

Summary
The ArsD metallochaperone delivers trivalent metalloids, As(III) or Sb(III), to the ArsA ATPase, the catalytic subunit of the ArsAB As(III) efflux pump. Transfer of As(III) increases the affinity of ArsA for As(III), allowing resistance to environmental arsenic concentrations. As(III) transfer is channeled from chaperone to ATPase, implying that ArsD and ArsA form an interface at their metal binding sites. A genetic approach was used to test this hypothesis. Thirteen ArsD mutants exhibiting either weaker or stronger interaction with ArsA were selected by either repressed transactivator yeast two-hybrid or reverse yeast two-hybrid assays. Additionally, Lys-37 and Lys-62 were identified as being involved in ArsD function by site-directed mutagenesis and chemical modification. Substitution at either position with arginine was tolerated, suggesting participation of a positive charge. By yeast two-hybrid analysis K37A and K62A mutants lost interaction with ArsA. All fifteen mutations were mapped on the surface of the ArsD structure, and their locations are consistent with a structural model generated by in silico docking. Four are close to metalloid binding site residues Cys-12, Cys-13 and Cys18, and seven are on the surface of helix 1. These results suggest that the interface involves one surface of helix 1 and the metalloid binding site.
Keywords: ArsD, metallochaperone, ArsA, antimonite, arsenite, resistance, transport ATPase
Introduction
Arsenic is the most prevalent environmental toxin, introduced from both geochemical and anthropogenic sources. As a result of its ubiquity and threat to human health, this metalloid has always ranked first on the U.S. Department of Health and Human Services and Environmental Protection Agency Superfund Priority List of Hazardous Substances <http://www.atsdr.cdc.gov/cercla/07list.html>. In humans arsenic exposure causes cancer, cardiovascular disease, peripheral neuropathies and diabetes mellitus (Abernathy et al., 1999, Beane Freeman et al., 2004). Most prokaryotes and eukaryotes, from E. coli to humans, have arsenic detoxification mechanisms (Bhattacharjee & Rosen, 2007). The arsRDABC operon of plasmid R773 encodes an arsenic detoxification system. The ArsAB complex is an ATP-driven As(III)/Sb(III) efflux pump, in which the 583 residue ArsA ATPase is the catalytic subunit (Hsu & Rosen, 1989).
ArsD is a metallochaperone for transfer of cytosolic As(III) or Sb(III) to ArsA (Lin et al., 2006, Lin et al., 2007a, Yang et al., 2010). Other metallochaperones include those for the transition metals copper and iron (Field et al., 2002, Rosenzweig, 2002). ArsD is a homodimeric 120-residue protein with three conserved cysteine residues, Cys-12, Cys-13 and Cys-18, that are required for chaperone activity and form a three-coordinate binding site for As(III). Wild type ArsD has two non-conserved cysteine pairs at Cys-112/Cys-113 and Cys-119/Cys-120 that are not required for ArsD chaperone activity (Lin et al., 2007a). While the wild type protein binds three As(III) per monomer, a derivative with a truncation at residue 109 that binds only a single As(III) was used in this and other studies (Yang et al., 2010). ArsA has two homologous halves, A1 and A2, connected by a short linker. Each half has a consensus nucleotide-binding domain (NBD) (Zhou et al., 2000). ArsA exhibits a low, basal rate of ATPase activity in the absence of As(III) or Sb(III) and a higher, activated rate when metalloid is bound. ArsA has a high affinity metalloid binding site composed of Cys-113 and Cys-422 (Ruan et al., 2006), and a third residue, Cys-172, that participates in high affinity binding and activation of ATP hydrolysis (Ruan et al., 2008). By transferring As(III) to ArsA, ArsD increases the affinity of ArsA for As(III), activating ArsA ATPase activity and conferring resistance to concentrations of arsenic found in many environmental settings (Lin et al., 2006).
We have proposed a model for metal transfer in which ArsA and ArsD interact at their metalloid binding sites, with As(III) or Sb(III) transferred in a step-wise manner from the cysteine residues of ArsD to those in ArsA (Lin et al., 2007b). In this report we use a genetic analysis to test this hypothesis and to identify ArsD residues that might interact with ArsA. Three types of mutant isolation were utilized: repressed transactivator yeast two-hybrid screens, reverse yeast two-hybrid screens and site directed mutagenesis. Many of the mutations clustered in two regions: near the metalloid binding site and on the surface of the helix 1. Recently the X-ray crystal structure of ArsD was solved at 1.4 Å, with two molecules in the asymmetric unit showing a dimer of 50 × 35 × 30 Å3 (Ye et al., 2010). Using this ArsD structure and the 2.1 Å crystal structure of ArsA (Zhou et al., 2000), an in silico analysis of ArsD-ArsA interaction was performed in parallel with the mutant selections (Ye et al., 2010). An ArsD monomer (38 × 35 × 15 Å3) fit well into the cavity (35 × 20 × 20 Å3) of the open form of ArsA. Overall, many of the ArsD residues found in the mutant analysis were also predicted to be within 3.2 Å of ArsA. The correspondence of these two independent lines of evidence, in vivo mutagenic data and in silico modeling of sites of interaction, supports our model of the interface between ArsD and ArsA.
Results
ArsD mutants exhibiting stronger interaction with ArsA
Using standard yeast two-hybrid analysis (Fields & Song, 1989), interaction of ArsD and ArsA was demonstrated (Lin et al., 2006). In this analysis the arsA or arsD genes were cloned into either the C-terminal DNA binding domain (BD) as “bait” or the C-terminal activation domain (AD) as “prey” of the split GAL4 transcriptional activator. Interaction of the two proteins resulted in expression of a HIS3 report gene, allowing growth of the yeast cells on media lacking histidine.
An extension of this procedure, called the repressed transactivator two-hybrid system, can be used to select for mutants with increased affinity for a binding partner (Hirst et al., 2001, Joshi et al., 2007). The HIS3 gene encodes imidazole glycerol phosphate dehydratase, catalyzing the sixth step in histidine biosynthesis. Histidine synthesis by this enzyme is competitively inhibited by 3-amino-1,2,4-triazole (3-AT) (Klopotowski & Wiater, 1965). If 3-AT is added to the medium, yeast cells must express higher levels of His3p to grow in the absence of histidine. Random mutagenesis of arsD was performed by error-prone polymerase chain reaction (PCR) induced with Mn2+ (Cadwell & Joyce, 1992) using a pair of primers homologous to the flanking regions of the arsD gene in plasmid pBD-D, in which arsD was fused to the sequence for the GAL4 DNA binding domain (see Supplemental Table 1 for strains and plasmids). The purified PCR product was mixed with vector plasmid pBD-k (addition of a kanamycin resistance gene) linearized with EcoRI and BamHI, co-transformed with pAD-D (the arsD sequence fused to the GAL4 activation domain sequence in vector plasmid pACT2) into S. cerevisiae strain AH109 and selected on SD-L-W-H plates with 10 mM 3-AT. By this method the PCR fragment integrates into the vector in vivo through homologous recombination (Hua et al., 1997). Approximately 30 colonies were isolated on SD-L-W-H plates with 10 mM 3-AT. The arsD gene from each colony was sequenced. Of the 30, some were duplicates, and 24 were unique. Of those 24, some contained single mutations, while others contained multiple mutations (Supplemental Table 2). Some substitutions such as S14R, T20I, Q24L, D28V and Q34R were found in a number of the mutants. In addition, multiple substitutions were found at residues 20 (Ile and Thr) and 28 (Thr, Asn, Val and His). Seventeen individual mutations were reintroduced by site-directed mutagenesis into the arsD gene to examine whether they increased the strength of interaction with ArsA. Other multiple mutations have not yet been analyzed in detail. From the results of yeast two-hybrid analysis, nine of the 17 mutations at eight positions (S14R, T20I, Q24L, D28T, D28V, T31A, Q34R, Q38R and V61A) exhibited apparently increased interaction with ArsA (Fig. 1A and Supplemental Table 2). AH109 yeast cell co-transformed with pBD-k-D and pAD-A (arsA sequence fused to the GAL4 activation domain sequence in plasmid pACT2) did not grow on SD-L-W-H plates containing 10 mM 3-AT, while AH109 yeast cell co-transformed with pBD-k-D mutants and pAD-A was able to grow in the presence of 30 - 60 mM 3-AT, except for Q38R, which exhibited growth in only 10 mM 3-AT. As a control, the interaction of each arsD mutation with pAD-D (arsD) was examined (Fig. 1B). Since ArsD is a homodimer, this estimates the ability of the mutants to dimerize with wild type ArsD. None of the mutants could grow in the presence of 3-AT, indicating that these mutants do not enhance dimerization with the wild type ArsD as a result of increased expression or protein folding/stability. In other words, these mutations have specific effects on interaction with ArsA.
Fig. 1. Yeast two-hybrid analysis of ArsD mutants exhibiting increased interaction with ArsA.

S. cerevisiae strain AH109 bearing pBD-k-D wild typeArsD or mutants (indicated at left) and either pAD-A (A) or pAD-D (B) plasmids was grown in SD medium overnight and inoculated with 10-fold serial dilutions on SD agar plates lacking histidine and supplemented with the indicated concentrations of 3-AT. The plates were incubated at 30 °C for 3 days and scored for growth.
ArsD mutants showing weaker interaction with ArsA
Using reverse yeast two-hybrid selection, five ArsD mutants with apparent weaker interaction with ArsA were isolated (Table 1). Reverse yeast two-hybrid selection was performed using URA3 as a reporter gene (Vidal et al., 1996). The URA3 gene encodes orotidine 5-phosphate decarboxylase, which metabolizes 5-fluoroorotic acid (5-FOA) to 5-fluorouridine, an analog of uridine, inhibiting cell growth. Selection for growth in the presence of 5-FOA yields for mutants with apparent weaker interactions, allowing for mapping the interface between interacting proteins (Dhayalan et al., 2008).
Table1.
Summary of ArsD mutants
| Residue | Location | Mutations | Phenotype/Interaction with ArsA |
Residues in 100 closest homologues |
|---|---|---|---|---|
| Cys-12 | Metal binding loop | C12A | Inactive | Cys |
| Cys-13 | Metal binding loop | C13A | Inactive | Cys |
| Ser-14 | Metal binding loop | S14R | Increased | Ser |
| Val-17 | Metal binding loop | V17A | Decreased | Val |
| Cys-18 | Metal binding loop | C18S | Inactive | Cys |
| Thr-20 | Metal binding loop | T20I | Increased | Thr/Ser/Val/Ala/Pro |
| Val-22 | Metal binding loop | V22A | Decreased | Val/Thr |
| Gln-24 | Helix 1 | Q24L | Increased | Gln/Pro |
| Val-27 | Helix 1 | V27D | Decreased | Val/Thr |
| Asp-28 | Helix 1 | D28T/V | Increased | Asp/Asn/Thr/Ser/Ala |
| Thr-31 | Helix 1 | T31A | Increased | Thr/Ala |
| Gln-34 | Helix 1 | Q34R | Increased | Asn/Asp/Glu |
| Lys-37 | Helix 1 | K37A/R | Decreased/None | Lys/Arg |
| Gln-38 | Helix 1 | Q38R | Increased | Gln/Ser/Thr/Lys |
| Gln-51 | Loop | Q51H | Decreased | Gln/Glu |
| Phe-55 | Helix 2 | F55L | Decreased | Phe |
| Val-61 | Helix 3 | V61A | Increased | Val/Ala |
| Lys-62 | Helix 3 | K62A/R | Decreased/None | Lys |
Strain MaV203 co-transformed with pAD-A and pBD-D could not grow on SD-L-W plates with 0.2 % 5-FOA, consistent with ArsD-ArsA interaction resulting in metabolism of 5-FOA. As described above, arsD in pBD-D was mutagenized by error-prone PCR. The purified PCR products were mixed with vector plasmid pBD-k linearized with EcoRI and BamHI, co-transformed with pAD-A into yeast strain MaV203 (URA3) and selected on SD-L-W plates with 0.2 % 5-FOA. 70 colonies were isolated with 5-FOA, compared with 1000 colonies on a plate without 5-FOA. 5-FOA resistance could result from reduced interaction of ArsD and ArsA, but other mutations producing chain termination or improperly folded ArsD, or even loss of the arsD from the plasmid would give the same phenotype. To distinguish between these possibilities, the ability of the ArsD mutants to dimerize with wild type ArsD by yeast two-hybrid analysis was examined. The 70 colonies were pooled, and the arsD genes amplified by PCR. The purified PCR products were mixed with vector plasmid pBD-k linearized with EcoRI and BamHI, co-transformed with either pAD-D into yeast strain AH109 and selected on plates lacking histidine. Only 300 colonies were isolated from plates lacking histidine, compared with 4000 colonies in the presence of histidine, indicating that fewer than 10% produced ArsD mutants able to dimerize with wild type ArsD. Of those 300, 30 colonies were purified, the arsD genes sequenced, and their ability to interact with either ArsA or ArsD examined by forward yeast two-hybrid assays in the absence of 5-FOA (Fig. 2). Five (V17A, V22A, V27D, Q51H and F55L) of the 30 did not interact with ArsA (Fig. 2A) but still interacted with wild type ArsD (Fig. 2B).
Fig. 2. Yeast two-hybrid analysis of ArsD mutants showing decreased interaction with ArsA.
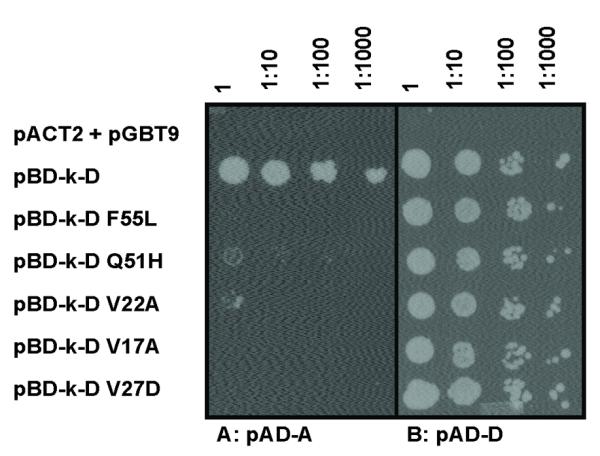
S. cerevisiae strain AH109 bearing pBD-k-D wild type or mutants (indicated at left) and either pAD-A (A) or pAD-D (B) plasmids was grown in SD medium overnight and inoculated on agar plates with SD lacking histidine with 10-fold serial dilutions. Plates were incubated at 30 °C for 3 days and scored for growth. As a negative control, the top row contained yeast cells co-transformed with vector plasmids pGBT9 and pACT2.
ArsD residues Lys-37 and Lys-62 are involved in interaction with ArsA
Alanine mutagenesis of lysine residues in ArsD was performed in an unsuccessful attempt to control reaction with an amine-specific crosslinker. There are six lysine residues in ArsD at positions 2, 37, 60, 62, 90, 104. A series of mutants were constructed by site directed mutagenesis, and the ArsD proteins purified. The purified proteins were screened for their ability to increase the affinity of ArsA for As(III) and hence stimulating ArsA ATPase activity at low metalloid concentrations (Lin et al., 2006). ArsD decreased the concentration of As(III) required for half-maximal stimulation of ArsA ATPase activity, i.e., the apparent affinity of ArsA for As(III), from approximately 540 to 20 μM (Fig. 3A). The K2A/K60A/K90A/K104A quadruple mutant stimulated ATPase activity similarly to the parental protein, indicating that those four lysine residues are not involved in interaction with ArsA. In contrast, the quadruple lysine mutant K2A/K37A/K62A/K104A was not able to stimulate ArsA ATPase activity. The K2A/K37R/K62R/K104A quadruple mutant was close to the parental ArsD in its ability to stimulate ArsA activity. These data suggest that an alanine substitution of Lys-37 and/or Lys-62 results in loss of stimulation of ArsA ATPase activity, while an arginine substitution is tolerated. Note that residue 62 is a lysine in the 100 closest homologues, and residue 37 is either lysine or arginine (Table 1).
Fig. 3. Activation of ArsA ATPase activity by ArsD lysine mutants and abolition of ArsD metallochaperone activity by acetylation.

Activated ArsA ATPase activity was determined at the indicated concentrations of sodium arsenite in the presence or absence of ArsD derivatives, as described under Materials and Methods. The concentration of ArsA was 0.3 μM, and ArsD was 6 μM. A. The concentration of As(III) required for half maximal activation is given in parentheses. (●), no ArsD (K1/2 = 540 μM); (○), ArsD (K1/2 = 20 μM); (Δ), K2A/K60A/K90A/K104A (K1/2 = 20 μM); (▼), K2A/K37R/K62R/K104A (K1/2 = 20 μM); (□), K2A/K37A/K62A/K104A (K1/2 = 520 μM). B. (●), no ArsD (K1/2 = 510 μM); (○), ArsD (K1/2 = 20 μM); (■),K2A/K104A (K1/2 = 10 μM); (□), K37A (K1/2 = 150 μM); (▼), K62A (K1/2 = 290 μM); (Δ), K37A/K62A (K1/2 = 430 μM). C. (●), no ArsD (K1/2 = 520 μM); (□), ArsD (K1/2 = 10 μM); (■), acetylated ArsD (K1/2 = 200 μM); (▲), K2A/K60A/K90A/K104A (K1/2 = 30 μM); (Δ), acetylated K2A/K60A/K90A/K104A (K1/2 = 280 μM); (▼), K2A/K37R/K62R/K104A (K1/2 = 10 μM); (◇), acetylated K2A/K37R/K62R/K104A (K1/2 = 10 μM). The data were fitted using SigmaPlot 9.0, with error bars representing the standard deviation from three assays.
To examine the role of lysine residues in more detail, single and double mutants were created. The double K2A/K104A mutant was able to stimulate ArsA ATPase activity as well as the parental ArsD, while the K37A/K62A double mutant exhibited almost no stimulation (Fig. 3B). The single lysine mutants, K37A and K62A, decreased the concentration of As(III) required for half-maximal stimulation of ArsA ATPase activity to 150 μM and 290 μM, respectively, showing that they retain some activity. They confer a much lower stimulation than the parental ArsD but higher than the double mutant. These results suggest that both Lys-37 and Lys-62 play roles during arsenic transfer from ArsD to ArsA and that a positive charge may be required at those two positions.
The requirement for a positive charge was further investigated by chemical modification of lysine residues with sulfosuccinimidyl acetate (Sulfo-NHS acetate), which acetylates amine groups and neutralizes the positive charge (Lewis et al., 1988). As expected, ArsD increased the apparent affinity of ArsA for As(III) from 520 μM to 10 μM before acetylation, and 200 μM after acetylation. Acetylation decreased ArsD activity dramatically although not completely, perhaps due to partial acetylation (Fig. 3C). The K2A/K60A/K90A/K104A quadruple mutant, with only lysine residues Lys-37 and Lys-62 remaining, increased the apparent affinity of ArsA for As(III) to 30 μM before acetylation, and 280 μM after acetylation. This suggests that acetylation of Lys-37 and Lys-62 causes loss of ArsD metallochaperone function. The K2A/K37R/K62R/K104A quadruple mutant, with only lysine residues Lys-60 and Lys-90 remaining, produced no increase in the apparent affinity of ArsA for As(III), with values of 10 μM before and after acetylation. This is consistent with proposal that Lys-60 and Lys-90 are not required for stimulating ArsA ATPase activity. It also shows that acetylation has no effect on ArsD except for neutralization of the charges on the lysine residues. It is not surprising that acetylation has no effect on K2A/K37R/K62R/K104A, in which residues 37 and 62 were substituted with arginines, since residues 37 and 62 retain positive charges after acetylation by Sulfo-NHS acetate. These results indicate that positively charged residues at positions 37 and 62 are involved in ArsD metallochaperone function.
There are several possible reasons for the loss of ability to stimulate ArsA. One possibility is that the mutant ArsD no longer binds metalloid. The ability of the K2A/K37A/K62A/K104A quadruple mutant to bind Sb(III) was determined (Supplemental Fig. 1). The C-terminally form of ArsD used in this study, which contains only a single metalloid binding site per monomer (Lin et al., 2007a, Yang et al., 2010), bound Sb(III) with a stoichiometry of approximately 0.8 Sb(III)/ArsD. The C12S/C13S/C18A triple mutant, which lacks the metalloid binding site, bound only low levels of Sb(III). The K2A/K37A/K62A/K104A quadruple mutant bound Sb(III) as well as the parental ArsD.
A second possibility is that the interaction of the mutant ArsD with ArsA has been altered. The lysine mutants were cloned into the yeast two-hybrid binding domain vector pBD-k and examined for interaction with either ArsA or wild type ArsD in the pACT2 activation domain vector (Fig. 4). All of the mutants retained interaction with wild type ArsD in pAD-D, indicating that they fold sufficiently normally to dimerize (Fig. 4B). Only the K2A/K37A/K62A/K104A was unable to interact with ArsA in pAD-A (Fig. 4A). The interaction of the K2A/K37R/K62R/K104A quadruple mutant was normal, consistent with the necessity for positive charges at positions 37 and 62. The K2A/K37A/K104A triple mutant had reduced interaction with ArsA, but the K2A/K62A/K104A triple mutant interacted normally. Thus, substitution of both Lys-37 and Lys-62 appears to be additive, consistent with the biochemical results. These results indicate that the effect of the lysine substitutions, particularly when both are changed, is to reduce interaction with ArsA without affecting metalloid binding.
Fig. 4. Yeast two-hybrid analysis of interaction between ArsD lysine mutants and ArsA or wild type ArsD.
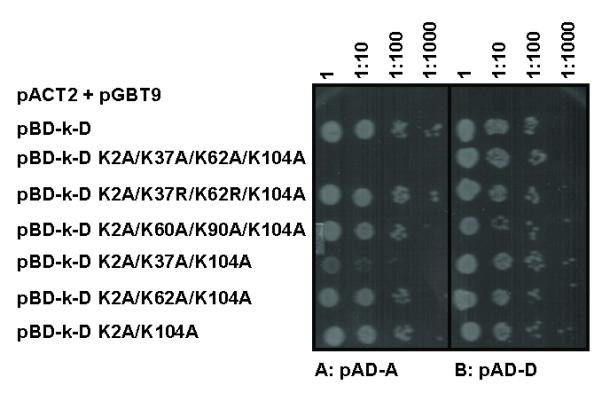
S. cerevisiae strain AH109 bearing pBD-k-D wild type or lysine mutants (indicated at left) and either pAD-A (A) or pAD-D (B) plasmids was grown in SD medium overnight and inoculated on agar plates with SD lacking histidine with 10-fold serial dilutions. Plates were incubated at 30 °C for 3 days and scored for growth. As a negative control, the top row contained yeast cells co-transformed with vector plasmids pGBT9 and pACT2.
Discussion
The ArsD chaperone transfers As(III) to ArsA, activating its ATPase activity and facilitating extrusion of the toxic metalloid (Lin et al., 2006). There has been considerable progress on the physiological and biochemical consequences of the interaction of the two partner proteins (Lin et al., 2007a, Yang et al., 2010). The 1.4 Å crystal structure of ArsD has been reported recently (Ye et al., 2010). The three residues, Cys-12, Cys-13 and Cys-18, that form the metalloid binding site are in a flexible loop that is connected to the helix 1 (residues 24-38). However, molecular details of the interaction and transfer reaction are lacking. In this study we analyzed the interaction genetically by the isolation of mutants in ArsD that either increased or decreased affinity for ArsA. The placement of the 16 mutations described above, as well as the three cysteines that form the As(III) binding site, on a monomer of ArsD are shown in Fig. 5A. The majority of the mutations are located from Ser-14 to Gln-38. These form a continuous span of residues from the metalloid binding loop (Ser-14, Val-17, Thr-20 and Val-22) to the end of the helix 1 (Gln-24, Val-27, Asp-28, Thr-31, Gln-34, Lys-37 and Gln-38) (Fig. 5B). S14R, T20I, Q24L, D28T/V, T31A, Q34R and Q38R each increase interaction with ArsA, while V17A, V22A, V27D and K37A decrease interaction. In addition, there are several mutations in other places. Q51H in the loop between strand 2 and helix 2, F55L in helix 2 and K62A in helix 3 each decrease interaction, while V61A in helix 3 increases interaction.
Fig. 5. Localization of mutations on the ArsD structure.
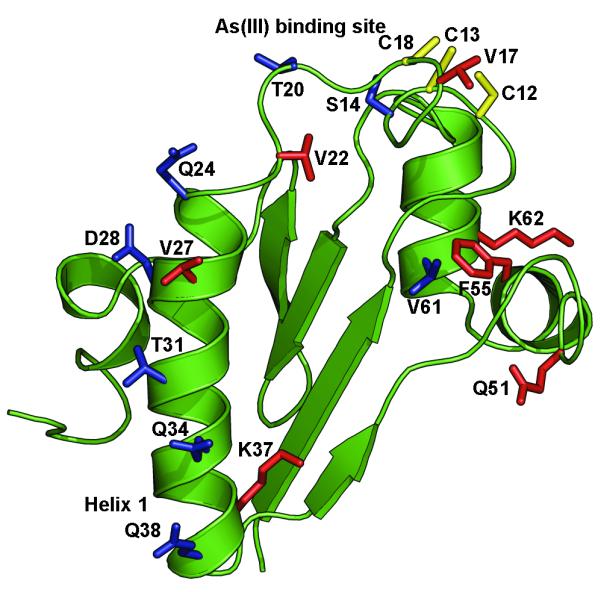

A. Cartoon of the ArsD structure in which the metalloid binding site was modeled (Ye et al., 2010) showing the location of residues mutated in this study. B. Residues mutated in this study are identified on the surface of ArsD. (a) Surface view showing the metalloid binding site; (b) the view was rotated along the x-axis to show the surface of helix 1. (c) and (d) show enlarged semi-transparent views of the metalloid binding site and helix 1 allowing visualization of the individual mutated residues. Residues in which mutations that increase interaction with ArsA are shown in blue. Those that result in decreased interaction with ArsA are shown in red. The cysteines of the metalloid binding site are shown in yellow.
The results clearly identify a surface of ArsD from the metalloid binding site to the end of helix 1 that is involved in interaction with ArsA. We have previously shown ArsD and ArsA could be crosslinked through the cysteines of their respective metalloid binding sites, so it is not surprising that residues in that loop (Ser-14, Val-17, Thr-20 and Val-22) would affect interaction; the two proteins almost certainly contact each other at this point during metalloid transfer. Comparison of these residues in the 100 closest homologues of R773 ArsD shows that the three cysteine residues, Ser-14 and Val-17 are conserved in all 100. In fact, most of the amino acids from residue 7 to 19 show little or no variability. The cysteines would be expected to be conserved since the thiolates are the direct arsenic ligands, but preservation of the others indicate that their side chains make essential contributions to ArsD function such as protein folding, metalloid binding and/or interaction with/transfer to ArsA.
Helix 1 is likely to be a major site of interaction with ArsA. Helices 2 and 3 may also contact ArsA at some point during metalloid transfer since several of the mutations (Q51H, F55L and V61A) are located in those helices. There is no clear explanation why some substitutions exhibit positive and others negative phenotypes. The effects could be indirect through conformational changes in ArsD. For example, the phenyl ring in Phe-55 in helix 2 pi-bonds with the Phe-7 and also interacts through weak hydrogen bonds with the carbonyl oxygens of Gln-51, Asn-47, Phe-46 and Asp-8. A leucine residue at position 55 would be predicted to lose these interactions and might cause a change in the secondary structure of helix 2 of ArsD. It is interesting to note that Phe-55 is absolutely conserved in the 100 closest homologues.
Lys-37 and Lys-62 may play a special role in ArsD function. Alanine substitutions abolish interaction with ArsA, but arginine substitutions are permitted. In the 100 closest homologues, residue 37 is either lysine or arginine, and residue 62 is always lysine. In the crystal structure, their side chains protrude into the solvent, giving ArsD a net positive charge at those surfaces. Lys-62 is in on the surface of helix 3 and forms a N-H...O hydrogen bond with side chain of the Glu-59. Glu-59 is the starting residue of the helix 3, and it would be predicted that an alanine residue at position 62 would be unable to hydrogen bond with Glu-59. Lys-37 is in helix 1, but its side chain protrudes from the opposite side of the helix from the residues in helix 1 identified in the mutant selection as increasing interaction with ArsA, and the K37A mutation exhibits decreased interaction. Other mutations that decrease interaction include V17A, V22A and V27D. Val-17 is conserved in the 100 closest homologues, and Val-22 and Val-27 are either a valine or threonine residues. Although threonine is more polar than valine, the two residues are isosteric, suggesting that the volumes of these residues may be involved in maintaining the ArsD conformation. To explore the role of the residues identified by random mutagenesis, a detailed analysis of the effects of site-directed substitutions with other residues will be conducted in future experiments. The purpose of this study was to identify residues that might form the interface with ArsA and not to identify the mechanisms of altered interaction with ArsA, which will require detailed biochemical and structural analysis of the altered proteins.
In parallel with this mutational analysis, the ArsD-ArsA interaction has been analyzed by in silico docking (Ye et al., 2010). In that analysis, an ArsD monomer fit into the cavity of the open form of ArsA at their metalloid binding sites. Although it is not known whether ArsD interacts with ArsA as a monomer or dimer, no reasonable solution to the docking analysis would accommodate a dimer. Overall, fifteen ArsD residues were predicted to be within 3.2 Å of 13 ArsA residues (Fig 6A). Comparison of those residues with those identified by mutational analysis (Fig. 6B) shows that all of the common residues are in the metalloid binding site and the outer surface of helix 1 (Fig. 6C). This agreement between two independent methods of analysis gives considerable confidence in the prediction that major contact areas between ArsD and ArsA take place at this surface of ArsD. This prediction can be further tested by site directed mutagenesis of those ArsD residues. In addition, the docking model predicts residues in ArsA that are 3-4 Å of those ArsD residues, which provides targets for further testing. The eventual goals are to determine the interacting surfaces of both proteins and the mechanism of transfer of As(III) from chaperone to ATPase.
Fig. 6. Comparison of ArsA residues identified from in silico docking with mutagenesis.

A. A model of ArsD interacting with ArsA was generated by in silico docking (Ye et al., 2010). ArsD residues predicted to be within of residues in ArsA are identified. B. Residues in which mutations that affect interaction with ArsA are identified. C. Residues common between the docking model and the results of mutagenesis are identified. In each case residues in which mutations that increase interaction with ArsA are shown in blue. Those that result in decreased interaction with ArsA are shown in red. The cysteines of the metalloid binding site are shown in yellow. Residues predicted from modeling to be near ArsA residues but which were not found in mutants are shown in cyan. Structural models were rendered using PYMOL (DeLano, 2001) (http://www.pymol.org).
Materials and Methods
Reagents
3-AT was purchased from MP Biomedicals (Solon, OH). 5-FOA was purchased from Fermentas (Burlington, Ontario). Yeast minimal SD base, amino acid Drop-out supplements (-Leu/-Trp and -Leu/-Trp/-His) were purchased from Clontech (Mountain view, CA). Sulfo-NHS acetate was purchased from Thermo Scientific. Unless otherwise mentioned, all other chemicals were obtained from Sigma.
Strains, plasmids and media
E. coli strain JM109 was used for molecular cloning. E. coli strain BL21(DE3) was used for protein expression and purification. ArsA with C-terminal His6 tag was cloned in the expression vector pAlter-1 as pAlter-dAhB (Li & Rosen, 2000). Two forms of ArsD were used in this study. The gene for full-length wild type ArsD was used in yeast two-hybrid analyses. All biochemical assays were performed with a fully active ArsD C-terminally truncated at residue 109 with a six-histidine tag (termed simply ArsD in this study). The gene for this ArsD was cloned in the expression vector pET28a as pET28a-ArsD109. E. coli cells were grown at 37°C in Luria-Bertani (LB) medium (Sambrook et al., 1989). Ampicillin (100 μg/ml), kanamycin (40 μg/ml) and 0.3 mM isopropyl-ß-D-thiogalactopyranoside were added as required. Yeast cells were grown in complete yeast extract-peptone-dextrose (YPD) or minimal synthetic dextrose (SD) media with the appropriate supplements at 30°C (Adams et al., 1998). Growth in liquid culture was estimated from the absorbance at 600 nm. Details of strain and plasmids are listed in Supplemental Table 1.
DNA manipulation and mutagenesis
Plasmid extraction, DNA restriction endonuclease analysis, ligation and other general molecular biological procedures were performed as described (Sambrook et al., 1989). Transformation of E. coli cells was carried out using a BIO-RAD MicroPluser (BIO-RAD, Hercules, CA). Transformation of yeast cells was performed using Fast Yeast Transformation™ kit from G-Biosciences (Maryland Heights, MO). DNA purification kits were obtained from QIAGEN (Valencia, CA). Either a Qiaprep Spin Miniprep kit or a Qiaquick gel extraction kit was used to prepare plasmid DNA for restriction enzyme digestion, sequencing, and recovering DNA fragments from agarose gels. Site-directed mutagenesis was performed using Quick-change Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The sequence of new plasmid constructs was confirmed by DNA sequencing using a CEQ2000 DNA sequencer (Beckman Coulter, Brea, CA).
Generation of a random mutated library of PCR fragments
Random mutagenesis of ArsD was performed using error-prone (ep)-PCR employing biased nucleotide composition of the PCR buffer, high concentration of Mg2+ and addition of Mn2+ (Cadwell & Joyce, 1992). The ep-PCR reaction mixture (50 μl) contained 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 7 mM MgCl2, 0.5mM MnCl2, 0.2 mM each of dATP and dGTP, 1 mM each of dCTP and dTTP, 25 pmol each of the oligonucleotide primers (forward, TAA AGA TGC CGT CAC AGA TAG ATTG; reverse, ACC TGA CCT ACA GGA AAG AGT TACT), 80 ng of template DNA, 2.5 U of Taq DNA polymerase (Invitrogen). PCR condition was 1 × 96 °C for 3 min, 30 × 94 °C for 30 s, 1 × 55 °C for 30 s, 1 × 72 °C for 100s, and, finally, 1 × 72 °C for 10 min. The ep PCR product was purified by agarose gel and quantified by absorption at 260 nm.
Yeast two-hybrid analysis
Plasmids pGBT9 and pACT2 were used as S. cerevisiae/E. coli shuttle vectors for yeast two-hybrid assay (Lin et al., 2007a). Two yeast stains, AH109 (Fields & Song, 1989) and MaV203 (Vidal et al., 1996), were used for yeast two-hybrid assays. Both are GAL4-based yeast two-hybrid systems. AH109 was used to analyze protein-protein interaction or for selection of mutants that retaining capability of interaction or showing stronger interaction in the presence of 3-AT. MaV203 was used to select for mutants with reduced capability of interaction through counter-selection in the presence of 5-FOA. pGBT9 was modified to pBD-k by inserting kanamycin resistant gene through AatII site in plasmid pGBT9. To determine protein-protein interaction by growth of serial dilutions, the transformed cells were cultured overnight in SD medium at 30°C and then washed, suspended and adjusted to an absorbance of 1 at A600nm in 20 mM Tris-HCl, pH 7.5. Portions of the cell suspensions (1 μl) were inoculated in serial 10-fold dilutions on SD agar plates lacking histidine or with 3-AT at the indicated concentration. The plates were incubated at 30°C for 3 days.
Sequencing of ArsD mutant genes in yeast colonies
Each yeast colony was grown in SD medium. The plasmids were isolated from yeast by Zymoprep II™ Yeast Plasmid Miniprep Kit (Zymo Research Cop. Orange, CA), transformed to E. coli JM109 and grown on LB plates with kanamycin. The plasmids were extracted from E. coli JM109, and arsD mutations in the pBD-k vector were sequenced with the GAL4-BD sequencing primer (GAG TAG TAA CAA AGG TCA A).
Protein expression and purification
Cells bearing the indicated plasmids were grown in LB medium overnight at 37°C and then diluted 50-fold into 1 L of the same medium. Proteins were expressed by induction with 0.3 mM isopropyl-ß-D-thiogalactopyranoside at A600 of 0.6-0.8 for 3 hrs. ArsA with a six histidine tag at the C-terminus was purified from cells of strain BL21(DE3) expressing pAlter-1-dAhB plasmid, as described (Zhou & Rosen, 1997). ArsD and its derivatives with a six histidine tag at the N-terminus were purified similarly. Purified proteins were stored at −80°C until use, and their concentrations were determined according to the method of Bradford (Bradford, 1976) or from the absorption at 280 nm (Gill & von Hippel, 1989).
ATPase activity assays
ATPase activity was estimated using a couple assay (Vogel & Steinhart, 1976), as described (Hsu & Rosen, 1989). The assay mixture in 0.2 ml final volume contained in Buffer A (50 mM MOPS-KOH, pH 7.5, 0.25 mM EDTA): 5 mM ATP, 1.25 mM phosphoenolpyruvate, 0.25 mM NADH, 2 units of pyruvate kinase (Sigma) and lactate dehydrogenase (Sigma), 0.3 μM ArsA, 6 μM ArsD, and increasing concentrations of sodium arsenite, all pre-warmed to 37 °C. The reaction was initiated by the addition of 2.5 mM MgCl2 and followed by monitoring the decrease in absorbance at 340 nm using a SpectraMax 340PC microplate reader (Molecular Devices). The linear steady state rates were used to calculate the specific activity.
Acetylation of lysine by sulfo-NHS acetate
The buffer of purified ArsD and derivatives was exchanged with a buffer containing 50 mM MOPS-KOH, 0.2 M NaCl, pH 7.8, 0.25 mM EDTA, 5mM tris(2-carboxyethyl)phosphine (TCEP) and 5 mM dithiolthreitol (DTT). Sulfo-NHS acetate dissolved in deionized water was diluted 100-fold into ArsD solutions to a final concentration of 1 mM, which represents at least a 10-fold excess over the amount of free amine in ArsD. The mixture was incubated for 60 min at room temperature and quenched with an excess of Tris-HCl, pH 6.8, final concentration, for 20 min. The solution was buffer exchanged and concentrated with an Amicon Ultra-15 Centrifugal Filter Unit. Glycerol was added to the concentrated protein at a final concentration of 10% (w/v), following which the protein was stored in small aliquots at −80°C until use.
Metalloid binding assay
The buffer of purified ArsD was exchanged with a buffer containing 50 mM MOPS-KOH, pH 7.5, 0.25 mM EDTA using a Bio-Gel P-6 Micro Bio-Spin column (Bio-Rad, Hercules, CA). Purified protein (100 μM) was incubated at 4 °C with 300 μM potassium antimonyl tartrate. After 1 h, each sample was passed through a Bio-Gel P-6 column equilibrated with the same buffer. The concentration of protein in the flow through was quantified from the absorption at 280 nm (Gill & von Hippel, 1989) or according to the method of Bradford (Bradford, 1976). The flow through was diluted with 2% HNO3, and the quantity of metalloid was measured by inductively coupled mass spectrometry (ICP-MS) with a PerkinElmer ELAN 9000. Antimony standard solutions in the range of 1-20 ppb in 2% HNO3 were obtained from Ultra Scientific, Inc. (North Kingstown, RI). From these data the molar ratio of antimony to protein was calculated.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health Grant AI43428.
References
- Abernathy CO, Liu YP, Longfellow D, Aposhian HV, Beck B, Fowler B, Goyer R, Menzer R, Rossman T, Thompson C, Waalkes M. Arsenic: health effects, mechanisms of actions, and research issues. Environ Health Perspect. 1999;107:593–597. doi: 10.1289/ehp.99107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A, Gottschling DE, Kaiser C, Stearns T. Methods in Yeast Genetics: A Cold Spring Harbor laboratory course manual. Cold Spring Harbor laboratory; Cold Spring Harbor, NY: 1998. p. 177. [Google Scholar]
- Beane Freeman LE, Dennis LK, Lynch CF, Thorne PS, Just CL. Toenail arsenic content and cutaneous melanoma in Iowa. Am J Epidemiol. 2004;160:679–687. doi: 10.1093/aje/kwh267. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee H, Rosen BP. Arsenic metabolism in prokaryotic and eukaryotic microbes. In: Nies DHS, Simon, editors. Molecular microbiology of heavy metals. Springer-Verlag; Heidelberg/New York: 2007. pp. 371–406. [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL user’s manual. DeLano Scientific; San Carlos, CA: 2001. [Google Scholar]
- Dhayalan A, Jurkowski TP, Laser H, Reinhardt R, Jia D, Cheng X, Jeltsch A. Mapping of protein-protein interaction sites by the ‘absence of interference’ approach. J Mol Biol. 2008;376:1091–1099. doi: 10.1016/j.jmb.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field LS, Luk E, Culotta VC. Copper chaperones: personal escorts for metal ions. Journal of bioenergetics and biomembranes. 2002;34:373–379. doi: 10.1023/a:1021202119942. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Hirst M, Ho C, Sabourin L, Rudnicki M, Penn L, Sadowski I. A two-hybrid system for transactivator bait proteins. Proc Natl Acad Sci U S A. 2001;98:8726–8731. doi: 10.1073/pnas.141413598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CM, Rosen BP. Characterization of the catalytic subunit of an anion pump. J Biol Chem. 1989;264:17349–17354. [PubMed] [Google Scholar]
- Hua SB, Qiu M, Chan E, Zhu L, Luo Y. Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast. Plasmid. 1997;38:91–96. doi: 10.1006/plas.1997.1305. [DOI] [PubMed] [Google Scholar]
- Joshi PB, Hirst M, Malcolm T, Parent J, Mitchell D, Lund K, Sadowski I. Identification of protein interaction antagonists using the repressed transactivator two-hybrid system. Biotechniques. 2007;42:635–644. doi: 10.2144/000112434. [DOI] [PubMed] [Google Scholar]
- Klopotowski T, Wiater A. Synergism of aminotriazole and phosphate on the inhibition of yeast imidazole glycerol phosphate dehydratase. Arch. Biochem. Biophys. 1965;112:562–566. doi: 10.1016/0003-9861(65)90096-2. [DOI] [PubMed] [Google Scholar]
- Lewis PN, Guillemette JG, Chan S. Histone accessibility determined by lysine-specific acetylation in chicken erythrocyte nuclei. Eur J Biochem. 1988;172:135–145. doi: 10.1111/j.1432-1033.1988.tb13865.x. [DOI] [PubMed] [Google Scholar]
- Li J, Liu S, Rosen BP. Interaction of ATP binding sites in the ArsA ATPase, the catalytic subunit of the Ars pump. J Biol Chem. 1996;271:25247–25252. doi: 10.1074/jbc.271.41.25247. [DOI] [PubMed] [Google Scholar]
- Li J, Rosen BP. The linker peptide of the ArsA ATPase. Mol Microbiol. 2000;35:361–367. doi: 10.1046/j.1365-2958.2000.01696.x. [DOI] [PubMed] [Google Scholar]
- Lin YF, Walmsley AR, Rosen BP. An arsenic metallochaperone for an arsenic detoxification pump. Proc Natl Acad Sci U S A. 2006;103:15617–15622. doi: 10.1073/pnas.0603974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Yang J, Rosen BP. ArsD residues Cys12, Cys13, and Cys18 form an As(III)-binding site required for arsenic metallochaperone activity. J Biol Chem. 2007a;282:16783–16791. doi: 10.1074/jbc.M700886200. [DOI] [PubMed] [Google Scholar]
- Lin YF, Yang J, Rosen BP. ArsD: an As(III) metallochaperone for the ArsAB As(III)-translocating ATPase. J. Bioenerget. Biomemb. 2007b;39:453–458. doi: 10.1007/s10863-007-9113-y. [DOI] [PubMed] [Google Scholar]
- Rosenzweig AC. Metallochaperones: bind and deliver. Chem Biol. 2002;9:673–677. doi: 10.1016/s1074-5521(02)00156-4. [DOI] [PubMed] [Google Scholar]
- Ruan X, Bhattacharjee H, Rosen BP. Cys-113 and Cys-422 form a high affinity metalloid binding site in the ArsA ATPase. J Biol Chem. 2006;281:9925–9934. doi: 10.1074/jbc.M600125200. [DOI] [PubMed] [Google Scholar]
- Ruan X, Bhattacharjee H, Rosen BP. Characterization of the metalloactivation domain of an arsenite/antimonite resistance pump. Mol Microbiol. 2008;67:392–402. doi: 10.1111/j.1365-2958.2007.06049.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory; New York: 1989. [Google Scholar]
- Vidal M, Brachmann RK, Fattaey A, Harlow E, Boeke JD. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc Natl Acad Sci U S A. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel G, Steinhart R. ATPase of Escherichia coli: purification, dissociation, and reconstitution of the active complex from the isolated subunits. Biochemistry. 1976;15:208–216. doi: 10.1021/bi00646a032. [DOI] [PubMed] [Google Scholar]
- Yang J, Rawat S, Stemmler TL, Rosen BP. Arsenic binding and transfer by the ArsD As(III) metallochaperone. Biochemistry. 2010;49:3658–3666. doi: 10.1021/bi100026a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ajees AA, Yang J, Rosen BP. The 1.4 A crystal structure of the ArsD arsenic metallochaperone provides insights into its interaction with the ArsA ATPase. Biochemistry. 2010;49:5206–5212. doi: 10.1021/bi100571r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Radaev S, Rosen BP, Gatti DL. Structure of the ArsA ATPase: the catalytic subunit of a heavy metal resistance pump. Embo J. 2000;19:1–8. doi: 10.1093/emboj/19.17.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Rosen BP. Tryptophan fluorescence reports nucleotide-induced conformational changes in a domain of the ArsA ATPase. J Biol Chem. 1997;272:19731–19737. doi: 10.1074/jbc.272.32.19731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.