

Abstract
The incorporation of 6-thioguanine (6-TG) into DNA increases the risk of 1O2-initiated skin cancer. We herein provide the first report on quantitative characterization of the photoactivity of 6-thioguanines including 6-TG and 6-thioguanosine. Time-resolved singlet oxygen luminescence was observed directly for the first time after UVA irradiation of 6-thioguanes in both CHCN3 and aqueous solutions. Their photosensitization was characterized by the quantum yield of singlet oxygen production, showing a dramatic decrease over time from initial 0.49–0.58 to zero. Experiments performed on both 6-TG and 6-thioguanosine did not show any significant difference in the quantum yield of singlet oxygen production, indicating that there was no potential participation of 7H- and 9H- tautomers. Our findings provide a primary basis for a better understanding of molecular events of thiopurine drugs in biological systems.
Introduction
The photophysics and photochemistry of thiopurine DNA bases are far less understood than those of normal DNA bases although some of them, such as azathioprine (Aza), 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG) have been used as cancer therapeutic and immunosuppressive agents for five decades.1,2 For normal single nucleotides, nonradiative decay occurs efficiently at subpicosecond timescale.3,4 This process is believed to enhance the photostability of DNA bases. The electronic properties of purines can, however, be greatly altered when oxygen atoms are replaced by sulfur. Unlike normal DNA bases, the formation yields of triplet states in thiocarbonyl compounds were found to be very high, e.g., 0.9 for thiouracils in H2O5, 0.99 for 6-thiopurine in THF,6 0.77 and 0.68 for 4-thiouridine in CH3CN, and an unity value for 4-thiothymidine in an ionic liquid.9 The interaction between triplet state and molecular oxygen can lead to the formation of singlet oxygen (1O2) via energy transfer and/or radicals (such as superoxide and hydroxyl radicals) via electron transfer processes. An understanding of the photoinduced activities of thiopurines became even more pronounced when researchers recognized the deleterious side effects with the use of thiopurine drugs. As a prodrug, Aza is cleaved to 6-MP that in turn is metabolized to 6-TG nucleotides (6-TGN). 6-TG is also directly converted to 6-TGN by hypoxanthine phosphoribosyltransferase (HPRT). 6-TGN is a precursor of DNA synthesis and 6-TG becomes incorporated into DNA.10,11 Their structures and metabolism are shown in scheme 1. The incorporation of 6-TG into DNA increased the risk of acute myeloid leukemia and skin cancer in thiopurine-treated patients.12–15 Accumulated evidence indicated that sunshine exposure and oxygen were the major contributors to this deleterious side effect.
Scheme 1.
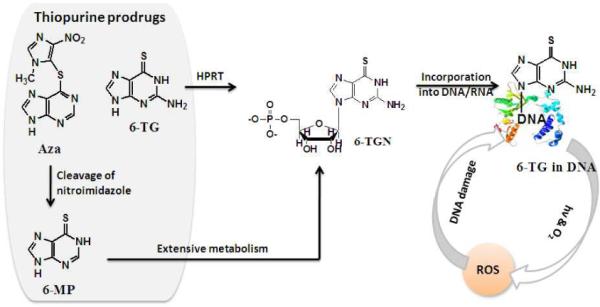
Structures and metabolism of thiopurine prodrug incorporation into DNA
It was, however, only until recently that 1O2 was acknowledged as a major risk factor for skin cancer from the patients with Aza treatment.16–18 Unlike normal DNA bases, 6-thioguanines have strong electronic transition in UVA region (315–400 nm). DNA substitution by 6-TG permits the absorption of UVA light, and subsequently introduces photoactive sites into DNA. An extensive interest has therefore been generated to elucidate the roles of thiopurines in skin sensitivity.12,19–21 Currently, researchers rely on indirect methods such as 1O2 trapping or biological damage tests to identify the formation of reactive oxygen species (ROS) from thiopurines. Direct evidence and quantitative information regarding the production of 1O2 is not available although this knowledge is crucial in elucidation of DNA damage mechanism. The extreme instability of thiopurines toward light and molecular oxygen complicates their physiochemical characterization.
The possibility that 6-TG may act as an endogenous 1O2 sensitizer and provide a source of DNA damage prompted us to explore quantitatively their photosensitization ability. Each base has a number of tautomers, formed by permuting hydrogen atoms among the set of heteroatoms.4 Tautomerism complicates the interpretation of photophysical experiments because electronic structure can differ dramatically for individual tautomers. Therefore, it is essential to determine what tautomers are actually present in a given experimental condition. It is more likely that 6-TG contains a mixture of 7H- and 9H- tautomers, while in 6-thioguanosine the N9-position is substituted with a sugar group. The experiments were performed on both 6-TG and 6-thioguanosine to test the potential participation of tautomers in the production of 1O2. The structures of 7H- and 9H-tautomers and 6-thioguanosine are given in scheme 2. Our results showed that both 6-TG and 6-thioguanosine were indeed efficient 1O2 sensitizers under UVA irradiation. The deactivation of 6-thioguanines occurred rapidly over time in the presence of oxygen and light. We herein provide a first report of direct observation and quantitative characterization of 1O2 production from 6-thioguanines. To some extent, our results explain the prevalence of photoactivity with thiopurine dose.
Scheme 2.

Structures of 6-TG 7H- and 9H-amino tautomers and 6-thioguanosine
Materials and Methods
Materials and instrumentation
Reagents and solvents were obtained commercially and used without further purification. meso-Tetra(4-carboxylphenyl) porphine (TCPP) was purchased from Frontier Scientific, Inc., [2-(dicyclohexyl phosphino) ethyl]trimethyl ammonium chloride (> 98%) from Strem Chemicals, Inc., hydrochloric acid (37.5%) from Fisher Scientific, 6-thioguanine (6-TG), 6-thioguanosine, Tris(hydroxymethyl)aminomethine (> 99.8%), Ethylenedinitrilo)tetraacetic acid, disodium salt dihydrate (EDTA, > 99%), sodium hydroxide, sodium azide (NaN3), deuterium acetonitrile-d3 (CD3CN, 99.8% of D) and deuterium oxide (D2O, 99% of D) from Sigma-Aldrich. Deionized water was obtained from a Nanopure Water (Barnsted System, USA). A Q-switched Nd:YAG laser with pulse duration of 3–4 ns and a maximum energy of 30 mJ at 532 nm (Polaris II, Electro Scientific Industries, Inc.), and a liquid N2-cooled germanium photodetector (Applied Detector Corporation) were used for time-resolved 1O2 luminescence measurements. Steady-state photooxidation was conducted in oxygen-saturated solution using a 150 W Xenon lamp (6255 Xenon lamp housed in 66907 Arc Lamp Source, Newport Oriel Instruments) equipped with an IR blocking filter (59042, Newport Oriel Instruments) and a monochromator with primary wavelength region 450–2000 nm (77250 1/8 m Monochromator and 77305 Grating, Newport Oriel Instruments), where the intensities in UVA range is below 15 W. Other instruments employed in this research include a BioMate 3 UV-Vis spectrophotometer (Thermo Scientific) and a Cary 300 UV-Vis spectrophotometer (Varian, Inc.) for taking absorbance and spectra. The determination of photooxidation products was done on a 300 MHz Bruker Spectrospin FT-NMR or a Varian Vnmrs 500 MHz NMR. All of the measurements were carried out at ambient temperature. Samples were protected from light when not being irradiated.
Direct observation of 1O2 after 355 nm irradiation of 6-thioguanines
Kinetics of 1O2 luminescence at 1270 nm was monitored by time-resolved Nd:YAG laser equipped with a low temperature cooled Ge detector, as previously described.22–25 6-thioguanines were dissolved in pH 7.4 D2O Tris buffer, pH 10 NaOH D2O or CD3CN solutions under dark, to avoid light-induced oxidation. The absorbances of the samples were controlled to be in the range of 0.1–0.4 at excitation wavelength of 355 nm. 1st-order kinetic fitting of 1O2 decay was calculated using Origin 6.1 program. 1O2 decay curves were corrected from control experiments by using the same but N2-saturated sample or the air-saturated sample in the presence of 1.5 mM NaN3. Data points of the initial ~10 μs were not used due to electronic interference signals from the detector.
Quantum yield of 1O2 production (ΦΔ)
ΦΔ was determined in air-saturated pH 7.4 D2O Tris buffer and pH 10 NaOH D2O solutions on a relative basis by steady-state photolysis using TCPP as a reference. A ΦΔ value of 0.53 for TCPP at pH 8–10 has been determined by our group.24 A water-soluble phosphine, [2-(dicyclohexylphosphino)ethyl]trimethylammonium chloride, was used as a 1O2 trap. A mixture of 3.00 mL of 6-thioguanines (OD = 0.1–1.0, 0.05–0.10 mM) and a phosphine (3.0–5.0 mM) trap were added into a 1-cm quartz cuvette and irradiated under UVA light for 20 minutes, followed by an immediate measurement of phosphine oxidation by 31P NMR.24 1O2 photooxidation of phosphine trap leads to the formation of a sole product phosphonate (see scheme 3 for chemical structures). The peaks at δ −6.45 (s, 1P) and δ 60.50 (s, 1P) represent phosphine and phosphonate, respectively. The percent yields of phosphonates were controlled below 15% and calculated by comparison of the integrated 31P NMR peaks of phosphine with those of phosphonate. The same trapping experiment was conducted for reference sensitizer TCPP under identical conditions. The absorbance of 6-thioguanines and TCPP at excitation wavelength of 350 nm was controlled the same. Control experiments in dark as well as in the absence of 6-thioguanines were carried out to ensure that there was no phosphine oxidation by heat or by ground-state oxygen molecules. ΦΔ was calculated according to the equation in scheme 3.
Scheme 3.

Measurement and calculation of ΦΔ
Results and Discussion
Spectroscopic properties of 6-thioguanines
6-thioguanosine is fairly soluble in aqueous solutions. The solubility of 6-TG in pure water is limited but can be largely enhanced at higher pH solutions due to the deprotonation of thiols or amines. The absorption spectra of 6-thioguanines were conducted in both pH 7.4 Tris buffer and pH 10 NaOH solutions. Figure 1 shows the changes in the extinction coefficient with absorbed light energy. Unlike normal DNA bases (e.g., guanine in figure 1) that absorb little UVA light (315–400 nm), 6-thioguanines have strong electronic transition in UVA region. The extinction coefficients at their maximum absorption wavelengths were calculated and summarized in table 1. The extinction coefficients for both 6-TG and 6-thioguanosine are in the order of 104 M−1 cm−1. 6-thioguanines in pH 7.4 Tris buffer absorb furthest to red at 340 nm. Apparently, incorporation of 6-TG into DNA introduces UVA sensitive sites into DNA.
Figure 1.
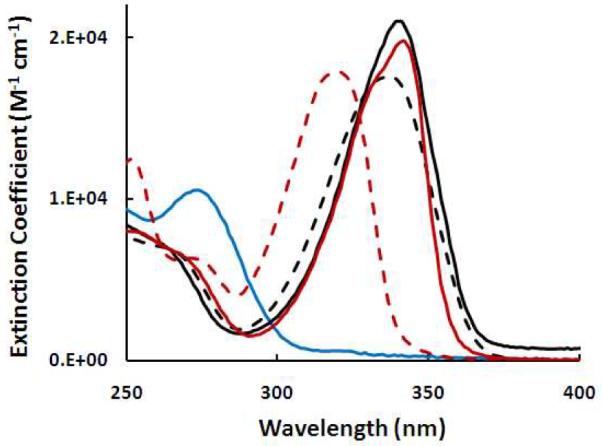
Extinction coefficient spectra of 6-TG in pH7.4 Tris buffer (solid black line) and in pH 10 NaOH (dash black line), 6-thioguanosine in pH 7.4 Tris buffer (solid red line) and pH 10 NaOH (dash red line), and guanine in pH 10 NaOH (blue line) solutions at ambient temperature
Table 1.
Wavelength maximum (λmax) and extinction coefficient maximum (εmax at λmax) determined from electronic absorption spectra of guanine and 6-thioguanines at ambient temperature
| Compound | Solvent | λmax, nm | εmax, M−1 Cm−1 |
|---|---|---|---|
| Guanine | pH 10 NaOH | 273 | 1.1×104 |
| 6-TG | pH 10 NaOH | 337 | 1.8×104 |
| 6-TG | pH 7.4 Tris buffer | 340 | 2.1×104 |
| 6-thioguanosine | pH 10 NaOH | 320 | 1.8×104 |
| 6-thioguanosine | pH 7.4 Tris buffer | 342 | 2.0×104 |
Direct observation of 1O2 luminescence at 1270 nm
Kinetic decay of 1O2 luminescence was observed directly for the first time at 1270 nm in air-saturated pH 7.4 D2O Tris buffer, pH 10 NaOH D2O and CD3CN solutions upon irradiation of 6-thioguanines at 355 nm, using previously reported experimental setup.22–25 Examples are shown in figure 2. The decay traces were assigned to 1O2 luminescence because both kinetics and intensities of 1O2 signals were sensitive to the concentrations of NaN3 (figures 2a) or molecular oxygen (figure 2b) in the solutions. NaN3 is a well-known efficient 1O2 quencher that reacts with 1O2 at a rate constant of 5.0×108 M−1 s−1 in water.26 Figures 2a indicates that azide ions quench not only the lifetime of 1O2 but also the initial intensity of 1O2 luminescence, which can be explained by its reactions with both 1O2 and excited states of a sensitizer.27 The kinetic simulation is shown in the insertions of figure 2. 6-thioguanines belong to the analogue of sulfides that are readily oxidized by 1O228 at a magnitude of rate constants 106–107 M−1 s−1.29 The rapid deactivation of 6-TG in the presence of oxygen molecules and light (short dash line in figure 2b) was due to the efficient self-quenching of 1O2, resulting in the oxidation of sulfur atoms. The total rate constants (kd) of 1O2 removal were measured to be 1.0×104 s−1 in pH 7.4 D2O Tris buffer, 5.6×103 s−1 in CH3CN and 7.2×104 s−1 in pH 10 NaOH D2O solutions, which are comparable with literature values of 1.5×104 s−1 in D2O,30 2.3×103 s−1 in CH3CN31 and 4.4×104 s−1 in pH 10 NaOH D2O24 within acceptable error limits respectively. Our results were also supported by the report that 6-TG in DNA was both the production source20 and target site32 of ROS under UVA irradiation. For accurate calculations, decay traces were corrected for the background from other rapid events synchronized with laser pulses such as electronic interference from the detector, by using the same sample but in the presence of NaN3 or a N2-saturated sample as a control.
Figure 2.

Time-resolved 1O2 luminescence recorded at 1270 nm upon pulsed-irradiation of 6-TG at 355 nm. 2a: 1O2 decay in an air-saturated pH 7.4 D2O Tris buffer solution in the absence (solid line) and presence of 1.5 mM NaN3 (dot line); insertion: 1st-order kinetic fitting of 1O2 decay after correction with a control in the presence of NaN3, dots: experimental data and red line: theoretical simulation. 2b: 1O2 decay in an air-saturated pH10 NaOH D2O solution at time intervals of zero minute (solid line) and 3 minutes (short dash line), dot line: N2-saturated sample; insertion: 1st-order kinetic fitting of 1O2 decay after correction with a N2-saturated control, dots: experimental data and red line: theoretical simulation.
Oxidation of thiocarbonyl chromophores contributes toward their instability and reactivity. Similar to guanine, thioguanine has thioketone and enethiol tautomers in addition to the 7H- and 9H-amino tautomers. Since the initial report by Gattermann and Schulze,33 the stability of several thioketones toward oxygen in the presence or absence of light has been investigated by several groups. The oxidation mechanisms of sulfides by 1O2 have also been extensively examined,28,34 suggesting a persulfoxide as a primary key intermediate.28 Ramamurthy and co-workers identified 1O2 as oxidizing species in light-induced oxidation of thioketones.35,36 The photooxidation of diaryl, aryl alkyl, and dialkyl thioketones by 1O2 generated via self-sensitization or other independent methods yielded the corresponding ketone and sulfine in varying amounts36 Recently Karran's group revealed that under UVA irradiation Aza-treated DNA contained 6-TG that was both production source20 and target site32 of ROS. They identified the formation of 1O218 and its photooxidation products as guanine sulphinate (GSO2),37 guanine-6-suflonate (GSO3) and guanine-6-thioguanine.16,18 Both 1O216,18,38–40 and GSO3 16,18 have been proven to play a key role in mutagenic oxidative DNA damage. Apparently, 1O2-initiated photooxidation of sulfur atoms in thioguanines led to not only deactivation of the compounds in photosensitization ability but also various intermediates and products that might be harmful to biological systems. The phototoxicity of those intermediates and final products, however, has not been fully understood and requires further investigations.
Determination of ΦΔ
ΦΔ is an important measure of photosensitization efficacy. It is usually determined on a relative basis that requires a reference. A well developed sensitizer, 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin (TCPP) was selected for this purpose. ΦΔ value of TCPP has been determined to be 0.53 in alkaline solutions of pH 8–10.24 A time-resolved 1O2 measurement is awkward to quantify photosensitization ability of 6-thioguanines due to the weak signals and rapid deactivation of 6-thioguanines. ΦΔ were therefore measured by steady-state photolysis using a water-soluble phosphine, [2-(dicyclohexylphosphino)ethyl]trimethylammonium chloride as a 1O2 acceptor. It has been reported that the photooxidation of phosphines by 1O2 leads to the formation of either phosphonate or a mixture of phosphonate and phosphinate.41,42 A sole product, phosphonate was observed and monitored by 31P NMR as previously reported (figure 3).24 1O2 photooxidation reaction of phosphine is shown in scheme 3. The percent yields of phosphine oxides were calculated according to the equation in scheme 3 (see discussion below), by comparison of the integrated 31P NMR peaks of phosphine δ -6.45 (s, 1P), with those of phosphonate δ 60.50 (s, 1P). Control experiments in the absence of 6-thiopurines as well as in dark did not show any increase in the conversion yield of phosphines to phosphonates although the initial phosphonate peaks were observed due to the slow phosphine oxidation by triplet oxygen in air (see examples in figure 4). Thus the formation of phosphine oxidation by heat or by ground-state oxygen molecules could be neglected under our experimental conditions.
Figure 3.
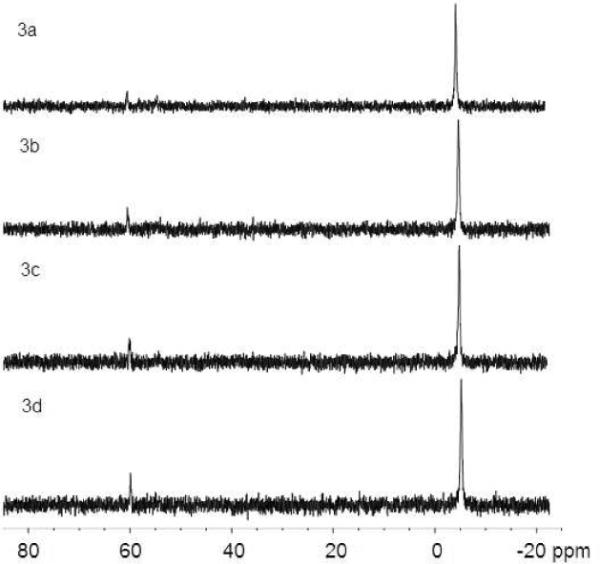
31P NMR spectra of 9.0 μmol of phosphine and 0.15 μmol 6-TG in O2-saturated pH 10 D2O solutions with irradiation time of 0 (3a), 20 min (3b), 60 min (3c) and 120 min (3d) at 350 nm
Figure 4.

31P NMR spectra from control experiments, 4a: 9.0 μmol of phosphine in O2-saturated pH 10 D2O solutions before irradiation, 4b: 20 min irradiation of 4a at 350 nm, 4c: 9.0 μmol of phosphine and 0.15 μmol 6-TG before irradiation, 4d: 4c kept in dark for 20 min
The bimolecular removal rate constants of 1O2 by both sulfides29 and phosphines41 are at a same magnitude of 106–107 M−1 s−1. 1O2 photooxidation of sulfur atoms in 6-thioguanines resulted in their deactivation, as shown in figure 2b. Under our experimental conditions, the concentrations of phosphine (3–5 mM) were controlled considerably higher than these of 6-thioguanines (0.05–0.10 mM) to ensure that the majority of 1O2 would react with phosphine trap, while the quenching of 1O2 by 6-thioguanines could be neglected. The percent yields of phosphonates were controlled below 15% to assure an efficient trapping condition. Dramatic decrease in photosensitization ability was evident by the trivial conversion of phosphine to phosphonate after 20 minutes irradiation at 350 nm as shown in figure 3. Taking this into account, the ΦΔ calculated for 6-thioguanines was approximated by multiplication of a factor of 2 when using TCPP, a conventional, stable sensitizer under UVA irradiation, as a reference. ΦΔ was calculated by comparing phosphonate yields from 6-thioguanines to that from TCPP reference. ΦΔ values thus obtained fell in the range of 0.49–0.58 (see table 2). Our results are comparable to literature values of some other thiocarbonyl compounds, e.g., 0.63 for 6-azauracil in CH3CN upon UVB irradiation43 and 0.50 for 4-thiothymidine in CH3CN upon UVA irradiation.44
Table 2.
ΦΔ measured upon UVA irradiation of 6-thioguanines
| Compound | Solvent | λirradiation, nm | Φ Δ |
|---|---|---|---|
| 6-TG | pH 10 NaOH | 337 | 0.58 ± 0.08 |
| 6-TG | pH 7.4 Tris buffer | 340 | 0.56 ± 0.18 |
| 6-thioguanosine | pH 10 NaOH | 320 | 0.49 ± 0.09 |
| 6-thioguanosine | pH 7.4 Tris buffer | 342 | 0.55 ± 0.08 |
Guanine has keto and enol tautomers in addition to the 7H- and 9H-amino tautomers found in adenine. The substitution of (deoxy)ribose at N9 position removes a heteroatom from the set that can accept hydrogen, reducing the number of possible tautomers in the nucleosides and nucleotides. Both of 6-TG and 6-thioguanosine were tested in this study to clarify the potential participation of 7H- or 9H-tautomers in the production of 1O2. As shown in table 2, no significant difference in ΦΔ from pH 10 or pH 7.4 aqueous solutions was observed from both compounds. Our data ruled out the effect of 7H- and 9H-tautomers on photosensitization ability of 6-thioguanines.
The results are also supported by the triplet excitation nature of thiol-DNA or -RNA bases. The photosensitized production of 1O2 is actually the quenching process of a sensitizer's triplet state by ground oxygen. To some extent the formation yield of triplet state can be a measure for 1O2 production. In general, the formation yields of triplet state in thiocarbonyl compounds were found to be very efficient, e.g., 0.9 for thiouracils in H2O5, 0.99 for 6-thiopurine in THF,6 0.77 and 0.68 for 4-thiouridine in CH3CN. Very recently Crespo-Hernandez's group reported an unity triplet yield upon UVA excitation of 4-thiothymidine in an ionic liquid,9 and theoretically predicted its phosphorescence energy of 2.23 eV (557 nm), which is in good agreement with experimental emission maximum at 542 nm.45 The energy difference between triplet and singlet states of molecular oxygen is 1270 nm or 0.98 eV. Obviously the triplet energy from thiol-DNA bases is high enough to excite molecular oxygen from triplet state to singlet state.
It is important to note that, in vivo, the levels of thioesters and thiols could be kept constantly in reduced forms in the presence of reductases. The possible regulation of thiopurine DNA bases in vivo could provide a constant phototoxicity site. Electron transfer between excited triplet thiopurines and molecular oxygen is another possible pathway that may generate superoxide radicals. Theoretical calculations and experimental studies indicated that the excited states of DNA bases might be reordered by solvation in terms of quantum yield and lifetime.4 The long-term risk associated with thiopurines can also result indirectly from photoactive products and metabolites. These factors should be taken into considerations in elucidation of biological damage. Obviously these are the areas that require further investigations.
Conclusion
By using photochemical techniques we were able to provide the first report of direct observation of 1O2 luminescence at 1270 nm after UVA irradiation of 6-TG and 6-thioguanosine. Their photoactivity toward UVA light and molecular oxygen was evaluated by ΦΔ in aqueous solutions. Our results showed that both 6-TG and 6-thioguanosne were efficient 1O2 sensitizers with ΦΔ values in the range of 0.49–0.58 but lost photoactivity rapidly over time. The effect of 7H- and 9H- tautomers on 1O2 production was not observed. These findings provide a primary basis for quantitative understanding of molecular events of thiopurines in biological systems and may lead to novel design of antioxidants toward thiopurine drugs. This research has the potential to add new perspective into material science as well when DNA construction is used.
ACKNOWLEDGEMENT
We thank the support from National Institutes of Health [NIH-RCMI program, G12RR013459) and National Science Foundation [NSF-PREM program, DMR-0611539).
References
- (1).Aarbakke Jarle, Janka-Schaub Gritta, Elion GB. Trends in Pharmacological Sciences. 1997;18:3–8. doi: 10.1016/s0165-6147(96)01007-3. [DOI] [PubMed] [Google Scholar]
- (2).Relling Mary V., Dervieux T. Nature Reviews Cancer. 2001;1:99–108. doi: 10.1038/35101056. [DOI] [PubMed] [Google Scholar]
- (3).Middleton Chris T., de La Harpe Kimberly, Su Charlene, Law Yu Kay, Crespo-Hernández Carlos E., Kohler B. Annual Review of Physical Chemistry. 2009;60:217–239. doi: 10.1146/annurev.physchem.59.032607.093719. [DOI] [PubMed] [Google Scholar]
- (4).Crespo-Hernández Carlos E., Cohen Boiko, Hare Patrick M., Kohler a. B. Chem. Rev. 2004;104:1977–2020. doi: 10.1021/cr0206770. [DOI] [PubMed] [Google Scholar]
- (5).Milder Steven J., Kliger DS. J. Am. Chem. Soc. 1985;107:7365–7373. [Google Scholar]
- (6).Alam Maksudul M., Fujitsuka Mamoru, Watanabe Akira, Ito O. J. Phys. Chem. A. 1998;102:1338–1344. [Google Scholar]
- (7).Foote CS, Dobrowolski DC. In: Oxygen Radicals Chem. Biol. Bors W, Saran M, Tait D, editors. Walter de Gruyter, Inc.; Berlin, Germany: 1984. pp. 465–472. [Google Scholar]
- (8).Heihoff K, Redmond RW, Braslavsky SE, Rougee M, Salet C, Favre A, Bensasson RV. Photochem. Photobiol. 1990;51:634–641. [PubMed] [Google Scholar]
- (9).Reichardt Christian, Crespo-Hernández CE. Chem. Commun. 2010;46:5963–5965. doi: 10.1039/c0cc01181a. [DOI] [PubMed] [Google Scholar]
- (10).Warren DJ, Andersen A, Slordal L. Cancer Res. 1995;55:1670–1674. [PubMed] [Google Scholar]
- (11).Cuffari C, Seidmen EG, Latour S, Theoret Y. Can J Physiol Pharmacol. 1996;74 [PubMed] [Google Scholar]
- (12).Karran P. Br. Med. Bull. 2006;79–80:153–170. doi: 10.1093/bmb/ldl020. [DOI] [PubMed] [Google Scholar]
- (13).Penn I. Transpl. Proc. XI. 1979:1047–1051. [PubMed] [Google Scholar]
- (14).Kinlen LJ, Sheil AGR, Peto J, Doll R. Br. Med. J. 1979;8:1461–1466. doi: 10.1136/bmj.2.6203.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Penn I. Transpl. Proc. 1979;XI:1047–1051. [PubMed] [Google Scholar]
- (16).O'Donovan Peter, Perrett Conal M., Zhang Xiaohong, Montaner Beatriz, Xu Yao-Zhong, Harwood Catherine A., McGregor Jane M., Walker Susan L., Hanaoka Fumio, Karran P. Science. 2005;309:1871–1874. doi: 10.1126/science.1114233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Perrett Conal M., Harwood Catherine A., McGregor JaneM., Karran P. Skin Cancer after Organ Transplantation, Cancer Treatment and Research. Vol. 146. Springer Science+Business Media, LLC; 2009. Carcinogenic Mechanisms Related to Immunosuppressive Therapy; pp. 123–132. [DOI] [PubMed] [Google Scholar]
- (18).Zhang Xiaohong, Jeffs Graham, Ren Xiaolin, O'Donovan Peter, Montaner Beatriz, Perrett Conal M., Karran Peter, Xu Y-Z. DNA Repair. 2007;6:344–354. doi: 10.1016/j.dnarep.2006.11.003. [DOI] [PubMed] [Google Scholar]
- (19).Cooke Marcus S, Duarte Tiago L, Cooper Deborah, Chen Jie, Nandagopal Sridevi, D EM. DNA repair. 2008;7:1982–1989. doi: 10.1016/j.dnarep.2008.08.007. [DOI] [PubMed] [Google Scholar]
- (20).Brem Reto, Li Feng, Karran P. Nucleic Acids Research. 2009;37:1951–1961. doi: 10.1093/nar/gkp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kelly Graham E., Meikle William D., Moore DE. Photochemistry and Photobiology. 1989;49:59–65. doi: 10.1111/j.1751-1097.1989.tb04078.x. [DOI] [PubMed] [Google Scholar]
- (22).Gandra Naveen, Frank Aaron T., Le Gendre Onica, Sawwan Nahed, Aebisher David, Liebman Joel F., Houk KN, Greer A, Gao R. Tetrahedron. 2006;62:10771–10776. [Google Scholar]
- (23).Li Wenbing, Gandra Naveen, Courtney Shavelle N., Gao R. ChemPhysChem. 2009;10:1789–1793. doi: 10.1002/cphc.200900155. [DOI] [PubMed] [Google Scholar]
- (24).Li Wenbing, Gandra Naveen, Ellis Erick, Cartney Shavelle, Gao R. ACS Appl. Mater. & Interface. 2009;1:1778–1784. doi: 10.1021/am9003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aebisher David, Azar Nikolay S., Zamadar Matibur, Gandra Naveen, Gafney Harry D., Gao Ruomei, Greer A. J. Phys. Chem. B. 2008;112:1913–1917. doi: 10.1021/jp709829z. [DOI] [PubMed] [Google Scholar]
- (26).Haag Werner R., Mill T. Photochemistry and Photobiology. 1987;45:317–321. [Google Scholar]
- (27).Hall Robert D., Chignell CF. Photochemistry and Photobiology. 1987;45:459–464. doi: 10.1111/j.1751-1097.1987.tb05403.x. [DOI] [PubMed] [Google Scholar]
- (28).Clennan EL. Acc. Chem. Res. 2001;34:875–884. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]
- (29).Wilkinson Francis, Helman W. Phillip, Ross AB. J. Phys. Chem. Ref. Data. 1995;24:663–1021. [Google Scholar]
- (30).Ogilby Peter R., Foote CS. J. Am. Chem. Soc. 1982;104:2069–2070. [Google Scholar]
- (31).Hurst John R., Schuster GB. J. Am. Chem. Soc. 1983;105:5756–5760. [Google Scholar]
- (32).Daehn Ilse, Karran P. Cancer Res. 2009;69:2393–2399. doi: 10.1158/0008-5472.CAN-08-4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gattermann L, Schulze H. Ber. 1896;29:2944. [Google Scholar]
- (34).Liang JJ, Gu CL, Kacher ML, Foote CS. J. Am. Chem. Soc. 1989;111:4717–4721. [Google Scholar]
- (35).Jayaraj Nithyanandhan, Maddipatla Murthy V. S. N., Prabhakar Rajeev, Jockusch Steffen, Turro NJ, Ramamurthy V. J. Phys. Chem. B. ASAP; 2010. [DOI] [PubMed] [Google Scholar]
- (36).Ramnath N, Ramesh V, Ramamurthy V. J. Org. Chem. 1983;48:214–222. [Google Scholar]
- (37).Ren Xiaolin, Li Feng, Jeffs Graham, Zhang Xiaohong, Xu Yao-Zhong, Karran P. Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. 2010;38:1832–1840. doi: 10.1093/nar/gkp1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ravanat J-L, Di Mascio P, Martinez GR, Cadet J. J. Biol. Chem. 2000;275:40601–40604. doi: 10.1074/jbc.M006681200. [DOI] [PubMed] [Google Scholar]
- (39).Ravanat J-L, Sauvaigo S, Caillat S, Martinez GR, Medeiros MHG, Di Mascio P, Favier A, Cadet J. Biol. Chem. 2004;385:17–20. doi: 10.1515/BC.2004.003. [DOI] [PubMed] [Google Scholar]
- (40).Montaner Beatriz, O'Donovan Peter, Reelfs Olivier, Perrett Conal M., Zhang Xiaohong, Xu Yao-Zhong, Ren Xiaolin, Macpherson Peter, Frith David, Karran P. EMBO Reports. 2007;8:1074–1079. doi: 10.1038/sj.embor.7401084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gao Ruomei, Ho David G., Dong Timothy, Khuu Daisy, Franco Nestor, Sezer Oemer, Selke M. Org. Lett. 2001;3:3719–3722. doi: 10.1021/ol010195v. [DOI] [PubMed] [Google Scholar]
- (42).Ho David G., Gao Ruomei, Celaje Jeff, Chung Ha-Yong, Selke M. Science. 2003;302:259–262. doi: 10.1126/science.1089145. [DOI] [PubMed] [Google Scholar]
- (43).Kobayashi Takashi, Harada Yosuke, Suzuki Tadashi, Ichimura T. J. Phys. Chem. A. 2008;112:13308–13315. doi: 10.1021/jp803096j. [DOI] [PubMed] [Google Scholar]
- (44).Harada Yosuke, Suzuki Tadashi, Ichimura Teijiro, Xu Y-Z. J. Phys. Chem. B. 2007;111:5518–5524. doi: 10.1021/jp0678094. [DOI] [PubMed] [Google Scholar]
- (45).Reichardt Christian, Crespo-Hernández CE. J. Phys. Chem. Lett. 2010;1:2239–2243. [Google Scholar]