

Abstract
We previously demonstrated the activation of Caspase-6 in the hippocampus and cortex in cases of mild, moderate, severe and very severe Alzheimer disease (AD). To determine whether Caspase-6 is also activated in familial AD, we performed an immunohistochemical analysis of active Caspase-6 and Tau cleaved by Caspase-6 in temporal cortex and hippocampal tissue sections from cases of familial AD. The cases included 5 carrying the amyloid precursor protein K670N, M671L Swedish mutation, 1 carrying the amyloid precursor protein E693G Arctic mutation, 2 each carrying the Presenilin I M146V, F105L, A431E, V261F, Y115C mutations, and 1 with the Presenilin II N141I mutation. Active Caspase-6 immunoreactivity was found in all cases. Caspase-6 immunoreactivity was observed in neuritic plaques or cotton wool plaques in some cases, neuropil threads and neurofibrillary tangles. These results indicate that Caspase-6 is activated in familial forms of AD, as previously observed in sporadic forms. Since sporadic and familial AD cases have similar pathological features, these results support a fundamental role of Caspase-6 in the pathophysiology of both familial and sporadic AD.
Keywords: Alzheimer disease, Arctic mutation, Casp6, Familial Alzheimer disease, Presenilin I mutation, Sporadic Alzheimer disease, Swedish mutation, Tau cleaved by Casp6
INTRODUCTION
Caspases, a group of cysteinyl endoproteases that cleave proteins after aspartic acid residues, are activated in inflammatory and apoptotic conditions (1). Caspase-dependent increased β-amyloid peptide production occurs in various cell types (2–4). For example, in primary cultures of human neurons, the caspase responsible for increasing β-amyloid peptide is Caspase-6 (Casp6) (3). We previously demonstrated that neoepitope antiserum to active Casp6 and to Tau protein cleaved by Casp6 (TauΔCasp6) highlights the presence of activated Casp6 in neuropil threads (NPTs), neurofibrillary tangles (NFTs) and neuritic plaques (NPs) in the hippocampus and temporal cortex in sporadic Alzheimer disease (AD) (5). Activation of Casp6 is observed in cases of mild cognitive impairment as well as in mild, moderate, severe and very severe AD (6). Furthermore, abundant active Casp6 and TauΔCasp6 were detected in the entorhinal cortex of a few non-cognitively impaired, aged individuals who had the lowest global cognitive scores of the group studied (6). These findings suggest that Casp6 activation is an early event in AD and that it may precede the development of frank lesions.
Because Casp6 is part of the group of effector caspases, it may cause apoptotic cell death when it is activated. Unlike the rapid induction of apoptosis by the effectors Caspase-3 or Caspase-7 (3, 7), however, Casp6 activation is associated with a protracted type of cell death in serum-deprived human primary neurons. Moreover, Casp6 activation in human embryonic kidney cells does not result in apoptotic cell death (8). In sporadic AD, Casp6 activation is present in neurons that lack the typical morphology of apoptotic neurons. In ischemic human fetal and adult brains Casp6 activation is both neuritic and nuclear and immunopositive neurons have the condensed chromatin appearance of apoptotic cells (5). Nuclear Casp6 is required for apoptotic cell death (9), whereas active Csp6 in sporadic AD is only present in non-nuclear compartments (i.e. cell body and neurites) (5). Taken together these data suggest that activation of Casp6 is more likely associated with neurodegeneration than with apoptotic cell death in sporadic AD. Furthermore, a proteomic study of sporadic AD identified several cytoskeletal and cytoskeleton-associated proteins as potential substrates of Casp6 (10). One of these, α-tubulin is cleaved by Casp6 and is present in neurons in sporadic AD. Recently, Casp6 has also been shown to regulate axonal pruning of sensory and retinocollicular axons and axonal degeneration in sensory and motor mouse neurons (11). Thus, since it can cleave important synaptic and cytoskeleton proteins (10), generate high levels of β-amyloid peptide (3, 12), and is activated very early in cognitive impairment and AD (5, 6), Casp6 activation may play a key role in the development of AD. Since sporadic and familial AD share similar pathological features, Casp6 activity would also likely be involved in the pathogenesis of familial AD. Here, we investigated familial AD brains for immunohistochemical evidence of active Casp6.
MATERIALS AND METHODS
Brain Tissue
Familial AD cases (8-02, 4-93, 25-98, 46-02, 69-01, 77-95, 140-96, and 397-94) were obtained from Huddinge Brain Bank at the Karolinska Institute, Stockholm, Sweden. The postmortem times ranged from 12 to 30 hours. After dissection, brain tissue was placed in buffered 4% formaldehyde for 1 month. The solicitation and storage of the brains were approved by the local ethical committee at Karolinska Institute; all patients (or their nearest relatives) had given informed consent to participate in donation according to the Huddinge Brain Bank routine procedures and provisions of the Helsinki declaration. Familial AD cases 2003-002, 2006-093, 2001-090, 2008-073, 2002-066, 2000-007, 2002-086, 2003-041, 2007-093 were obtained with informed consent from the patients or their nearest relative by the Indiana Alzheimer Disease Center (Indiana University Medical Center, Indianapolis, IN). The postmortem interval for these cases ranged from 2.5 to 8 hours in all cases except 2001-090 (27 hours) and 2002-086 (26 hours). The brains were fixed in formalin and paraffin-embedded. Ages of onset and death are provided in Table 1. Sporadic AD (female/76 years old) and non-AD (female/85 years old) cases, used as a positive and negative controls, respectively, were obtained from Dr Catherine Bergeron (University of Toronto, Toronto, Canada) and processed as described previously (5, 6).
Table 1.
Clinical Data
| Case # | Mutation | Age of onset (years) |
Age at Death (years) |
|---|---|---|---|
| 4–93 | Swedish APP K670N, M671L | 57 | 68 |
| 397–94 | Swedish APP K670N, M671L | 61 | 66 |
| 69–01 | Swedish APP K670N, M671L | 52 | 62 |
| 140–96 | Swedish APP K670N, M671L | 49 | 62 |
| 77–95 | Swedish APP K670N, M671L | 44 | 56 |
| 8–02 | Arctic APP E693G | 57 | 70 |
| 46–02 | PSEN I M146V | 39 | 50 |
| 25–98 | PSEN I M146V | 38 | 48 |
| 2003–002 | PSEN I A431E | 35 | 43 |
| 2006–093 | PSEN I A431E | 41 | 42 |
| 2001–090 | PSEN I F105L | 43 | 73 |
| 2008–073 | PSEN I F105L | 58 | 67 |
| 2000–007 | PSEN I V261F | 36 | 43 |
| 2002–086 | PSEN I V261F | 36 | 47 |
| 2003–041 | PSEN I Y115C | 41 | 51 |
| 2007–093 | PSEN I Y115C | 56 | 68 |
| 2002–066 | PSEN II N141I | 42 | 57 |
APP = amyloid precursor protein; PSEN = presenilin.
Immunohistochemistry
The temporal cortex, hippocampus, and entorhinal cortex were immunostained; in some cases, only 1 of these areas was available. Paraffin-embedded tissues were cut at 4 µm thickness and processed for immunoreactivity with anti-active Casp6 and Tau cleaved by Casp6 (TauΔCasp6) antisera on a Ventana Automated immunostainer (Ventana Medical Systems, Tucson, AZ), as previously described (5, 6). The anti-active Casp6 antiserum 1277 was used at a dilution of 1/1,000 and the anti-TauΔCasp6 antiserum 10635 was used at a 1/12,000 dilution. Detection of immunostaining was done with diaminobenzidine using the iview-DAB kit (Ventana). The PHF-1 antibody was a kind gift from Dr Peter Davis (Department of Pathology, Albert Einstein College of Medicine, Bronx, NY) and was used at a dilution of 1/100. For double staining, anti-active Casp6 was first applied and detected with diaminobenzidine using the iview-DAB kit and PHF-1 followed and was detected with Fast Red A an B using the Enhanced V-Red kit (Ventana).
Semiquantitative Scoring of Immunostaining
NFTs, NPTs, and NPs were identified using conventional neuropathological diagnostic criteria and tau immunostaining by S.A. Flame-shaped filamentous tau-positive neuronal inclusions were considered to represent NFTs. Diffuse, finely granular or homogeneous perikaryal tau positivity were considered pre-tangles and tau-positive thin linear profiles in the neuropil were considered NPTs. NPs were identified as an amyloid core surrounded by a halo of radially arranged, thickened and beaded Tau-positive neurites. So-called cotton wool plaques in presenilin (PSEN) mutant brains were included in the assessment. We considered structures to be Tau-positive when they were stained with either the anti-TauΔCasp6 antiserum or the PHF-1 antibody; there were no identifiable differences in the structures observed with either. Semiquantitative scoring was done based on the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) protocol (13) by neuropathologist S.A. in a blinded manner. The densities of NFTs, NPs, and NPTs stained with anti-active Casp6, TauΔCasp6, or PHF-1 were scored as absent [0], low/mild [1], moderate [2], high/severe [3] or extremely high [4].
Collection of Photomicrographs
The superior/medial temporal gyrus (STG) and the CA2/CA3 area of the hippocampus were chosen as representative areas to assess the intensity and density of the staining. Micrographs were taken with a Nikon CoolPix 4500 digital camera on an Olympus BX41 microscope with 2x and 40×objectives. The same area was photographed in each of the different immunostains. Micrographs were imported and assembled into Canvas 9.0 (ACD Systems of America Inc, Miami, FL) and scaled down to 35% to show the entire image. Micrographs showing anti-active Casp6 and PHF-1 were taken with the 40x or 60×objectives, imported and scaled down by 50%. Otherwise, no other manipulation was done to the micrographs.
RESULTS
Sections from the hippocampus/entorhinal cortex and the STG were immunostained with either the anti-active Casp6 antiserum or TauΔCasp6 antiserum or were double stained with anti-active Casp6 antiserum and anti-paired helical filament PHF-1 antibody (Fig. 1). Low magnification micrographs of a representative case showed a similar distribution of immunopositivity with the 2 antisera and the combination. Anti-active Casp6 staining was less intense than that of the TauΔCasp6 or PHF-1 stains, consistent with a lower abundance of enzymes relative to cytoskeleton proteins in the cells. Figure 1 also indicates specificity of the immunostains since some areas are unstained.
Figure 1.

Immunohistochemical staining with 1277 anti-active Casp6 antiserum, anti-TauΔCasp6 antiserum, and anti-active Casp6 antiserum + anti-PHF-1 antibodies in the superior temporal gyrus (STG) and hippocampus of a representative familial Alzheimer disease (AD) case. Micrographs were taken with the 2x objective and reduced by 50% after importing the image. Detection of anti-active Casp6 and TauΔCasp6 was with diaminobenzidine; PHF-1 immunoreactivity was detected with Fast Red. The squares represent areas shown in Figures 2 and 3. Scale bars = 1 mm.
Active Casp6
The sporadic AD positive control shows positive active Casp6 in NFTs, NPs and NPTs (Fig. 2A); the non-cognitively impaired negative control confirmed the specificity of the anti-active Casp6 immunoreactivity (Fig. 2B). Immunoreactivity was observed in all familial AD cases but differed among them (Fig. 2C–J; Table 2). Immunoreactivity was easily detected in most PSEN I mutants and the PSEN II mutant but was less intense and frequent in the APP mutants, especially in the hippocampus.
Figure 2.
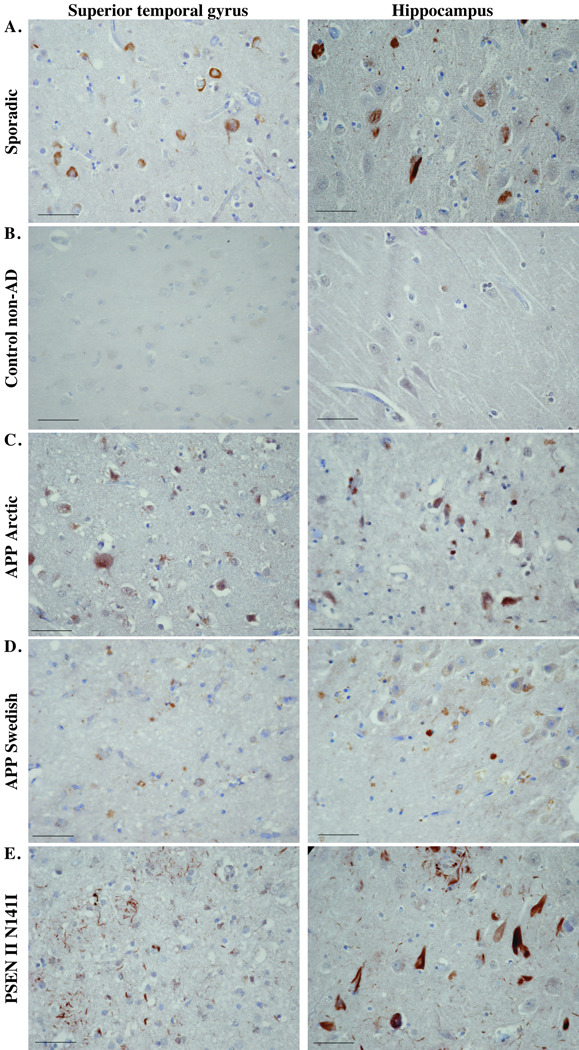

Immunohistochemical staining with anti-active Casp6 antiserum. Micrographs of anti-Casp6 immunostained superior temporal gyrus or hippocampus in the areas indicated in Figure 1 from a sporadic Alzheimer disease (AD) case (A), a non-AD case (B), an AD with the Arctic mutation (C), a representative AD APP Swedish case (D), an AD with the PSEN II N141I AD case (E), and a representative AD case of each of the PSEN I mutants indicated (F–J). Micrographs were taken with the 40x objective and reduced to 35%. Scale bars = 50 µm.
Table 2.
Anti-Active Caspase 6 Immunohistochemistry
| Case | Mutation | Region | NP | NFT | NPT |
|---|---|---|---|---|---|
| 4–93 | Swedish APP K670N, M671L | Cortex | 0 | 2 (pre) | 0 |
| 397–94 | Swedish APP K670N, M671L | Hippocampus | 0 | 2 | 1 |
| 69–01 | Swedish APP K670N, M671L | Cortex | 0 | 1 | 0 |
| 140–96 | Swedish APP K670N, M671L | S/MTG | 0 | 1 (pre) | 0 |
| 77–95 | Swedish APP K670N, M671L | S/MTG | 0 | 1 | 1 |
| 46–02 | PSEN I M146V | S/MTG | 0 | 3 | 0 |
| 25–98 | PSEN I M146V | S/MTG | 0 | 1 | 0 |
| 8–02 | Arctic APP E693G | S/MTG | 0 | 0 | 0 |
| Hippocampus | 0 | 1 | 0 | ||
| ERC | 0 | 0 | 0 | ||
| 2003–002 | PSEN I A431E | S/MTG | 2 | 2 | 2 |
| Hippocampus | 3 | 3 | 2 | ||
| ERC | 2 | 2 | 2 | ||
| 2006–093 | PSEN I A431E | S/MTG | 2 | 3 | 1 |
| Hippocampus | 2 | 3 | 1 | ||
| ERC | 3 | 3 | 1 | ||
| 2001–090 | PSEN I F105L | S/MTG | 3 | 1 | 1 |
| Hippocampus | 1 | 1 | 1 | ||
| ERC | 2 | 1 | 1 | ||
| 2008–073 | PSEN I F105L | S/MTG | 3 | 2 | 2 |
| Hippocampus | 2 | 3 | 1 | ||
| ERC | 3 | 2 | 1 | ||
| 2000–007 | PSEN I V261F | S/MTG | 2–3 | 2–3 | 1 |
| Hippocampus | 0 | 2 | 1 | ||
| ERC | 2 | 2 | 1 | ||
| 2002–086 | PSEN I V261F | S/MTG | 3 | 1–2 | 1 |
| Hippocampus | 2 | 3 | 1 | ||
| ERC | 3 | 3 | 1 | ||
| 2003–041 | PSEN I Y115C | S/MTG | 2 | 1 | 1 |
| Hippocampus | 3 | 3 | 1 | ||
| ERC | 2 | 3 | 1 | ||
| 2007–093 | PSEN I Y115C | S/MTG | 3 | 2 | 1 |
| Hippocampus | 3 | 3 | 1 | ||
| ERC | 2–3 | 2 | 1 | ||
| 2002–066 | PSEN II N141I | S/MTG | 3 | 1 | 1 |
| Hippocampus | 2 | 3 | 1 | ||
| ERC | 3 | 2 | 1 |
Numerical scores indicate relative abundance of staining.
APP = amyloid precursor protein; PSEN = presenilin; NFT = neurofibrillary tangles; NP = neuritic plaques; NPT – neuropil threads; ERC = entorhinal cortex; S/MTG = superior/medial temporal gyrus.
All cases showed some anti-active Casp6-positive structures with the shape of NFTs (Fig. 2C–J). In most cases, equivalent amounts were present in the hippocampus and entorhinal cortex and these were similar or greater than the amount present in the STG (Table 2). Cases with the same mutation mostly showed similar staining. One PSEN I F105L (2001-090) mutant had relatively less staining for active Casp6 than the other case. This individual lived 30 years with the disease compared to 9 years in the F105L mutant with more active Casp6 staining. Similarly, the PSEN I A431 mutant with a disease duration of 1 y (2006-093) showed strong immunopositive NFT staining, whereas the other patient who lived relatively longer (8 years) had only moderate NFT immunoreactivity in the STG and entorhinal cortex.
Immunopositivity for active Casp6 was also present in NPs. This was not observed in the APP mutants or in the PSEN I V261F (2000-007) case hippocampus but was scored as moderate and severe in the other areas in the other PSEN I mutants and in the PSEN II mutant. In the STG of the PSEN I V261F mutant (Fig. 2H), these had the appearance of cotton wool plaques (14–16). In the PSENI F105L, Y115C and the PSEN II N141I mutants there were more Casp6-immunopositive NPs than NFTs in the STG and the entorhinal cortex, and often more NFTs than NPs in the hippocampus. Low levels of anti-active Casp6 immunoreactivity was also observed in NPTs of 2 of the APP Swedish cases, in most PSEN I mutants and in the PSEN II mutant.
Although there was more immunoreactive active Casp6 in the PSEN mutants than in the APP mutants, this difference could simply be the result of an artifact of epitope conservation. Furthermore, since the anti-active Casp6 detects only the large subunit of the active form of Casp6, it is possible that the active form of Casp6 would be inhibited. Therefore, to confirm Casp6 activity in these brains, we used another antiserum directed against a protein cleaved by Casp6, i.e. TauΔCasp6.
TauΔCasp6
As observed previously in sporadic AD, immunoreactivity with the TauΔCasp6 antiserum was much stronger than with the anti-active Casp6 antiserum (Fig. 3). This was expected since a cytoskeleton protein such as Tau is more abundant and stable than the Casp6 enzyme. Almost all cases showed “frequent” NPs, NFTs, and NPTs labeled with the anti-TauΔCasp6 antiserum (Fig. 3; Table 3). Since TauΔCasp6 must be generated from active Casp6, these results indicate that the lower abundance of active Casp6 immunoreactivity in the APP mutants was likely due to loss of the active Casp6 epitope. Nevertheless, all APP and PSEN mutants were scored 2 or 3 for NFTs and in all 3 areas, the APP Arctic case showed sparse TauΔCasp6 immunoreactivity in the STG but retained moderate to severe immunopositive reactivity in the hippocampus and entorhinal cortex. Furthermore, the PSEN I F105L (2001-090) mutant had low levels of immunoreactive NPs and NPTs in the hippocampus; the PSEN I V261F (2000-007) mutant had negligible TauΔCasp6 immunopositive NPs in the hippocampus and the PSEN I A431E (2006-093) mutant showed low levels of TauΔCasp6-positive NPTs in the STG. These results indicate that there is significant Casp6 activity in the STG, the hippocampus and the entorhinal cortex in most of the familial AD cases.
Figure 3.
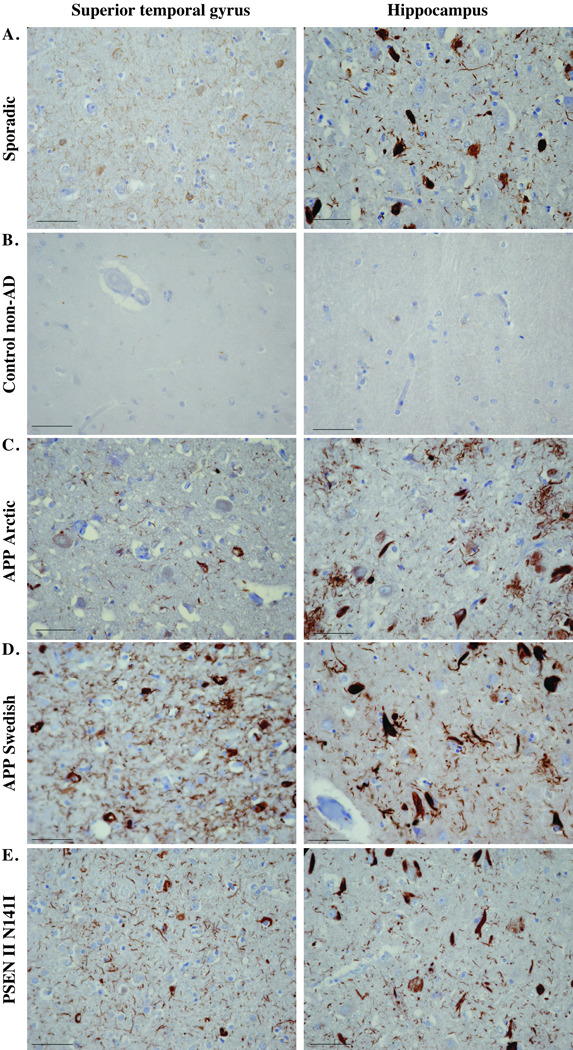

Immunohistochemical staining with 10635 anti-TauΔCasp6 antisera. Micrographs of TauΔCasp6 immunostained superior temporal gyrus or hippocampus in the areas indicated in Figure 1 from a sporadic Alzheimer disease (AD) case (A), a non-AD case (B), an AD with the Arctic mutation (C), a representative AD APP Swedish case (D), an AD with the PSEN II N141I AD case (E), and a representative AD case of each of the PSEN I mutants indicated (F–J). Micrographs were taken with the 40x objective and reduced to 35%. Scale bars = 50 µm.
Table 3.
Anti-TauΔ Casp6 Immunohistochemistry
| Case | Mutation | Region | NP | NFT | NPT |
|---|---|---|---|---|---|
| 4–93 | Swedish APP K670N, M671L | Cortex | 2 | 2 | 2 |
| 397–94 | Swedish APP K670N, M671L | Hippocampus | 3 | 3 | 3 |
| 69–01 | Swedish APP K670N, M671L | Cortex | 3 | 3 | 3 |
| 140–96 | Swedish APP K670N, M671L | S/MTG | 3 | 3 | 3 |
| 77–95 | Swedish APP K670N, M671L | S/MTG | 2–3 | 2–3 | 2–3 |
| 46–02 | PSEN I M146V | S/MTG | 3 | 3 | 3 |
| 25–98 | PSEN I M146V | S/MTG | 3 | 3 | 3 |
| 8–02 | Arctic APP E693G | S/MTG | 1 | 1 | 1 |
| Hippocampus | 3 | 3 | 2–3 | ||
| ERC | 2–3 | 2–3 | 2–3 | ||
| 2003–002 | PSEN I A431E | Hippocampus | 3 | 3 | 3 |
| ERC | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2006–093 | PSEN I A431E | S/MTG | 3 | 3 | 1 |
| Hippocampus | 3 | 3 | 2 | ||
| ERC | 3 | 3 | 2 | ||
| 2001–090 | PSEN I F105L | S/MTG | 3 | 3 | 3 |
| Hippocampus | 1 | 3 | 1 | ||
| ERC | 3 | 3 | 3 | ||
| 2008–073 | PSEN I F105L | S/MTG | 3 | 3 | 3 |
| Hippocampus | 2–3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2000–007 | PSEN I V261F | S/MTG | 3 | 3 | 3 |
| Hippocampus | 0 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2002–086 | PSEN I V261F | S/MTG | 3 | 3 | 2 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2003–041 | PSEN I Y115C | S/MTG | 3 | 3 | 3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2007–093 | PSEN I Y115C | S/MTG | 3 | 3 | 3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2002–066 | PSEN II N141I | S/MTG | 3 | 3 | 3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 |
Numerical scores indicate relative abundance of staining.
APP = amyloid precursor protein; PSEN = presenilin; NFT = neurofibrillary tangles; NP = neuritic plaques; NPT – neuropil threads; ERC = entorhinal cortex; S/MTG = superior/medial temporal gyrus.
Double Staining of Active Casp6 and PHF-1
To determine if the active Casp6 colocalized with NFTs, NPs and NPTs, we performed a double immunostaining with anti-active Casp6 and PHF-1 in the cases from which we had both temporal cortex and hippocampus/entorhinal cortex sections. PHF-1 immunoreactivity was very strong and often overpowered that of the active Casp6. PHF-1 immunostaining was strong in all areas of each case, except in PSEN I F105L (2001-090) in which staining of NPs and NPTs was low in the hippocampus and in the PSEN I V261F (2000-007) mutant in which there was no staining of NPs in the hippocampus (Table 4). This was consistent with the TauΔCasp6 immunostaining results. PHF-1 immunoreactivity was strong in the other 2 cases that had lower TauΔCasp6 (APP Arctic and PSEN I A431 2006-093 S/MTG), however. Most NFTs and many NPTs were stained with both the anti-active Casp6 and the PHF-1 antibodies (Fig. 4A, B) but NFTs and NPTs with only PHF-1 and some with only active Casp6 were also observed (Fig. 4B). In the Arctic case, many neurons had prominent punctate Casp6 immunoreactivity throughout the cytoplasm, which was distinct from diffuse staining of pre-tangles. On double immunolabeling, a few of the neurons showed punctate staining with both anti-Casp6 and the PHF-1 antibodies (Fig. 4C).
Table 4.
Paired Helical Filament-1 Immunohistochemistry
| Case | Mutation | Region | NP | NFT | NPT |
|---|---|---|---|---|---|
| 8–02 | Arctic APP E693G | S/MTG | 2–3 | 2–3 | 2–3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2003–002 | PSEN I A431E | S/MTG | 3 | 3 | 4 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 4 | ||
| 2006–093 | PSEN I A431E | S/MTG | 3 | 3 | 3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2001–090 | PSEN I F105L | S/MTG | 3 | 3 | 3 |
| Hippocampus | 1 | 3 | 1 | ||
| ERC | 3 | 3 | 3 | ||
| 2008–073 | PSEN I F105L | S/MTG | 3 | 3 | 4 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 4 | ||
| 2000–007 | PSEN I V261F | S/MTG | 3 | 3 | 3 |
| Hippocampus | 0 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2002–086 | PSEN I V261F | S/MTG | 3 | 3 | 3–4 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3–4 | ||
| 2003–041 | PSEN I Y115C | S/MTG | 3 | 3 | 3–4 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3–4 | ||
| 2007–093 | PSEN I Y115C | S/MTG | 3 | 3 | 3–4 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 | ||
| 2002–066 | PSEN II N141I | S/MTG | 3 | 3 | 3 |
| Hippocampus | 3 | 3 | 3 | ||
| ERC | 3 | 3 | 3 |
Numerical scores indicate relative abundance of staining.
APP = amyloid precursor protein; PSEN = presenilin; NFT = neurofibrillary tangles; NP = neuritic plaques; NPT – neuropil threads; ERC = entorhinal cortex; S/MTG = superior/medial temporal gyrus.
Figure 4.

Co-immunostaining of active Casp6 with PHF-1. Micrographs show colocalization of active Casp6 (brown) and PHF-1 (red) in (A) neurofibrillary tangles (black arrow), (B) neuropil threads (black arrowhead) and a structure resembling a tangle stained only with anti-active Casp6 (white arrow). Left panel is a lower magnification indicating the area where the picture was taken (black arrow). Scale bar = 1 mm. Right panel is a higher magnification. Scale bar = 50 µm. (C) Micrograph showing punctate cytoplasmic anti-active Casp6 immunoreactivity in the APP Arctic case. The right panel represents a magnified image of the anti-active Casp6 immunopositive neurons with more or less PHF-1 immunoreactivity.
DISCUSSION
To provide evidence in support of the hypothesis that activated Casp6 is an important upstream component of AD pathogenesis, we investigated whether Casp6 was activated in familial forms of AD. Our results indicate that Casp6 is activated in the brains of familial AD cases due to a variety of distinct mutations. This was shown using the neoepitope antiserum 1277, which detects the large p20 subunit necessary for the active form of Casp6 and by showing immunoreactivity with the 10635 neoepitope antiserum against Tau protein truncated at amino acid 402 (VSGD) by Casp6 (5); this neoepitope antiserum does not detect full length Tau protein.
The immunoreactivity with anti-active Casp6 was less intense in the APP and the PSEN I M146V mutants than in the other PSEN I mutants and the PSEN II mutant. Since all cases showed considerable immunoreactivity against TauΔCasp6, a more abundant and stable protein than Casp6, this indicates that the Casp6 was activated in these cases. Loss of the active Casp6 epitope might be due to several factors. Since the PSEN I mutant (2002-086) showed strong immunoreactivity despite having a postmortem interval of 27 hours, it is unlikely to be an artifact due to postmortem delay. There was also no effect of disease duration on the level of active Casp6 immunoreactivity. It is possible, however, that Casp6 was activated earlier in these cases and became attenuated at the end stage of disease. Consistent with this idea, Casp6 is activated early in sporadic AD (as shown in mild cognitively impaired brains) and decreases in the end stage of disease (6). Alternatively, the active Casp6 epitope was less well preserved in the APP and PSEN I M146V familial AD cases.
As in sporadic AD, anti-active Casp6 and TauΔCasp6 immunoreactivity was not associated with the chromatin condensation in neurons characteristic of apoptotic cells death. Therefore, it is likely that in both sporadic and familial AD cases, the active Casp6 performs a different function. Since several of the Casp6 substrates are cytoskeleton and cytoskeleton-associated proteins (10) and Casp6 activity results in axonal pruning and degeneration in mice neurons in culture (11), active Casp6 may play a role in neurodegeneration rather than in cell death in AD. Exceptions were the 1 Arctic case and 1 APP Swedish case (69-01), both of which had anti-active Casp6 nuclear staining in numerous neurons. These neurons had small, condensed nuclei and their cytoplasm was shrunken often leaving a pericellular halo; they were, therefore, most likely ischemic neurons. Indeed, nuclear active Casp6 immunoreactivity was previously observed in ischemic neurons (5). The active Casp6 in the nucleus of cells is associated with apoptotic cell death as Casp6 cleaves the lamins, especially lamin A, resulting in chromatin condensation (9). Since these occurred only in a few cases, ischemia was not likely directly related to the familial AD mutations.
There was considerable variability amongst the different cases. For example, in the Arctic case, TauΔCasp6 immunoreactivity in neuropil threads that occasionally arranged in a circular fashion was reminiscent of the coreless ring-like amyloid plaques observed in these cases (17). In a few of the PSEN cases there were cotton wool plaques that stained with both the anti-active Casp6 and the TauΔCasp6 antibodies. In the Arctic case, punctate active Casp6 immunoreactivity that often did not colocalize with PHF-1 immunoreactivity was observed throughout the cytoplasm although some punctate staining was observed with both antibodies in some neurons.
Active Casp6 staining in neuronal round cytoplasmic inclusions was obvious in the Swedish and Arctic cases but was also seen in PSEN I and PSEN II cases; this was unexpected and differed from our previous observations of active Casp6 in mild and moderate forms of sporadic AD (5, 6). Similar inclusions were, however, previously observed in PHF-1-positive NFTs in very severe sporadic AD (6) and such inclusions are generally considered to represent end stage pathological events (18). Therefore, the presence of active Casp6 inclusions could be due to more widespread simultaneous neuronal involvement in familial AD than in sporadic AD. Accumulation of active Casp6 in inclusions is also not unique to this caspase. Active Caspase-3 is abundant in ubiquitin- and cytokeratin-positive inclusions that form in apoptotic epithelial cells in culture and these inclusions might sequester active caspases thereby delaying apoptosis (19, 20). Similar sequestration of active Casp6 in degenerating neurons might prevent the complete demise of the affected neurons since complete destruction of the neurites would have a severe irreversible impact on brain function.
The amyloid hypothesis predicts oligomeric β-amyloid peptide to be responsible for the activation of Casp6 in familial AD. Although primary neurons resist extracellular β-amyloid peptide toxicity unless they are co-incubated with the phosphoinositide 3-kinase inhibitor, wortmannin, the intracellular β-amyloid peptide is highly toxic to human neurons and this toxicity is dependent on Casp6 activation (21) . Consistent with this hypothesis, Casp6 is more strongly activated in the APP Swedish and PSEN 1 hippocampus, which increase the levels of β-amyloid peptide compared to the Arctic case, which simply increases the propensity of the β-amyloid peptide to aggregate (22, 23). The other areas of the Arctic case did not differ significantly from the APP Swedish, PSEN I and PSEN II cases, however. Consequently, we cannot exclude the possibility that the familial AD-linked mutations initially disrupt cellular homeostasis to result in Casp6 activation independently of the production of β-amyloid peptide. Indeed, serum deprivation in human primary neurons results in Casp6-dependent increase of β-amyloid peptide (3, 12). Furthermore, pro-Casp6 expression, modulated by p53 activation, increases in AD (24, 25). Over-expression of pro-Casp6 in the human embryonic kidney 293 cell line results in Casp6 self-activation (8), indicating that over-expression of Casp6 is sufficient to result in its activation. Therefore, a disruption of cellular homeostasis by familial AD mutant proteins that would increase pro-Casp6 protein levels could result in the activation of Casp6 in all of these cases.
In conclusion, the presence of Casp6 activity in familial AD APP, PSEN I and PSEN II mutants supports the hypothesis that Casp6 activity is involved in the pathophysiology of AD.
ACKNOWLEDGMENTS
The authors thank Dr Catherine Bergeron (U. Toronto, Canada) for the sporadic AD and control tissues. The authors gratefully acknowledge the technical assistance of Jocelyne Jacques from the Department of Pathology at the JGH. The authors also thank Dalia Halawani and Andrea Lee from the LeBlanc laboratory for reading the manuscript. Several cases were provided by the Indiana Alzheimer Disease Center (Indiana University, Indianapolis).
This work was supported by CIHR MOP81146 and FRSQ to ALB, Swedish Research Council to NB and BW, and NIA P30 AG10133 to BG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Creagh EM, Conroy H, Martin SJ. Caspase-activation pathways in apoptosis and immunity. Immunol Rev. 2003;193:10–21. doi: 10.1034/j.1600-065x.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 2.Gervais F, Xu D, Robertson G, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's ß-amyloid precursor protein and amyloidogenic ß-peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 3.LeBlanc AC, Liu H, Goodyer C, et al. Caspase-6 role in apoptosis of human neurons, amyloidogenesis and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- 4.Tesco G, Koh YH, Tanzi RE. Caspase activation increases beta-amyloid generation independently of caspase cleavage of the beta-amyloid precursor protein (APP) J Biol Chem. 2003;278:46074–46080. doi: 10.1074/jbc.M307809200. [DOI] [PubMed] [Google Scholar]
- 5.Guo H, Albrecht S, Bourdeau M, et al. Active caspase-6 and caspase-6 cleaved tau in neuropil threads, neuritic plaques and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol. 2004;165:523–531. doi: 10.1016/S0002-9440(10)63317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albrecht S, Bourdeau M, Bennett D, et al. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol. 2007;170:1200–1209. doi: 10.2353/ajpath.2007.060974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Goodyer C, LeBlanc A. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and -8. J Neurosci. 2000;20:8384–8389. doi: 10.1523/JNEUROSCI.20-22-08384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaiman G, Champagne N, LeBlanc AC. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim Biophys Acta. 2009;1793:592–601. doi: 10.1016/j.bbamcr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Ruchaud S, Korfali N, Villa P, et al. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. Embo J. 2002;21:1967–1977. doi: 10.1093/emboj/21.8.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klaiman G, Petzke TL, Hammond J, et al. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics. 2008;7:1541–1555. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikolaev A, McLaughlin T, O'Leary DD, et al. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.LeBlanc A. Increased production of 4 kDa amyloid beta peptide in serum deprived human primary neuron cultures: possible involvement of apoptosis. J Neurosci. 1995;15:7837–7846. doi: 10.1523/JNEUROSCI.15-12-07837.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD) .2. Standardization of the Neuropathologic Assessment of Alzheimer's Disease. Neurol. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 14.Houlden H, Baker M, McGowan E, et al. Variant Alzheimer's disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol. 2000;48:806–808. [PubMed] [Google Scholar]
- 15.Miravalle L, Calero M, Takao M, et al. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochem. 2005;44:10810–10821. doi: 10.1021/bi0508237. [DOI] [PubMed] [Google Scholar]
- 16.Tabira T, Chui DH, Nakayama H, et al. Alzheimer's disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70:367–372. doi: 10.1002/jnr.10392. [DOI] [PubMed] [Google Scholar]
- 17.Basun H, Bogdanovic N, Ingelsson M, et al. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008;65:499–505. doi: 10.1001/archneur.65.4.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10 Suppl:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 19.MacFarlane M, Merrison W, Dinsdale D, et al. Active caspases and cleaved cytokeratins are sequestered into cytoplasmic inclusions in TRAIL-induced apoptosis. J Cell Biol. 2000;148:1239–1254. doi: 10.1083/jcb.148.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinsdale D, Lee JC, Dewson G, et al. Intermediate filaments control the intracellular distribution of caspases during apoptosis. Am J Pathol. 2004;164:395–407. doi: 10.1016/S0002-9440(10)63130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, McLaughlin R, Goodyer C, et al. Selective cytotoxicity of intracellular amyloid ß peptide1-42 through p53 and bax in cultured primary human neurons. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng IH, Scearce-Levie K, Legleiter J, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 23.Cheng IH, Palop JJ, Esposito LA, et al. Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- 24.MacLachlan TK, El-Deiry WS. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci U S A. 2002;99:9492–9497. doi: 10.1073/pnas.132241599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raina AK, Hochman A, Zhu X, et al. Abortive apoptosis in Alzheimer's disease. Acta Neuropathol (Berl) 2001;101:305–310. doi: 10.1007/s004010100378. [DOI] [PubMed] [Google Scholar]