |
< 0.3 |
| 13 | 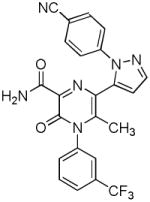 |
2.2 |
| 14 | 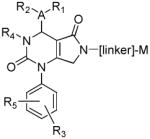 |
1 – 100 |
| 15 | 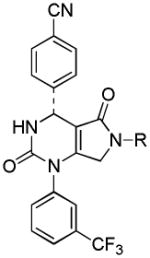 |
< 100 |
| 16 | 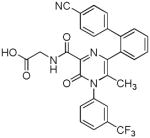 |
0.7 |
| 17 | 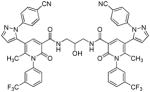 |
0.26 |
| 18 | 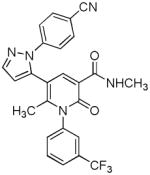 |
0.21 |
| 19 | 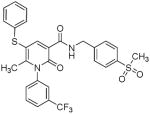 |
30000 |
| 20 | 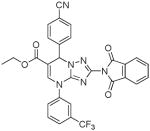 |
_c |
| 21 |  |
<30 |
| 22 | 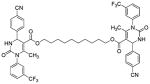 |
_c |
KI value;
kinact/KI (M−1s−1);
unavailable.
The lungs are highly perfused and enzyme inhibitors are generally hydrophobic in nature, consequently, oral or intratracheal administration of HNE inhibitors results in limited or no efficacy due to rapid clearance. To circumvent this problem, a transition state inhibitor of HNE was attached to a 25-amino acid fragment of human surfactant peptide B, yielding a construct 1 with a long lung residence time and minimal immunogenicity. Intratracheal administration of construct 1 prevented HNE-induced emphysema in a rodent model (77). In contrast, administration of the peptidyl difluoroketone inhibitor by itself failed to protect the lungs from the action of HNE.
A peptidyl trifluoromethyl ketone transition state inhibitor of HNE with a cyclic urea amide bridged carboxyl group at the N-terminus 2 has shown noteworthy efficacy in a lung hemorrhage hamster model when administered intravenously by infusion or by bolus injection (78). Compound 2 has high solubility and stability in water and shows no overt toxic effects in mice at high doses.
Substituted benzoxazinones have attracted intense interest as inhibitors of HNE in particular (79) and proteases in general (80–83). The interaction of HNE with this class of compounds involves acylation of the active site serine followed by partitioning of the resulting acyl enzyme leading to the formation of 1H,3H-quinazoline-2,4-dione or the deacylation product (Figure 3). The preferred pathway is dependent on the nature of R (79).
Figure 3.

Mechanism of inhibition of a serine protease by substituted benzoxazinones.
A recent study has described the synthesis of benzoxazinone analogs 3 (84) which exhibit a dual mode of action by inhibiting superoxide anion generation and HNE release. Inhibition of HNE was found to be critically dependent on ring A substitution by chlorine. The results of preliminary structure-activity relationship studies involving benzoxazinone derivatives 4–5 have also been reported (85). A 5-ethyl-7-methoxy substitution pattern conferred an optimal balance between chemical stability and potency. Furthermore, rat plasma stability was found to be dependent on the nature of the piperazine and piperidine substituent.
Substituted azetidine-2, 4-diones 6 have been shown to be highly potent and selective inhibitors of HNE (86–87). Optimal inhibitory activity was observed with R1 = R2 = ethyl and R3 = N1-aryl moiety having a heteroarylthiomethyl group at the para position. A “double-hit” mechanism of action involving rapid formation of a tetrahedral adduct followed by collapse to form a transient conjugated system (a Michael acceptor) has been postulated. Subsequent reaction with an active site nucleophilic residue was proposed to result in the formation of a fairly stable enzyme-inhibitor adduct. The high reactivity of compound 6, as evidenced by its low stability in blood plasma and off-target effects, may impact adversely the therapeutic utility of this class of inhibitors.
N-Benzoylpyrazoles 7 constitute a new class of inhibitors of HNE that inhibit the enzyme by rapidly acylating the active site serine (88). Deacylation rates were found to be dependent on ring substitution. These compounds showed variable stability in phosphate buffer (pH 7.3) that was dependent on ring substitution.
The HNE acylating agent par excellence continues to be Sivelestat (ElaspolR) (Figure 4) (89), marketed in Japan and Korea for the treatment of acute lung injury associated with systemic inflammatory response syndrome and is currently explored for a range of other indications, including sepsis associated with acute respiratory distress syndrome and disseminated intravascular coagulation (90–92).
Figure 4.

Interaction of Sivelestat with human neutrophil elastase.
A new class of carbamylating agents 8, namely where the R3NH(C=O) portion of the inhibitor is covalently attached to the active site serine, based on the cyclosulfamide scaffold that are selective for HNE has been reported (93). The deacylation rates of carbamylated enzymes are much lower than those of acylated enzymes. The structural motif, robustness, synthetic tractability, and multiple points of diversity embodied in the cyclosulfamide scaffold are well-suited to the design of selective inhibitors of HNE and related proteases.
Mechanism-based inhibitors have been extensively used in drug design and discovery. A mechanism-based inhibitor behaves as a substrate that is processed by the catalytic machinery of an enzyme generating a reactive electrophilic species that, upon further reaction with a nucleophilic active site residue, leads to irreversible inactivation of the enzyme. Inhibitors of this type offer the promise of greater enzyme selectivity. A series of mechanism-based inhibitors 9 designed to interact with the S′ subsites of HNE has been reported (94). Inhibitor 9 is selective for HNE and was proposed to generate a Michael acceptor via an enzyme-induced 3-aza Grob fragmentation process, initiated by the attack of the active site serine at the ring carbonyl carbon.
A low micromolar reversible competitive inhibitor 10 of HNE based on the N-amino-4-imidazolidinone scaffold has been reported (95). Inhibitor 10 is devoid of any inhibitory activity toward proteinase 3 because it exploits differences in the S′ subsites of the two enzymes.
Several new classes of structurally-diverse heterocyclic inhibitors of HNE have been reported in the patent literature. 1, 4-Diarylpyrimidopyridazinyldiones 11 (96) and the structurally similar 4-(4-cyano-2-thioaryl)-dihydropyrimidinones 12 (97) inhibited HNE potently with IC50s in the low nM or pM range, respectively and were tested in a rat acute lung injury model. Suitably-functionalized 2-pyrazinone derivatives such as 13 (98) have nM IC50 values toward HNE. 3,4,6,7-Tetrahydro-1H-pyrrolo[3,4-d]pyrimidine-2,5-diones 14 inhibit HNE and also exhibit muscarinic M3 antagonist activity (99), while the related 4-(4-cyanophenyl)-1-(3-trifluoromethylphenyl)-3,4,6,7-tetrahydro-1H-pyrrolo[3,4-d]pyrimidine-2,5-diones 15 inhibit HNE with IC50 values < 100 nM and were also found to reduce blood hemorrhage in lungs of rats exposed to HNE (100). Burkamp et al have described the use of functionalized 2-pyrazinone derivatives, such as 16, in the inhibition of HNE (101). Dimeric 2-pyridinone derivatives 17 inhibit HNE with IC50s in the nM to pM range, reduced HNE-induced damage in rat lungs, and inhibited rat neutrophil elastase activity in bronchoalveolar lavage fluid (102). 2-Pyridone 18 (103) is a nM inhibitor of HNE and related structural variants 19 inhibit HNE at μM levels (104). Other heterocyclic inhibitors that have been disclosed include tetrazolopyrimidines 20 (105), dihydropyridones 21 having IC50s < 30 μM (106), and dihydropyrimidone multimers 22 which were also shown to have HNE inhibitory activity in rat lungs (107). It is not clear what the current status of many of these classes of compounds is.
Although the exploration of low molecular weight inhibitors of HNE as potential therapeutics has traditionally received the most attention, recently there has been an increasing interest in the use of cyclic peptides in the design of enzyme inhibitors, including inhibitors of HNE (108–109). Of particular interest are the plant-derived mini-proteins cyclotides (110). In contrast to linear peptides, these macrocyclic, cystine-knotted peptides exhibit exceptional enzymatic and non-enzymatic stability, suggesting that the cyclotide scaffold could provide an effective means of engineering inhibitors of HNE that are devoid of the shortcomings associated with linear peptides. The potential of this approach is significantly augmented by the introduction of zicotonide, a cystine knot molecule, into the clinic (111).
5. Expert Opinion
Significant progress has been made toward our understanding of the key biochemical and cellular events associated with COPD. The multiple pathogenic mechanisms involved in the disorder are currently better clarified, and the relative significance and contribution of the mediators involved are better defined, than in the past. Nevertheless, our knowledge regarding the inter-relationships among the various processes and mediators and how they impact the initiation and progression of the disorder, is still incomplete. The problem is further compounded by the unavailability of suitable validated biomarkers (112) for assessing the efficacy of potential therapeutic interventions. The complexity of COPD suggests that effective therapeutic interventions may require the administration of more than one agent such as, for instance, an HNE or MMP-12 inhibitor with an anti-inflammatory agent such as a phosphodiesterase-4 inhibitor, or a dual function agent capable of disrupting the cycle of proteolysis, apoptosis, inflammation and oxidative stress (113–116) (Figure 1). This is a fast moving area of research that, despite the challenges that lie ahead, will continue to illuminate our understanding of the disorder, as well as identify new therapeutic targets. Exploration of these targets is likely to lead to the emergence of disease-modifying therapeutics for COPD in the not too distant future.
Article highlights box.
COPD is the fourth leading cause of death in the US. Currently, there are no drugs on the market that arrest or slow the relentless progression of COPD.
COPD involves the confluence and interplay of oxidative stress, a protease/antiprotease, apoptosis, and chronic inflammation.
The release of serine proteases (HNE, Pr3) by neutrophils, and cysteine (cathepsin S) and metalloproteases (MMP-12) by macrophages is believed to account for most matrix destruction in the lungs.
In addition to their degradative action, proteases released by inflammatory cells play an important role in modulating inflammation and activating pro-inflammatory cytokines.
An array of selective neutrophil elastase inhibitors have been reported/patented recently. A peptidyl transition state inhibitor of HNE showed noteworthy efficacy in a lung hemorrhage hamster model without overt toxic effects. Several heterocyclic inhibitors of HNE with IC50s in low nM to pM range have been reported and shown to abrogate the deleterious effects of HNE in the rat acute lung injury model.
The complexity of COPD suggests that effective therapeutic interventions may require the administration of more than one agent to disrupt the cycle of proteolysis, apoptosis, inflammation and oxidative stress.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (HL57788).
Footnotes
Declaration of interest
The authors declare no conflict of interest.
References
Papers of special note have been highlighted as:
• of special interest
•• of considerable interest
- 1.Calverley PM, Walker P. Chronic obstructive pulmonary disease. Lancet. 2003;362:1053–1061. doi: 10.1016/s0140-6736(03)14416-9. [DOI] [PubMed] [Google Scholar]
- 2.Punturieria A, Croxton TL, Weinmann G, Kiley JP. The changing face of COPD. Am Fam Physician. 2007;75:315–316. [PubMed] [Google Scholar]
- 3.Stockley RA, Mannino D, Barnes PJ. Burden and Pathogenesis of Chronic Obstructive Pulmonary Disease. Proc Am Thor Soc. 2009;6:524–526. doi: 10.1513/pats.200904-016DS. [DOI] [PubMed] [Google Scholar]
- 4•.Punturieria A, Croxton TL, Weinmann G, Kiley JP. Chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:441–443. doi: 10.1164/rccm.200807-1128OE. An insightful overview of COPD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seifert S, Vogelmeier C. Emerging drugs in chronic obstructive pulmonary disease. Expert Opin Emerging Drugs. 2009;14:181–194. doi: 10.1517/14728210902798055. [DOI] [PubMed] [Google Scholar]
- 6••.Barnes PJ. New therapies for chronic obstructive pulmonary disease. Med Princ Pract. 2010;19:330–338. doi: 10.1159/000316368. A comprehensive state-of-the-art review focusing on COPD therapeutics. [DOI] [PubMed] [Google Scholar]
- 7.Ohbayashi H. Neutrophil elastase inhibitors as treatment for COPD. Expert Opin Investig Drugs. 2002;11:965–980. doi: 10.1517/13543784.11.7.965. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra S, Paul Man SF, Sin DD. Emerging drugs for the treatment of chronic obstructive pulmonary disease. Expert Opin Emerging Drugs. 2006;11:275–291. doi: 10.1517/14728214.11.2.275. [DOI] [PubMed] [Google Scholar]
- 9.Kelly E, Greene CM, McElvaney NG. Targeting neutrophil elastase in cystic fibrosis. Expert Opin Ther Targets. 2008;12:145–157. doi: 10.1517/14728222.12.2.145. [DOI] [PubMed] [Google Scholar]
- 10.Sussan TE, Rangasamy T, Blake DJ, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA. 2009;106:250–255. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 12.Reeves AP, Lu H, Jacobs HL, et al. Killing activity of neutrophils is mediated through the activation of proteases by K+ flux. Nature. 2002;416:291–296. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 13.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nature Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 14.Hager M, Cowland JB, Borregaard N. Neutrophil granules in health and disease. J Intern Med. 2010;268:25–34. doi: 10.1111/j.1365-2796.2010.02237.x. [DOI] [PubMed] [Google Scholar]
- 15.Malech HL, Gallin JI. Neutrophils in human diseases. New Engl J Med. 1987;320:365–376. doi: 10.1056/NEJM198709103171107. [DOI] [PubMed] [Google Scholar]
- 16.Quinn DJ, Weldon S, Taggart CC. Antiproteases as therapeutics to target inflammation in cystic fibrosis. Open Respir Med J. 2010;30:20–31. doi: 10.2174/1874306401004010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voynow JA, Fischer BM, Zheng S. Proteases and cystic fibrosis. Int J Biochem Cell Biol. 2008;40:1238–1245. doi: 10.1016/j.biocel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moraes TJ, Zurawska JH, Downey GP. Neutrophil granule contents in the pathogenesis of lung injury. Curr Opin Hematol. 2006;13:21–27. doi: 10.1097/01.moh.0000190113.31027.d5. [DOI] [PubMed] [Google Scholar]
- 19.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, et al. Neutrophil elastase-mediated degration of IRS-1- accelerates lung tumor growth. Nat Med. 2010;16:219–223. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Nadel JA. Role of neutrophils in mucous hypersecretion in COPD and implications for therapy. Trends Respir Med. 2004;3:147–159. doi: 10.2165/00151829-200403030-00003. [DOI] [PubMed] [Google Scholar]
- 21.Witko-Sarsat V, Halbwachs-Mecarelli L, Schuster A, et al. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am J Respir Cell Mol Biol. 1999;20:729–736. doi: 10.1165/ajrcmb.20.4.3371. [DOI] [PubMed] [Google Scholar]
- 22.Abrahamson DR, Irbin MH, Blackburn WD, Heck LW. Degradation of basement laminin by human neutrophil elastase. Am J Pathol. 1990;136:1267–1274. [PMC free article] [PubMed] [Google Scholar]
- 23.Kafienah W, Buttle D, Burnett D, Hollander AP. Cleavage of native type I collagen by human neutrophil elastase. Biochem J. 1998;330:897–902. doi: 10.1042/bj3300897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janucz MJ, Doherty NS. Degradation of matrix proteoglycan by human neutrophils involves both elastase and cathepsin G. J Immunol. 1991;146:3922–3928. [PubMed] [Google Scholar]
- 25.Rubio F, Accurso FJ, Remold-O’Donnell E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax. 2004;59:318–323. doi: 10.1136/thx.2003.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:S29–S32. doi: 10.1164/ajrccm.160.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- 27.Palmgren MS, de Shazo RD, Carter RM, et al. Mechanisms of neutrophil damage to human alveolar extracellular matrix: the role of serine and metalloproteases. J Allergy Clin Immunol. 1992;89:905–915. doi: 10.1016/0091-6749(92)90447-a. [DOI] [PubMed] [Google Scholar]
- 28.Chapman HA, Shi G-P. Protease injury in the development of COPD. Chest. 2000;117:S295–S298. doi: 10.1378/chest.117.5_suppl_1.295s. [DOI] [PubMed] [Google Scholar]
- 29.Ohnishi K, Tagaki M, Kurokawa Y, et al. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest. 1998;78:1077–1087. [PubMed] [Google Scholar]
- 30.Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1998;152:1666–1672. doi: 10.1164/ajrccm.152.5.7582312. [DOI] [PubMed] [Google Scholar]
- 31.Saetta M, DiStephano A, Turato G, et al. CD8+ T lymphocytes in peripheral airways of smokers with COPD. Am J Respir Crit Care Med. 1998;157:822–826. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 32.Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in COPD. The New England J Med. 2005;352:1967–1977. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 33.Shapiro SD. COPD Unwound. The New England J Med. 2005;352:2016–2019. doi: 10.1056/NEJMe058044. [DOI] [PubMed] [Google Scholar]
- 34.Wouters EFM. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:26–33. doi: 10.1513/pats.200408-039MS. [DOI] [PubMed] [Google Scholar]
- 35.Rennard SI. Inflammation and repair processes in COPD. Am J Respir Crit Care Med. 1999;160:S12–S16. doi: 10.1164/ajrccm.160.supplement_1.5. [DOI] [PubMed] [Google Scholar]
- 36.Harada A, Sekido N, Akahoshi T, et al. Essential involvement of IL-8 in acute inflammation. J Leuk Biol. 1994;56:559–564. [PubMed] [Google Scholar]
- 37.Taub DT, Anver M, Oppenheim JJ, et al. T lymphocyte recruitment by IL-8. J Clin Invest. 1996;97:1931–1941. doi: 10.1172/JCI118625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richman-Eisenstadt JB, Jorens PG, Hebert CA, et al. Interleukin 8: an important chemoattractant in sputum of patients with COPD. Am J Physiol. 1993;264:L413–L418. doi: 10.1152/ajplung.1993.264.4.L413. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto C, Yoneda T, Yoshikawa M, et al. Airway inflammation in COPD assessed by sputum levels of IL-8. Chest. 1997;112:505–510. doi: 10.1378/chest.112.2.505. [DOI] [PubMed] [Google Scholar]
- 40.Meyer-Hoffert U. Neutrophil-derived serine proteases modulate innate immune responses. Frontiers Biosci. 2009;14:3409–3418. doi: 10.2741/3462. [DOI] [PubMed] [Google Scholar]
- 41.Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann NY Acad Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- 42••.Abboud RT, Vimalanathan S. Pathogenesis of COPD. The role of the protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2010;12:361–367. An excellent review focused on the multiple roles of proteases in COPD. [PubMed] [Google Scholar]
- 43.Owen CA. Roles of proteinases in the pathogenesis of chronic obstructive pulmonary disease. Intl J COPD. 2008;3:253–268. doi: 10.2147/copd.s2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham CT. Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol. 2008;40:1317–1333. doi: 10.1016/j.biocel.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rangasamy T, Misra V, Zhen L, et al. Cigarette-smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol. 2009;296:L888–L900. doi: 10.1152/ajplung.90369.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Boutten A, Goven D, Boczkowski J, Bonay M. Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin Ther Targets. 2010;14:329–346. doi: 10.1517/14728221003629750. A state-of-the-art assessment of the Nrf2 pathway as a validated target in COPD. [DOI] [PubMed] [Google Scholar]
- 47.MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thor Soc. 2005;2:50–60. doi: 10.1513/pats.200411-056SF. [DOI] [PubMed] [Google Scholar]
- 48.Luppi F, Hiemstra PS. Epithelial responses to oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:527–531. doi: 10.1164/rccm.200612-1821ED. [DOI] [PubMed] [Google Scholar]
- 49.Tuder RM, Petrache I, Elias JA, et al. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol. 2003;28:551–554. doi: 10.1165/rcmb.F269. [DOI] [PubMed] [Google Scholar]
- 50••.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J COPD. 2009;4:19–31. A critical evaluation of the relationship of apoptosis to emphysema. [PMC free article] [PubMed] [Google Scholar]
- 51.Giordano RJ, Lahdenranta J, Zhen L, et al. Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem. 2008;283:29447–29460. doi: 10.1074/jbc.M804595200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52••.Gorska K, Maskey-Warzechowska M, Krenke R. Airway inflammation in chonic obstructive pulmonary disease. Curr Opin Pulm Med. 2010;16:89–96. doi: 10.1097/MCP.0b013e3283341ba0. An excellent assessment of inflammation and COPD. [DOI] [PubMed] [Google Scholar]
- 53•.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. An excellent comprehensive review of COPD pathogenesis. [DOI] [PubMed] [Google Scholar]
- 54.MacNee W, Tuder RM. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thor Soc. 2009;6:527–531. doi: 10.1513/pats.200905-027DS. [DOI] [PubMed] [Google Scholar]
- 55.Wright TL, Churg A. Current concepts in mechanisms of emphysema. Tox Pathol. 2007;35:111–115. doi: 10.1080/01926230601059951. [DOI] [PubMed] [Google Scholar]
- 56••.Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. 2008;294:L612–L631. doi: 10.1152/ajplung.00390.2007. An insightful review of COPD pathogenesis from animal models. [DOI] [PubMed] [Google Scholar]
- 57.Taraseviciene-Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest. 2008;118:394–402. doi: 10.1172/JCI31811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bode W, Meyer EF, Powers JC. Human leukocyte elastase and porcine pancreatic elastase: X-ray crystal structures, mechanism, substrate specificity, and mechanism-based inhibitors. Biochemistry. 1989;28:1951–1963. doi: 10.1021/bi00431a001. [DOI] [PubMed] [Google Scholar]
- 59.Stein RL, Strimpler AM, Ho H, Powers JC. Catalysis by human leukocyte elastase. Mechanistic insights into specificity requirements. Biochemistry. 1987;26:1301–1305. doi: 10.1021/bi00379a015. [DOI] [PubMed] [Google Scholar]
- 60.Bachovchin WW. 15N NMR spectroscopy of hydrogen-bonding interactions in the active site of serine proteases:evidence for a moving histidine mechanism. Biochemistry. 1986;25:7751–7759. doi: 10.1021/bi00371a070. [DOI] [PubMed] [Google Scholar]
- 61.Ekeowa U, Gooptu B, Belorgey D, et al. Alpha-1-Antitrypsin deficiency, chronic obstructive pulmonary disease and the serpinopathies. Clin Sci (London) 2009;116:837–850. doi: 10.1042/CS20080484. [DOI] [PubMed] [Google Scholar]
- 62.Silverman GA, Bird PI, Carrell W, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 63•.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. A state-of-the-art review of TIMPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turk V, Stoka V, Turk D. Cystatins: biochemical and structural properties, and medical relevance. Front Biosci. 2008;13:5406–5420. doi: 10.2741/3089. [DOI] [PubMed] [Google Scholar]
- 65.Carrell RW, Jeppsson J-O, Laurell CB, et al. Structure and variation of human α-1-antitrypsin. Nature. 1982;298:329–334. doi: 10.1038/298329a0. [DOI] [PubMed] [Google Scholar]
- 66.Cooley J, Takayama TK, Shapiro SD, et al. The serpin MN/EI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry. 2001;40:15762–15770. doi: 10.1021/bi0113925. [DOI] [PubMed] [Google Scholar]
- 67.Thomson RC, Ohlsson K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor: a potent inhibitor of leukocyte elastase. Proc Natl Acad Sci USA. 1986;83:6692–6696. doi: 10.1073/pnas.83.18.6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wiedow O, Schroder J-M, Gregory H, et al. Elafin: an elastase-specific inhibitor of human skin. J Biol Chem. 1990;265:14791–14795. [PubMed] [Google Scholar]
- 69.Wiedow O, Lidemann J, Utecht B. Elafin is a potent inhibitor of proteinase 3. Biochem Biophys Res Comm. 1991;174:6–10. doi: 10.1016/0006-291x(91)90476-n. [DOI] [PubMed] [Google Scholar]
- 70.Boudier C, Bieth J. Mucous proteinase inhibitor: a fast acting inhibitor of leukocyte elastase. Biochem Biophys Acta. 1989;995:36–41. doi: 10.1016/0167-4838(89)90230-6. [DOI] [PubMed] [Google Scholar]
- 71.Churg A, Wang RD, Xie C, Wright JL. α-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2003;168:199–207. doi: 10.1164/rccm.200302-203OC. [DOI] [PubMed] [Google Scholar]
- 72.Taggart C, Cervantes-Laurean D, Kim G, et al. Oxidation of either methionine 351 or methionine 358 in α-1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 73.Beatty K, Matheson N, Travis J. Kinetics and chemical evidence for the inability of oxidized α-1-proteinase inhibitor to protect lung elastin from elastolytic degradation. Biol Chem Hoppe-Zeiler. 1984;365:731–736. doi: 10.1515/bchm2.1984.365.2.731. [DOI] [PubMed] [Google Scholar]
- 74.Li Z, Alam S, Wang J, et al. Oxidized α-1-antitrypsin stimulates the release of monocyte chemotactic protein-1 from lung epithelial cells: potential role in emphysema. Am J Physiol Lung Cell Mol Physiol. 2009;297:L358–L400. doi: 10.1152/ajplung.90373.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhong J, Groutas WC. Recent developments in the design of mechanism-based and alternate substrate inhibitors of serine proteases. Curr Top Med Chem. 2004;4:1203–1216. doi: 10.2174/1568026043387971. [DOI] [PubMed] [Google Scholar]
- 76.Maryanoff BE, Constanzo MJ. Inhibitors of proteases and amide hydrolases that employ α-ketoheterocycles as a key enabling functionality. Bioorg Med Chem. 2008;16:1562–1595. doi: 10.1016/j.bmc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 77.Guarnieri F, Spencer JL, Lucey EC, et al. A human surfactant peptide-elastase inhibitor construct as a treatment for emphysema. Proc Nat Acad Sci USA. 2010;107:10661–10666. doi: 10.1073/pnas.1001349107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Inoue Y, Omodani T, Shiratake R, et al. Development of a highly water-soluble peptide-based human neutrophil elastase inhibitor: AE-3763 for treatment of acute organ injury. Bioorg Med Chem. 2009;17:7477–7486. doi: 10.1016/j.bmc.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 79.Krantz A, Spencer RW, Tam TF, et al. Design and synthesis of 4H-3,1-benzoxazin-4-ones as potent alternate substrate inhibitors of human leukocyte elastase. J Med Chem. 1990;33:464–479. doi: 10.1021/jm00164a002. [DOI] [PubMed] [Google Scholar]
- 80.Neumann U, Schechter NM, Gutschow M. Inhibition of human chymase by 2-amino-3,1-benzoxazin-4-ones. Bioorg Med Chem. 2001;9:947–954. doi: 10.1016/s0968-0896(00)00310-2. [DOI] [PubMed] [Google Scholar]
- 81.Gutschow M, Kuerschner L, Neumann U, Pietsch M, Koglin N, Eger K. 2-(Diethylamino)thieno[1,3]oxazin-4-ones as stable inhibitors of human leukocyte elastase. J Med Chem. 1999;42:5437–5447. doi: 10.1021/jm991108w. [DOI] [PubMed] [Google Scholar]
- 82.Neumann U, Gutschow M. 3,1-Benzothiazin-4-ones and 3,1-benzoxazin-4-ones: Highly different activities in chymotrypsin inactivation. Bioorg Med Chem. 1995;23:72–88. [Google Scholar]
- 83.Mistuhashi H, Nonaka T, Hamamura I, Kishimoto T, Muratani E, Fujii K. Pharmacological activities of TEI-8362, a novel inhibitor of human neutrophil elastase. Brit J Pharmacol. 2009;126:1147–1152. doi: 10.1038/sj.bjp.0702425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hsieh P-W, Yu H-P, Chang Y-J, Hwang TL. Synthesis and evaluation of benzoxazinone derivatives on activity of human neutrophil elastase and on hemorrhagic shock-induced lung injury in rats. Eur J Med Chem. 2010;45:3111–3115. doi: 10.1016/j.ejmech.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 85.Shreder KR, Cajica J, Du L, et al. Synthesis and optimization of 2-pyridyl-3-yl-benzo[d][1,3]oxazin-4-one based inhibitors of human neutrophil elastase. Bioorg Med Chem Lett. 2009;19:4743–4746. doi: 10.1016/j.bmcl.2009.06.053. [DOI] [PubMed] [Google Scholar]
- 86.Mulchande J, Oliveira R, Carrasco M, et al. 4-Oxo-β-lactams (Azetidine-2, 4-diones) are potent and selective inhibitors of human leukocyte elastase. J Med Chem. 2010;53:241–253. doi: 10.1021/jm901082k. [DOI] [PubMed] [Google Scholar]
- 87.Mulchande J, Simoes SI, Gaspar MM, et al. Synthesis, stability, biochemical and pharmacokinetic properties of a new potent and selective 4-oxo-β-lactam inhibitor of human leukocyte elastase. J Enz Inh Med Chem. 2010 doi: 10.3109/14756366.2010.486794. posted online on June 14, 2010. [DOI] [PubMed] [Google Scholar]
- 88.Schepetkin IA, Khlebnikov AI, Quinn MT. N-Benzoyl pyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J Med Chem. 2007;50:4928–4938. doi: 10.1021/jm070600+. [DOI] [PubMed] [Google Scholar]
- 89.Nakayama Y, Odagaki Y, Fujita S, et al. Clarification of the mechanism of human sputum elastase inhibition by a new inhibitor, ONO-5046, using electron-spray ionization mass spectrometry. Bioorg Med Chem Lett. 2002;12:2349–2353. doi: 10.1016/s0960-894x(02)00393-1. [DOI] [PubMed] [Google Scholar]
- 90.Hayakawa M, Ketabami K, Wada T, et al. Silevestat (selective neutrophil elastase inhibitor) improves the mortality rate of sepsis associated with both acute respiratory distress syndrome and disseminated intravascular coagulation patients. Shock. 2010;33:14–18. doi: 10.1097/SHK.0b013e3181aa95c4. [DOI] [PubMed] [Google Scholar]
- 91.Sakashita A, Nishimura Y, Nishiuma T, Takenaka K, Kobayashi K, Kotani Y, Yokoyama M. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur J Pharmacol. 2007;571:62–71. doi: 10.1016/j.ejphar.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 92.Yoshikawa S, Tsushima K, Koizumi T, Kubo K. Effects of a synthetic protease inhibitor (gabexate mesylate) and a neutrophil elastase inhibitor (sivelestat sodium) on acid-induced lung injury in rats. Eur J Pharmacol. 2010;641:220–225. doi: 10.1016/j.ejphar.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 93.Yang Q, Dou D, Gan X, et al. Inhibition of serine proteases by a new class of cyclosulfamide-based carbamylating agents. Arch Biochem Biophys. 2008;475:115–120. doi: 10.1016/j.abb.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, Dou D, He G, et al. Mechanism-based inhibitors of serine proteases with high selectivity through optimization of S′ subsite binding. Bioorg Med Chem. 2009;17:3536–3542. doi: 10.1016/j.bmc.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.He G, Dou D, Wei L, et al. Inhibitors of human neutrophil elastase based on a highly functionalized N-amino-4-imidazolidinone scaffold. Eur J Med Chem. 2010;45:4280–4287. doi: 10.1016/j.ejmech.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Von Nussbaum F, Karthaus D, Klein M, et al. WO135599 A1 20091112. Preparation of 1,4-diarylpyrimidopyridazinyldiones as neutrophil elastase inhibitors. 2009
- 97.Von Nussbaum F, Karthaus D, Anlauf S, et al. WO080199 A1 20090702. 4-(4-Cyano-2-thioaryl)-dihydropyrimidones as neutrophil elastase inhibitors and their preparation and use in the treatment of diseases of the lung and cardiovascular system. 2009
- 98.Ainge D, Chapman D, Lindsjoe M, et al. WO061271 A1 20090514. Preparation of 2-pyrazinone derivatives as inhibitors of neutrophil elastase. 2009
- 99.Finch H, Ray NC, Edwards C. WO060203 A1 20090514. Preparation of 3,4,6,7-tetrahydro-1H-pyrrolo[3,4-d]pyrimidine-2,5-diones having dual neutrophil elastase inhibiting and muscarinic M3 antagonist activity. 2009
- 100.Ray NC, Finch H, Edwards C, et al. WO060158 A1 20090514. Preparation of 4-(4-cyanophenyl)-1-(3-trifluoromethylphenyl)-3,4,6,7-tetrahydro-1H-pyrrolo[3,4-d]pyrimidine-2,5-diones as human neutrophil elastase inhibitors. 2009
- 101.Burkamp F, Hansen P, Hossain N, et al. WO058076 A1 20090507. Preparation of 2-pyrazinone derivatives and their use as inhibitors of neutrophil elastase. 2009
- 102.Bergstroem L, Lundvkist M, Loenn H, Sjoe P. WO030158 A1 20080313. Preparation of 1-phenyl-6-methyl-2-pyridinone derivatives as neutrophil elastase inhibitors. 2008
- 103.Hansen P, Lawitz K, Lepistoe M, Ray A. WO129962 A1 20071115. Preparation of 2-pyridone derivatives as neutrophil elastase inhibitors. 2007
- 104.Brimert T, Lawitz K, Loenn H, et al. WO098684 A1 20080921. Preparation of 2-pyridone derivatives as neutrophil elastase inhibitors and their use for treating inflammatory diseases and conditions. 2006
- 105.Von Nussbaum F, Karthaus D, Anlauf S, et al. WO078953 A1 2010715. Preparation of tetrazolopyrimidines as human neutrophil elastase inhibitors in treatment of pulmonary and cardiovascular diseases. 2010
- 106.Klingstedt T, Lepistoe M, Lundkvist M, Loenn H. WO104752 A1 20080904. Preparation of dihydropyridones as human neutrophil elastase inhibitors. 2008
- 107.Finch H, Edwards C, Ray NC, Fitzgerald MF. WO136857 A1 20061228. Preparation of dihydropyrimidone multimers as human neutrophil elastase inhibitors. 2006
- 108.Thongyoo P, Bonomelli C, Leatherbarrow RJ, Tate EW. Potent inhibitors of β-tryptase and human leukocyte elastase based on the MCoTI-II scaffold. J Med Chem. 2009;52:6197–6200. doi: 10.1021/jm901233u. [DOI] [PubMed] [Google Scholar]
- 109.Sisay MT, Hautmann S, Mehner C, Konig GM, Bajorath J, Gutschow M. Inhibition of human leukocyte elastase by brunsvicamides A-C: cyanobacterial cyclic peptides. ChemMedChem. 2009;4:1425–1429. doi: 10.1002/cmdc.200900139. [DOI] [PubMed] [Google Scholar]
- 110.Craik DJ, Cemazar M, Daly NL. The cyclotides and related macrocyclic peptides as scaffolds in drug design. Curr Opin Drug Discov Dev. 2006;9:251–260. [PubMed] [Google Scholar]
- 111.Schmidtko A, Lotsch S, Freynhagen R, Geisslinger G. Zicotonide for treatment of severe chronic pain. Lancet. 2010;375(9725):1569–1577. doi: 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- 112.Sin DD, Vestbo J. Biomarkers in chronic obstructive pulmonary disease. Proc Am Thor Soc. 2009;6:543–545. doi: 10.1513/pats.200904-019DS. [DOI] [PubMed] [Google Scholar]
- 113.Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. J Chron Obstr Pulm Dis. 2010;7:141–153. doi: 10.3109/15412551003758304. [DOI] [PubMed] [Google Scholar]
- 114.Le Quement C, Guenon I, Gillon JY, et al. The selective MMP-12 inhibitor, AS111793 reduces airway inflammation in mice exposed to cigarette smoke. Brit J Pharmacol. 2008;154:1206–1215. doi: 10.1038/bjp.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Banner KH, Press NJ. Dual PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br J Pharmacol. 2009;157:892–906. doi: 10.1111/j.1476-5381.2009.00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116•.Morjaria JB, Malerba M, Polosa R. Biologic and pharmacologic therapies in clinical development for the inflammatory response in COPD. Drug Discov Today. 2010;15:396–405. doi: 10.1016/j.drudis.2010.03.001. An excellent and informative review related to inflammation and COPD. Includes a discussion of new therapies for COPD. [DOI] [PubMed] [Google Scholar]