

Abstract
Neurofibromatosis type 1 (NF1) or von Recklinghausen neurofibromatosis is a genetic disorder that occurs in 1 of 4000 births and is characterized by benign and malignant tumors. Cardiovascular defects also contribute to NF1, though the pathogenesis is still unclear. Deficiency in neurofibromin (encoded by Nf1) in mice results in mid-embryonic lethality owing to cardiac abnormalities previously thought to be secondary to cardiac neural-crest defects. Using tissue-specific gene inactivation, we show that endothelial-specific inactivation of Nf1 recapitulates key aspects of the complete null phenotype, including multiple cardiovascular abnormalities involving the endocardial cushions and myocardium. This phenotype is associated with an elevated level of ras signaling in Nf1−/− endothelial cells and greater nuclear localization of the transcription factor Nfatc1. Inactivation of Nf1 in the neural crest does not cause cardiac defects but results in tumors of neural-crest origin resembling those seen in humans with NF1. These results establish a new and essential role for Nf1 in endothelial cells and confirm the requirement for neurofibromin in the neural crest.
Neurofibromin has ras GTPase-activating activity and can downregulate ras signaling. The absence of this activity is thought to underlie some or all aspects of NF1 in humans. NF1 is characterized by proliferation and malignant transformation of neural-crest derivatives. Affected individuals also have disorders that seem unrelated to the neural crest, including hypertension, renal artery stenosis, higher incidence of congenital heart disease, especially valvular pulmonic stenosis, and vascular abnormalities in the central nervous system known as moya-moya1,2. NF1 functions as a tumor-suppressor gene, and loss of heterozygosity in somatic tissues has been associated with tumor formation3.
Attempts to produce animal models of NF1 have been hampered by the fact that inactivation of Nf1 in mice leads to mid-gestation lethality from cardiovascular abnormalities. These defects include structural malformations of the outflow tract of the heart and enlarged endocardial cushions3–5, which are the anlage of cardiac valves. Normally, neural-crest cells migrate to the outflow region of the heart and contribute to the endocardial cushions as early as embryonic day (E) 10.5 (Fig. 1a,b). Neural-crest defects can result in abnormal morphogenesis of the heart6, and cardiac abnormalities in Nf1−/− embryos have been attributed to the loss of neurofibromin function in cardiac neural-crest cells3,4. But the majority of cells in the forming endocardial cushions are derived from underlying endothelial cells that undergo epithelial–mesenchymal transformation (Fig. 1c–e). Hearts of Nf1−/− embryos have abnormal epithelial–mesenchymal transformation5. This could result from defective signaling by the neural crest or myocardium or from abnormal differentiation of the endothelium.
Fig. 1.

Tissue-restricted transgenic expression of Cre recombinase in neural crest and endothelium. E10.5 embryos (a,c) and hearts (b,d) resulting from a cross of either Wnt1–cre (a,b) or Tek–cre (c,e) mice to the Z/EG reporter mouse. Cre activates expression of GFP in derivatives of the neural crest (a,b) and of endothelial cells (c–e). A transverse section through the heart of a Tek–cre × Z/EG embryo (e) shows GFP expression in both endothelial cells lining the endocardial cushions (yellow arrows) and the endothelial-derived mesenchymal cells within the cushion (white arrows). f, PCR was used to detect the recombined allele (Rcre) as described previously7 in genomic DNAs isolated from Nf1flox embryonic hearts either positive or negative for Tek–cre. PCR amplification of the Nf1flox allele from both samples is shown below as control. pa, pharyngeal arches; nt, neural tube; rv, right ventricle; lv, left ventricle; nc, neural crest; h, heart; ec, endocardial cushions.
We used a conditional gene inactivation approach to ablate neurofibromin function specifically in the endothelium. A conditional allele of Nf1 (Nf1tm1Par, herein called Nf1flox) is susceptible to Cre-mediated recombination in vivo (Fig. 1f) and is a phenocopy of the null allele7. We crossed Nf1flox mice with Tek–cre transgenic mice (also called Tie2-Cre) in which expression of Cre recombinase is driven by an endothelial-specific promoter/enhancer8. Endothelial-specific inactivation of Nf1 (Nf1ec) recapitulates the cardiovascular phenotype observed in Nf1−/− embryos. We examined cardiac anatomy of wild-type, Nf1−/− and Nf1ec embryos from eight litters (Fig. 2 and Table 1). In comparison with wild-type embryos (Fig. 2a,e), Nf1−/− (Fig. 2b,f) and Nf1ec (Fig. 2c,g) embryos had enlarged endocardial cushions. Hearts of Nf1ec embryos also had ventricular septal defects (Fig. 2g), double outlet right ventricle (data not shown), thinned myocardium (Fig. 2k) and pericardial effusions (Fig. 2c and data not shown), which were similar to those seen in Nf1−/− embryos.
Fig. 2.
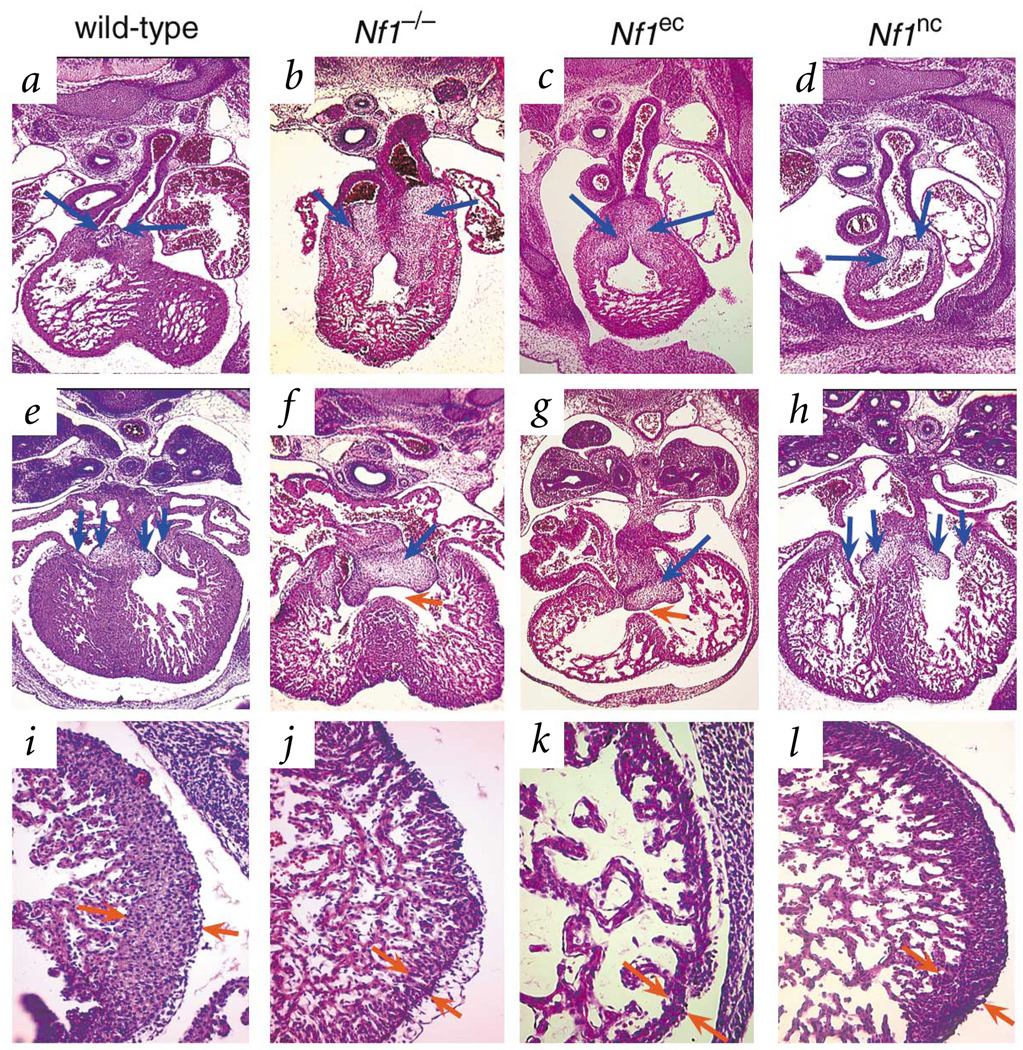
Endothelial-specific Nf1 inactivation disrupts heart development. Sections of hearts from E13.5 wild-type (a,e,i), Nf1−/− (b,f,j), Nf1ec (c,g,k) and Nf1nc (d,h,l) embryos. Outflow tract endocardial cushions (a–d, blue arrows) and atrioventricular cushions (e–h, blue arrows) and high-power magnification of left ventricular myocardium (i–l) are shown. The endocardial cushions of hearts from Nf1ec embryos (c,g) were enlarged and appeared similar to those of Nf1−/− embryos (b,f). A ventricular septal defect was seen in hearts of both Nf1−/− (f) and Nf1ec (g) embryos. The compact layer of the myocardium of Nf1ec embryos (k) was thinned, as it was in Nf1−/− embryos (j), whereas Nf1nc embryos (l) resembled wild-type (i).
Table 1.
Cardiac phenotypes in Nf1 knockout mice
| Cardiac phenotype | |||||
|---|---|---|---|---|---|
| Genotype | Number analyzed | DORV | VSD | Enlarged AV cushion | Thinned myocardium |
| wild-type | 10 | 0 | 1 | 0 | 0 |
| Nf1−/− | 10 | 10 | 10 | 10 | 10 |
| Nf1ec | 11 | 8 | 10 | 8 | 8 |
Frequency of specific cardiac embryonic phenotypes in wild-type, Nf1−/− and Nf1ec embryos at E13.5. DORV, double outlet right ventricle; VSD, ventricular septal defect; AV, atrioventricular.
The myocardial defects that we observed were secondary to endothelial dysfunction and were not reproduced by tissue-specific inactivation of Nf1 in myocardial cells (data not shown). Signaling between the endocardium and myocardium is required for proper myocardial development9. Likewise, an endothelial-derived neurofibromin-dependent paracrine signal may be required by neural-crest cells for proper development of neural-crest-derived structures. Nevertheless, our data indicate a genetic requirement for neurofibromin in endothelial cells. These observations may explain why individuals with NF1 have a higher incidence of valvular pulmonic stenosis, hypertension and other cardiovascular disorders2.
Neurofibromin deficiency in endocardial cells results in altered ras signaling. Immunohistochemistry identified phosphorylated MAPK (P-MAPK) in the hearts of wild-type and neurofibromin-deficient embryos at E11.5. Occasional endothelial cells positive for P-MAPK were present in the region of the endocardial cushions of hearts of wild-type embryos (Fig. 3a,c). Expression was more robust and was present in more endothelial cells lining the endocardial cushions in hearts of Nf1−/− embryos (Fig. 3b,d). Activation of MAPK suggests that Nf1 deficiency results in elevated ras signaling in endocardial cells, though other regulators of MAPK phosphorylation and other ras-GAPs exist. Using a ras activation assay, we determined that endothelial cell–specific loss of neurofibromin leads to ras activation in Nf1ec embryos (Fig. 3e). These data suggest that ras signaling is normally downregulated as endothelial cells mature into valve leaflets. Neurofibromin is required for this downregulation. It has been suggested that defects in erbB3 and PDGFα receptor signaling can lead to developmental defects of the endocardial cushions10,11. But the genes encoding these receptors as well as erbB2, erbB4 and neuregulin are expressed normally in Nf1-deficient embryos (ref. 5 and M.M. Lakkis and J.A.E., unpublished results). Likewise, mutations in Sox4, Pax3, Sema3c, Itga4 and members of the Tgfβ family have been implicated in developmental cardiac defects that include endocardial-cushion abnormalities12–16. These genes also seem to be expressed normally in Nf1−/− embryos (E10.5–13.5; M.M. Lakkis, A.D.G., M.M.L. and J.A.E., unpublished results).
Fig. 3.

Elevated ras signaling and increased Nfatc1 nuclear localization in hearts of Nf1−/− embryos. a–d, Activated MAPK (arrows) detected by immunohistochemistsry in sections of outflow tract (a,b) and atrioventricular (c,d) endocardial cushions from E11.5 wild-type (a,c) and Nf1−/− (b,d) embryos. e, Higher levels of activated ras were present in Nf1ec embryos as detected by pull-down assay with the ras binding domain of raf. wt, wild-type. f,g, Nfatc1 was localized to the cytoplasm of endothelial cells from E9.5 wild-type (yellow arrow, f) embryos and to the nucleus in Nf1−/− littermates (yellow arrow, g). h,i, Nfatc1 was in the cytoplasm of endothelial cells cultured from wild-type endocardial-cushion explants infected with adenovirus expressing lacZ (h) but in the nucleus of cells infected with rasV12 virus (i). j,k, Human umbilical vein endothelial cells were transfected with Nfatc1 alone (j) or Nfatc1 plus rasV12 (k), which resulted in nuclear localization of Nfatc1 (arrow, k).
Recently, several mouse lines have been generated that exhibit defects in endocardial-cushion morphogenesis14,17–20. Nfatc1 knockout mice die in utero with hypoplastic valves17,20. Nfatc1 activity is regulated by phosphorylation and sub-cellular localization. The ras signaling pathway can affect nuclear localization of Nfat isoforms in myocytes21 and T cells22. An elevated level of ras signaling in Nf1−/− endocardial cells affects Nfatc1 nuclear localization. Nfatc1 was located in the cytoplasm of wild-type endocardial cells at E9.5 (Fig. 3f). At later time points, it becomes localized to the nucleus17,20. Nfatc1 was prematurely localized to the nucleus in Nf1−/− endocardial cells at E9.5 (Fig. 3g). Expression of a constitutively active form of ras in endothelial cells from embryonic endocardial-cushion explants (Fig. 3h,i) or human umbilical vein endothelial cells (Fig. 3j,k) was sufficient to enhance Nfatc1 nuclear localization. We propose that increased ras activity in neurofibromin-deficient endocardial cells leads to Nfatc1 activation. This may contribute to the cardiovascular defects seen in mouse embryos lacking Nf1 in endothelial cells and to vascular defects observed in individuals with NF1, though our data do not exclude the possible contribution of other downstream pathways regulated by Nf1.
These results show that neurofibromin is required in endothelial cells, but do not rule out a simultaneous requirement in the neural crest during cardiac development. Because neural-crest derivatives are affected in individuals with NF1 and in mice with disruption of Nf1 (refs. 3,4,23,24), we used a cre–lox fate-mapping strategy to examine cardiac neural-crest migration in wild-type and Nf1 mutant embryos25,26. Cardiac neural-crest cells populated the outflow tract of hearts from E12.5 wild-type and Nf1 mutant embryos (Fig. 4a,b). Although the gross morphology of hearts from Nf1−/− embryos is abnormal3–5, the abundance and location of neural-crest cells was similar to that seen in hearts of wild-type embryos (Fig. 4). Although it is possible that subtle abnormalities of the neural crest exist, cardiac defects in Nf1−/− mice occurred despite apparently normal neural-crest migration.
Fig. 4.

Fate mapping of cardiac neural crest. a,b, Hearts of E12.5 wild-type (a) and Nf1−/− (b) embryos stained for β-galactosidase activity. Cardiac neural-crest cells are labeled blue by Cre-mediated activation of lacZ in R26R reporter mice30. Red arrows point to labeled cells populating the outflow tract of the developing heart. c,d, Cross-sections through hearts at the level of the truncus show labeled blue cells forming the septum between the aorta and pulmonary artery in both wild-type (c) and Nf1−/− (d) embryos. e,f, Red arrows point to labeled neural-crest cells invading the outflow tract endocardial cushions in the conus region of the right ventricular outflow tract in wild-type (e) and Nf1−/− (f) embryos. pa, pulmonary artery; ao, aorta; rv, right ventricle; lv, left ventricle.
To determine if Nf1 function is required in the neural crest, we carried out tissue-specific Nf1 gene inactivation. We used three independent lines of transgenic mice expressing Cre recombinase in neural-crest cells. In each case, previous fate-mapping experiments have confirmed expression of these transgenes in neural-crest cells fated to populate the heart and other neural-crest derivatives25–27. We examined embryos derived from appropriate crosses between Nf1flox mice and Mpz–cre (three litters at E13.5), Pax3–cre (six litters at E13.5, five newborn litters) and Wnt1–cre (five litters at E13.5, six newborn litters). Because we obtained similar results with all neural-crest transgenic cre lines, we refer to them collectively as Nf1nc.
Nf1nc embryos were viable throughout gestation, though Nf1−/− littermates are not viable after E13.5 (refs. 3,4). At birth, 9 of 67 pups (13% versus the expected 12.5%) were Nf1nc. At E13.5, cardiac development in Nf1nc embryos was normal (Fig. 1d,h,l), but newborn Nf1nc pups were not normal. Newborn Nf1nc pups were smaller than wild-type (Fig. 5a,b) and did not initiate normal respiration. Consistent with the tissue-specificity of our Cre-expressing transgenic mice, there was hyperplasia of tissues of neural-crest origin, including massively enlarged sympathetic ganglia (Fig. 5d). These abnormal structures, seen in all five Nf1nc pups examined, consisted of axons that stained for neurofilament and small cell bodies with large nuclei that expressed tyrosine hydroxylase (data not shown). This morphology is consistent with ganglioneuroma or ganglioneurosarcoma, which can develop into malignant peripheral nerve sheath tumors28. We observed hyperplastic tissues with similar appearance adjacent to the lumbar spine and infiltrating the abdominal omentum and the uterus. In all cases, the adrenal glands were enlarged (Fig. 5f). There was a marked increase in the number of adrenal medullary cells, suggesting pheochromocytoma, and a thinning and distortion of the adrenal cortex (Fig. 5e,f). The adrenal medulla is of neural-crest origin, and the abnormally abundant cells stained for tyrosine hydroxylase (Fig. 5h). In some cases, we observed cells positive for tyrosine hydroxylase traversing the cortical boundaries (Fig. 5h). Hence, Nf1nc pups showed hyperplasia of tissues that are affected in individuals with NF1 and provide a useful model for the study of Nf1 and cancer. We conclude that Nf1 function in the neural crest is not required for normal development of the heart.
Fig. 5.

Nf1nc mice live longer than Nf1−/−embryos and develop tumors of neural-crest origin. a,b, Comparison of newborn wild-type (a) and Nf1nc (b) littermates. c,d, Sagittal sections of newborn pups showed normal sympathetic ganglia in wild-type embryos (arrow, c) and massively enlarged ganglia in Nf1nc embryos (arrows, d). e–h, Adrenal gland (ad) of wild-type (e,g) and Nf1nc (f,h) embryos showed hyperplastic adrenal medullary cells reminiscent of pheochromocytoma in Nf1nc pups. The cells formed a large mass extrinsic to the adrenal gland (yellow arrows, f). Immunohistochemistry showed medullary (m) cells positive for tyrosine hydroxylase surrounded by cortex (c) in wild-type embryos (g), whereas cells positive for tyrosine hydroxylase were seen invading the cortex and surrounding tissues in Nf1nc embryos (yellow arrows, h).
Methods
Mouse breeding and genotyping
We genotyped Nf1+/−, Nf1flox, Pax3–cre, Wnt1–cre, Tek–cre, Mpz–cre, Z/EG and Gt(ROSA)26Sortm1Sor (R26R) mice as described previously4,7,8,25–27,29,30. The institutional animal care and use committee of the University of Pennsylvania approved all animal protocols.
Histology and immunohistochemistry
Fixation and immunohistochemistry protocols are available at http://www.uphs.upenn.edu/mcrc. For detection of neurofilament, we used monoclonal antibody 2H3 (Developmental Studies Hybridoma Bank) at a dilution of 1:25. We used Phosphop44/42 MAPK (Thr202/Tyr204) monoclonal antibody E10 (Cell Signaling Technology) at a dilution of 1:100. To detect Nfatc1, we used monoclonal antibody 7A6 at a dilution of 1:50 (BD PharMingen).
Endothelial cell culture
We carried out endocardial-cushion explants from E10.5 mouse embryos and subsequent adenovirus infection as described previously5. We cultured human umbilical vein endothelial cells in endothelial growth medium (Clonetics) according to the manufacturer’s guidelines and co-transfected them with 1 µg of pCDNA3-Nfatc1 and either 2 µg of pCMV-rasV12 or 2 µg of pCMV using Lipofectin (Invitrogen). We analyzed Nfatc1 localization 48 h after transfection.
Ras activation assay
We used the Ras Activation Assay Kit (Upstate) according to the manufacturer’s guidelines and incubated 1 µg of lysate from E11.5 wild-type or Nf1ec mice with 5 µl of a 50% slurry of Raf-1 RBD (ras assay reagant)–agarose.
Acknowledgments
We are grateful to M. Yanagisawa for providing Tek–cre mice; B. Zhou for providing the Nfatc1 expression plasmid; L. Rorke for reviewing pathologic specimens; and M. Weiss, C. Thompson, C. Simon, E. Morrisey, M. Kahn and members of J.A.E.’s laboratory for critically reading the manuscript and for helpful discussions. This work was supported by grants from the WW Smith Foundation, the American Heart Association and the US National Institutes of Health to J.A.E. L.F.P. is supported by grants from the US National Institute of Neurological Disorders and Stroke and the US Department of Defense. A.D.G. is supported by the Department of Cell and Developmental Biology predoctoral training grant from the US National Institutes of Health. Y.Z. is a recipient of a Young Investigator Award from the National Neurofibromatosis Foundation.
Footnotes
Competing interests statement
The authors declare that they have no competing financial interests.
References
- 1.Friedman JM, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet. Med. 2002;4:105–111. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Lin AE, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am. J. Med. Genet. 2000;95:108–117. doi: 10.1002/1096-8628(20001113)95:2<108::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 3.Jacks T, et al. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 4.Brannan CI, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 5.Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development. 1998;125:4359–4367. doi: 10.1242/dev.125.22.4359. [DOI] [PubMed] [Google Scholar]
- 6.Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kisanuki YY, et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 9.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 10.Erickson SL, et al. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2- and heregulin-deficient mice. Development. 1997;124:4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- 11.Morrison-Graham K, Schatteman G, Bork T, Bowen-Pope D, Weston J. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development. 1992;115:133–142. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- 12.Epstein J. Pax3, neural crest and cardiovascular development. Trends Cardiovasc. Med. 1996;6:255–261. doi: 10.1016/S1050-1738(96)00110-7. [DOI] [PubMed] [Google Scholar]
- 13.Feiner L, et al. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- 14.Galvin KM, et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000;24:171–174. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- 15.Ya J, et al. Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ. Res. 1998;83:986–994. doi: 10.1161/01.res.83.10.986. [DOI] [PubMed] [Google Scholar]
- 16.Yang JT, Rayburn H, Hynes RO. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development. 1995;121:549–560. doi: 10.1242/dev.121.2.549. [DOI] [PubMed] [Google Scholar]
- 17.de la Pompa JL, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 18.Camenisch TD, et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen B, et al. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 2000;24:296–299. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- 20.Ranger AM, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 21.Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. J. Biol. Chem. 2001;276:3524–3530. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- 22.Woodrow MA, Rayter S, Downward J, Cantrell DA. p21ras function is important for T-cell antigen receptor and protein kinase C regulation of nuclear factor of activated T cells. J. Immunol. 1993;150:3853–3861. [PubMed] [Google Scholar]
- 23.Vogel KS, et al. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cichowski K, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 25.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 27.Yamauchi Y, et al. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 1999;212:191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- 28.Ricci A, Jr., et al. Malignant peripheral nerve sheath tumors arising from ganglioneuromas. Am. J. Surg. Pathol. 1984;8:19–29. doi: 10.1097/00000478-198401000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- 30.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]