

Abstract
Clinical phenotype of individuals with spinocerebellar ataxia 2 (SCA2) is characterised by cerebellar ataxia and cognitive impairment. Although L-dopa-responsive Parkinsonism is considered as a rare clinical presentation in SCA2, it has been brought to the attention of many neurologists in several studies. The authors report an autopsy case of SCA2 with Parkinsonism from a Japanese family using archival materials of our Brain Bank to describe unique neuropathologic findings. The individual clinically showed Parkinsonism as a predominant phenotype instead of cerebellar ataxia. Besides the classic SCA2 neuropathologic alterations, Lewy bodies and Lewy neurites were present in the brainstem nuclei. Genetic analysis revealed shorter abnormal expansion of CAG repeats (less than 39). In contrast, the authors could not find α-synuclein pathology in two SCA2 cases without Parkinsonism. The present case will provide a neuropathologic evidence of correlation between α-synucleinopathy and Parkinsonism of SCA2 as well as shed light on understanding the pathomechanism of Parkinsonism in SCA2.
Background
Spinocerebellar ataxia 2 (SCA2) (OMIM #183090) is an autosomal dominant cerebellar degeneration associated with expanded CAG trinucleotide repeat in ATXN2 gene (MIM ID *601517).1 Neuropathologic phenotype of SCA2 is characterised by degeneration of the olivopontocerebellar system as well as substantia nigra (SN), striatum and globus pallidus.2 In addition, an abnormal aggregation of poly-glutamine is observed as 1C2 (antibody specific for poly-glutamine) immunoreactive intranuclear inclusions. Although common clinical presentations of SCA2 are cerebellar ataxia, nystagmus, slow eye movement, hyporeflexia and cognitive dysfunction,3 recent studies have pointed out that L-dopa-responsive Parkinsonism is core clinical symptom in a subset of individuals with SCA2.4–8 According to epidemiological and genetic studies of patients with autosomal dominant parkinsonism, an up to 10% of them showed abnormal CAG expansion of ATXN2 gene.5 7 9 However, the neuropathologic substrate of SCA2 with parkinsonism remains unresolved. Because the neuronal loss of the SN is commonly observed in SCA2, this kind of classic pathologic change does not sufficiently explain the cause of L-dopa responsive parkinsonism of SCA2. In this paper, we provide new insight and evidence of pathologic basis of L-dopa responsive parkinsonism of SCA2.
Case presentation
The individual, that of a 40-year-old man, developed unstable gait and difficulty on driving a car at age 40. However, he was not seen by neurologist for approximately 20 years. Neurological examination at age 63 showed dysarthria, dysphasia, directional nystagmus as well as dysdiadochokinesis, dysmetria and truncal ataxia. Tendon reflexes were brisk in the all extremities with Babinski signs. As the most remarkable symptoms, he had severe rigidity and bradykinesia as well as stooped posture and freezing on gait. No tremor, either resting or postural, was observed. At age 66, there was severe rigidity in the neck as well as upper and lower extremities. He was able to stand and walk with full assistance. However, his gait was affected by severe bradykinesia and frozen gait. No involuntary movement was observed during his clinical course. He died of aspiration pneumonia at age 67. Anti-Parkinsonian medication was not prescribed. Because there were no neuroimages of the patient, brain MRI (figure 1) was carried out immediately after the patient’s death. It showed atrophy of the cerebellum and pons with hot-cross bun sign.10 The genetic study was permitted by the family only for the genes associated with SCA genes. Using frozen brain tissue, genetic analysis showed that he had an expanded allele with 22/37 in ATXN2 gene.
Figure 1.
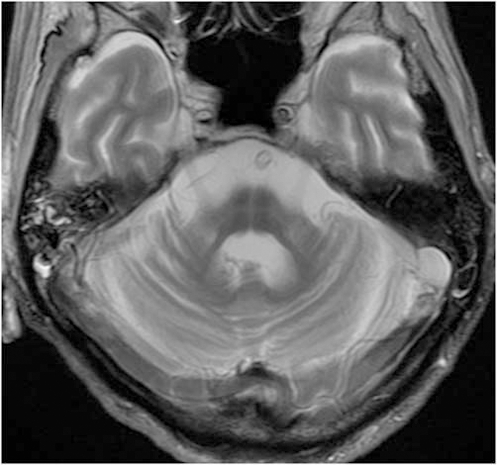
Brain MRI (T2 weighted image) shows severe atrophy of the cerebellum and pons with hot-cross bun sign.
His mother, brother and cousin had similar clinical presentation. His second daughter developed gait disturbance at age 18, followed by cognitive impairment, ataxia and choreic movement.
Neuropathology
Gross pathology
The weight of the fresh brain was 1220 g. The atrophy was severe in the basis pontis and middle cerebellar peduncle as well as moderate in the cerebellum. The SN and locus coeruleus appeared to have lost its pigment (figure 2A).
Figure 2.
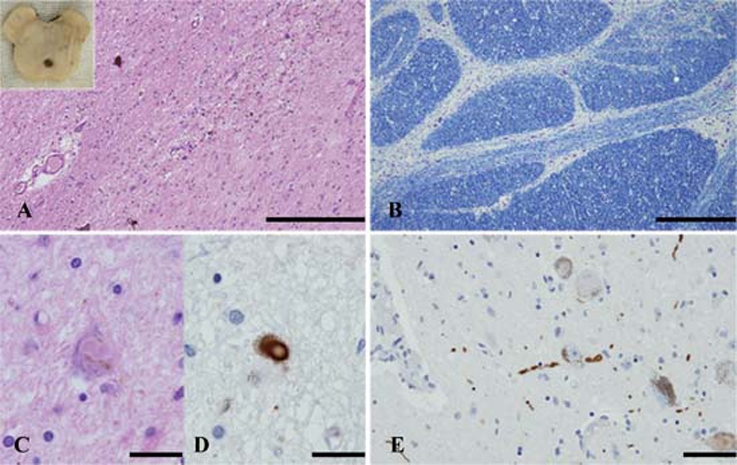
Neuropathologic results. Sections of the substantia nigra (A, C, D), basis pontis (B) and locus coeruleus (E). (A) Severe neuronal loss is evident in the substantia nigra. Note: The inset shows severe depigmentation at the level of the substantia nigra. (B) A photomicrograph shows severe loss of neurons and transverse fibres at the level of basis pontis. (C, D) Lewy bodies are seen in a remaining neuron. Lewy body is immunopositive using antibody raised against α-synuclein. (E) A-synuclein immunopositive Lewy neurites are seen. H&E stain (A, C), Klüver-Barrera stain (B), immunohistochemistry using monoclonal antibody specific for α-synuclein phosphorylated at Ser129 (D, E). Scale bars: A=200 μm, B=500 μm, C, D, E=25 μm.
Lewy body pathology
On microscopic examination, the SN and locus coeruleus showed severe loss of pigment and neuronal loss (figure 2A). The most striking pathologic finding was presence of α-synuclein immunoreactive Lewy bodies and neuritis in the several anatomical regions (figure 2C–E). Lewy bodies and neuritis were present in the basal nucleus of Meynert, hypothalamus, amygdala, SN, locus coeruleus and dorsal nucleus of vagus. However, neither Lewy bodies nor Lewy neurites were observed in the transentorhinal, cingulate, frontal, temporal and parietal cortex. The distribution of α-synuclein immunoreactive Lewy body pathology was consistent with stage 3 (case 2) according to Braak’s staging methodology.11
SCA2 pathology
In the cerebellum, there was moderate neuronal loss of the Purkinje cells. Mild neuronal loss and gliosis were present in the molecular and granular cell layers. Dentate nucleus was relatively well preserved. The interlobular white matter showed severe loss of myelinated fibres. In the brainstem, there was moderate to severe neuronal loss in the basis pontis (figure 2B) and inferior olivary nucleus. The 1C2 antibody depicted intranuclear immunopositive deposits in the pontine nucleus.
In order to confirm our results, we analysed two genetically confirmed SCA2 cases without Parkinsonism using α-synuclein immunohistochemistry. Both cases showed age at onset 20 and 29 years old and died at age 50 and 55, respectively. The former had an expanded allele with 22/43 and the latter had 22/42 in ATXN2 gene. Although there is neuronal loss in the SN in both cases, no α-synuclein immunopositive deposits were observed.
Discussion
Our results give a new perspective on pathogenesis of SCA2 with Parkinsonism. Clinical phenotype of individuals with SCA2 is usually cerebellar ataxia, dysarthria, slow saccadic eye movement, hyporeflexia and cognitive impairment.12 13 Although parkinsonism is considered as a variant clinical presentation in SCA2,14 15 it has been brought to the attention of neurologist in several studies. In addition, L-dopa may be effective medication to improve their Parkinsonism.4 16 17
Because neuropathologic studies always show severe neuronal loss of the SN regardless of symptoms,2 13 the degeneration of SN might not be a simple cause of Parkinsonism in SCA2. In the present case, it is still problematic whether or not the distribution of LBs/LNs is enough to cause Parkinsonism. In addition, we have to leave open the possibility that the α-synucleinopathy (Lewy body pathology) of the present cases is associated with ageing instead of SCA2.18 However, we reported another case with SCA2 in association with Parkinsonism and Lewy pathology from an unrelated family in Japanese neurology journal.19 Briefly, the individual was diagnosed as having Parkinson’s disease in the sixth decade of life. He was started on L-dopa therapy and showed mild improvement of his neurological conditions. Thereafter, genetic analysis revealed that he had an expanded allele with 22/38 in ATXN2 gene. Neuropathologic examination showed the presence of LBs/LNs in the brainstem nuclei as well as the olivopontocerebellar degeneration.
From the genetic study point of view, the frequency of CAG expansion of ATXN2 ranges from 0% to 10% in families of autosomal dominant Parkinsonism.5 7 9 In particular, Parkinsonism of SCA2 is strongly associated with a shorter abnormal expansion of CAG repeats (less than 39)5. At present, a direct interaction between α-synuclein accumulation and a shorter expansion of CAG repeats on ATXN2 is undetermined. It might be interesting that both the present case and reported one had shorter CAG repeats expansion. In addition, we could not find LBs/LNs in genetically confirmed two SCA2 cases without Parkinsonism (longer expanded allele with 22/42 and 22/43 in ATXN2).
Based on our results, we suggest that the neuropathologic substrate of SCA2 with Parkinsonism is associated with Lewy related α-synuclein pathology in the brainstem. Further analysis may warrant genetic and pathologic correlation of SCA2 with Parkinsonism.
Learning points.
-
▶
L-dopa-responsive Parkinsonism is considered as a rare but important clinical presentation in SCA2.
-
▶
We presented an individual of SCA2 with clinically prominent Parkinsonism instead of cerebellar ataxia.
-
▶
Neuropathologic findings revealed the presence of Lewy bodies and neurites in the various anatomical regions.
-
▶
The present case provides a neuropathologic evidence of correlation between α-synucleinopathy (Lewy body pathology) and Parkinsonism of SCA2 as well as shed light on understanding the pathomechanism of Parkinsonism in SCA2.
Acknowledgments
The authors gratefully acknowledge Aoyagi Shinichi, Tano, Suwabe Katsura, Aikyo Naoo, Harada Mieko, Kimura Yuki for expert technical assistance. Kunimasa Arima gave them opportunity to see SCA cases without Parkinsonism. Kuniaki Tsuchiya gave them an insightful comment. The authors gratefully acknowledge Robert W Smith for editing the manuscript. This study was supported in part by the Japan Society for the Promotion of Science 19500311 (MT). Grant-in-Aid for Scientific Research (KAKENHI B) (20390248) (SM), The Specified Disease Treatment Research Program (SM) and Comprehensive Brain Science Network (SM, MT).
Footnotes
Competing interests None.
Patient consent Obtained.
References
- 1.Lastres-Becker I, Rüb U, Auburger G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008;7:115–24 [DOI] [PubMed] [Google Scholar]
- 2.Estrada R, Galarraga J, Orozco G, et al. Spinocerebellar ataxia 2 (SCA2): morphometric analyses in 11 autopsies. Acta Neuropathol 1999;97:306–10 [DOI] [PubMed] [Google Scholar]
- 3.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–94 [DOI] [PubMed] [Google Scholar]
- 4.Shan DE, Soong BW, Sun CM, et al. Spinocerebellar ataxia type 2 presenting as familial levodopa-responsive parkinsonism. Ann Neurol 2001;50:812–15 [DOI] [PubMed] [Google Scholar]
- 5.Lu CS, Wu Chou YH, Kuo PC, et al. The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol 2004;61:35–8 [DOI] [PubMed] [Google Scholar]
- 6.Hardy J, Lees AJ. Parkinson’s disease: a broken nosology. Mov Disord 2005;20(Suppl 12):S2–4 [DOI] [PubMed] [Google Scholar]
- 7.Charles P, Camuzat A, Benammar N, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology 2007;69:1970–5 [DOI] [PubMed] [Google Scholar]
- 8.Furtado S, Payami H, Lockhart PJ, et al. Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov Disord 2004;19:622–9 [DOI] [PubMed] [Google Scholar]
- 9.Gwinn-Hardy K, Chen JY, Liu HC, et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 2000;55:800–5 [DOI] [PubMed] [Google Scholar]
- 10.Takao M, Kadowaki T, Tomita Y, et al. ‘Hot-cross bun sign’ of multiple system atrophy. Intern Med 2007;46:1883. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211 [DOI] [PubMed] [Google Scholar]
- 12.Dürr A, Smadja D, Cancel G, et al. Autosomal dominant cerebellar ataxia type I in Martinique (French West Indies). Clinical and neuropathological analysis of 53 patients from three unrelated SCA2 families. Brain 1995;118:1573–81 [DOI] [PubMed] [Google Scholar]
- 13.Orozco G, Estrada R, Perry TL, et al. Dominantly inherited olivopontocerebellar atrophy from eastern Cuba. Clinical, neuropathological, and biochemical findings. J Neurol Sci 1989;93:37–50 [DOI] [PubMed] [Google Scholar]
- 14.Sasaki H, Wakisaka A, Sanpei K, et al. Phenotype variation correlates with CAG repeat length in SCA2–a study of 28 Japanese patients. J Neurol Sci 1998;159:202–8 [DOI] [PubMed] [Google Scholar]
- 15.Cancel G, Dürr A, Didierjean O, et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet 1997;6:709–15 [DOI] [PubMed] [Google Scholar]
- 16.Wilkins A, Brown JM, Barker RA. SCA2 presenting as levodopa-responsive parkinsonism in a young patient from the United Kingdom: a case report. Mov Disord 2004;19:593–5 [DOI] [PubMed] [Google Scholar]
- 17.Infante J, Berciano J, Volpini V, et al. Spinocerebellar ataxia type 2 with Levodopa-responsive parkinsonism culminating in motor neuron disease. Mov Disord 2004;19:848–52 [DOI] [PubMed] [Google Scholar]
- 18.DelleDonne A, Klos KJ, Fujishiro H, et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol 2008;65:1074–80 [DOI] [PubMed] [Google Scholar]
- 19.Yomono HS, Kurisaki H, Hebisawa A, et al. Autopsy case of SCA2 with Parkinsonian phenotype. Rinsho Shinkeigaku 2010;50:156–62 [DOI] [PubMed] [Google Scholar]