

Abstract
Although metal free cycloadditions of cyclooctynes and azides to give stable 1,2,3-triazoles have found wide utility in chemical biology and material sciences, there is an urgent need for faster and more versatile bioorthogonal reactions. We have found that nitrile oxides and diazocarbonyl derivatives undergo facile 1,3-dipolar cycloadditions with cyclooctynes. Cycloadditions with diazocarbonyl derivatives exhibited similar kinetics compared to azides whereas the reaction rates of cycloadditions with nitrile oxides were much faster. Nitrile oxides could conveniently be prepared by direct oxidation of the corresponding oximes with BAIB and these conditions made it possible to perform oxime formation, oxidation and cycloaddition as a one-pot procedure. The methodology was employed to functionalize the anomeric center of carbohydrates with various tags. Furthermore, oximes and azides provide an orthogonal pair of functional groups for sequential metal free click reactions and this feature makes it possible to multi-functionalize biomolecules and materials by a simple synthetic procedure that does not require toxic metal catalysts.
Keywords: click chemistry, cycloaddition, carbohydrates, bioorthogonal, multifunctional
Introduction
Metal free click cycloadditions of cyclooctynes with azides1 to give stable 1,2,3-triazoles have found wide utility in labeling glycans,2 proteins3 and lipids4 of living cells, glycoprotein enrichment for proteomics,5 protein6 and oligonucleotide modification7 and tissue reengineering.8 These reactions, which have been coined “Strain-Promoted Alkyne-Azide Cycloadditions (SPAAC)” have also made entry in material sciences and have for example been employed for the assembly, crosslinking9 and surface modification of dendrimers,10 derivatization of polymeric nanostructures,11 and patterning of surfaces.12
Density functional theory (B3LYP) calculations of the transition states of cycloadditions of phenyl azide with acetylene and cyclooctyne indicate that the fast rate of the “strain promoted” cycloaddition is actually due to a lower energy required for distorting the 1,3-dipole and alkyne into the transition-state geometry.13 The first generation of cyclooctynes proceeded with relatively slow rates of reaction, however, it has been found that significant increases in the rate of strain-promoted cycloaddition can be accomplished by appending electron-withdrawing groups to the propargylic position of cyclooctyne.2 For example, difluorinated cyclooctyne (DIFO, 1)14 reacts with azides approximately sixty-times faster than similar cycloadditions with an unsubsituted cyclooctyne. We have reported that derivatives of 4-dibenzocyclooctynol (DIBO, 2) react fast with azido-containing saccharides and amino acids and can be employed for visualizing metabolically labeled glycans of living cells.15 Attractive features of DIBO include easy access to the compound by a simple synthetic approach, non-toxicity and the possibility of straightforward attachment of a variety of probes. Furthermore, the structure of DIBO is amenable to analog synthesis and derivatives (3 and 4) have been introduced that exhibit even higher rates of reaction than the parent compound.16
Our finding that cyclooctynes can undergo fast cycloadditions with nitrones has further expanded the scope of metal free click reactions,17 and the usefulness of this approach has been demonstrated by site specific protein modification by a three-step protocol entailing periodate oxidation of an N-terminal serine to give an aldehyde, which could easily be converted into a nitrone and then reacted with probe-modified dibenzocyclooctynes.
As part of a program to develop metal free click reactions, we report here that in addition to azides and nitrones, nitrile oxides and diazocarbonyl derivatives readily undergo cycloadditions with dibenzocyclooctyne to give stable isoxazoles and pyrazoles, respectively. It has been found that the various 1,3-dipoles exhibit distinct levels of reactivity making it possible to perform sequential cycloadditions. In addition, we have shown, for the first time, that an oxime can function as a latent 1,3-dipole for a nitrile oxide, which is fully orthogonal with cycloadditions of azides. These findings make it possible to employ strain promoted cycloadditions for the assembly of complex multifunctional and bio-inspired materials without the need of employing a toxic metal catalyst.
Result and Discussion
Nitrile oxides can undergo cycloadditions with terminal alkynes to give 3,5-isoxazoles,18 however, the success of these reactions is often compromised by a slow rate of reaction and competing dimerization of nitrile oxides.19 3,5-Disubstituted isoxazoles have been prepared in high yield by intramolecular cycloadditions,20 the use of activated dipolarophiles21 such as benzyne and norbornenes or by employing a Cu(I) catalyst.22 Furthermore, diazocarbonyl reagents, which are sufficiently stable for use in chemical synthesis, have been employed in 1,3-dipolar cycloadditions with substituted alkynes and benzynes to give pyrazoles and indazoles, respectively.23 These findings inspired us to explore strain-promoted alkyne-nitrile oxide cycloadditions (SPANOC) and alkyne-diazocarbonyl (SPADC) with DIBO (2) and compare the rates of reactions with similar cycloadditions with azides (SPAAC) and nitrones (SPANC) (Scheme 1).
Scheme 1.

Rate constants of cycloadditions of DIBO (2) with various 1,3-dipoles: nitrile oxide, azide, nitrone and diazocarbonyl derivatives. k’ is the relative rate with benzyl azide set at 1. The second order rate constant for nitrone 9 was determined by using equimolar mixture of reagents due to a strong absorbance at 305 nm.
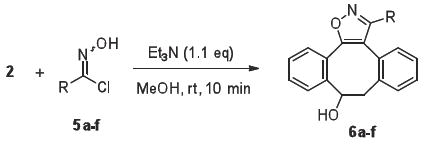
A range of imidoyl chlorides (5a-f), which can easily be converted into nitrile oxides by treatment with a mild base, was prepared by reactions of the corresponding aldehydes with hydroxylamine24 followed by chlorination of the resulting oximes with N-chlorosuccinimide.25 Addition of the imidoyl chlorides 5a-f to a solution of DIBO in methanol in the presence of triethylamine led, within several minutes, to the quantitative formation of isoxazoles 6a-f (Table 1).
Table 1.
Rate constants and yields for the cycloadditions of DIBO (2) with various nitrile oxides
 | |||
|---|---|---|---|
| Entry | R | k (M-1 s-1) | Yield (%)a |
| 1 | C6H5b, c, d (5a) | 3.38 ±0.03g | 93 |
| 2 | C6H5b, c, d (5a) | 2.46 ± 0.03h | ND |
| 3 | 4-MeO-C6H4b, e (5b) | 2.15 ± 0.02g | 89 |
| 4 | 4-O2N-C6H4b, f (5c) | 8.47 ± 0.03g | 93 |
| 5 | 4-F-C6H4b, c, d (5d) | 3.99 ± 0.05g | 90 |
| 6 | 4-Cl-C6H4b, c, d (5e) | 3.42 ± 0.03g | 90 |
| 7 | 4-Br-C6H4b, c, d (5f) | 3.31 ± 0.06g | 93 |
Isolated yields of combined isomers.
Second order rate constants were determined from pseudo first order rate constants at various concentrations of in situ formed nitrile oxides at 25 ± 0.1°C.
Pseudo first order kinetics were determined using UV-Vis spectroscopy by following the decay of the absorbance of compound 2 at 305 nm.
[2] = 6.0 × 10-5 M; for details on the concentrations of nitrile oxides, see supporting information.
[2] = 3.0 × 10-5 M; [5b] = 2.5-5.0 × 10-4 M.
Pseudo first order kinetics were determined by UV-Vis spectroscopy following the decay of the absorbance of 5c at 325 nm; [5c] = 6.0 × 10-5 M, [2] = 7.0-17.5 × 10-4 M.
Reaction was performed in methanol.
Reaction was performed in acetonitrile.
Accurate rate measurements of the cycloaddition reactions were conducted by UV spectroscopy following the growth of the decay of the characteristic absorbance of the acetylene of DIBO (2) at 305 nm. The rates were measured in methanol or acetonitrile solutions at 25±0.1 °C. The kinetics of the cycloadditions was studied under pseudo first order conditions by maintaining a fixed concentration of DIBO (2) while the concentration of the dipoles was varied. Consumption of starting material followed a first order equation and the pseudo first order rate constants were obtained by least-squares fitting of the data to a single exponential equation. The observed rate constants were linearly dependent on the concentration of dipoles,26 and second order cycloaddition rate constant calculated from the concentration dependencies of observed rates are listed in Table 1. As can be seen, the cycloadditions with the nitrile oxides are exceptionally fast and the substituent exert only small influence. It appears that strongly electron-withdrawing substituents, such as a nitro group (entry 4), somewhat increase the rate of reaction. Furthermore, the use of methanol or acetonitrile had only marginal influence on the reaction rate (entry 1 and 2).
Next, we compared the reaction rates of 1,3-dipolar cycloadditions of DIBO (2) with a nitrile oxide derived from imidoyl chlorides 5a, benzyl derived azide 7, nitrone 9 and diazocarbonyl derivative 11 to give isoxazole 6a, triazole 8, N-methyl isoxazoline 10 and pyrazole 12, respectively (Scheme 1 and Figure 2). It was found that the azide, nitrone and diazocarbonyl exhibit similar rates of reaction. However, the rate of cycloaddition of the nitrile oxide was fifty seven times faster than a similar reaction with benzyl azide.
Figure 2.

Consumption of DIBO 2 (6.0 × 10−5 M in methanol at 25 °C) in the presence of of various dipoles (3 mM). The lines shown were drawn using parameters obtained by least-squares fitting of single exponential equation. The inset shows reaction of DIBO with 5a at a different time scale.
Having established that nitrile oxides react exceptionally fast with DIBO (2), attention was focused on streamlining the process of nitrile oxide formation and cycloaddition. It was expected that the number of reaction steps could be reduced by a direct oxidation of oximes to nitrile oxides by using a mild oxidant such as [bis(acetoxy)iodo]benzene (BAIB).27 Furthermore, a one-pot multistep sequence in which oxime formation, oxidation and cycloaddition are performed by sequential addition of reagents was expected to reduce the number of workup and purification steps thereby increasing the efficiency and overall yield of the transformation. Thus, a reaction of benzaldehyde (13a) with hydroxylamine in methanol gave, after a reaction time of 2 h, an intermediate benzaldehyde oxime, which was treated with DIBO and BAIB and after an additional reaction time of 10 min, TLC and MS analysis indicated complete conversion of the oxime into isoxazole 6a, highlighting that the oxidation and cycloaddition step proceed with exceptionally high reaction rates (Table 2, entry 1). Additional experiments demonstrated that DIBO (2) is stable when exposed to BAIB alone and thus the oxidation and cycloaddition could be performed as a tandem reaction sequence. However, hydroxylamine decomposes DIBO (2) probably by a nucleophilic attack at the strained alkyne. Thus, the success of the transformation required the use of either an equimolar quantity of aldehyde and hydroxylamine or more conveniently the addition of acetone prior to cycloaddition to convert the excess hydroxylamine into ketoxime, which can react with BAIB but does not provide a 1,3-dipole. Alternatively, the addition of an excess of BAIB before administering DIBO (2) also led to complete consumption of the remaining excess of hydroxylamine and resulted in smooth formation of the desired isoxazoles 6a,g-h. Furthermore, this experimental approach made it possible to prepare isoxazoles in high yield, which have unstable corresponding imidoyl chlorides, notably in the aliphatic series (Table 2, entry 3).
Table 2.
One-pot oxime formation and SPANOC with DIBO (2)
 | |||
|---|---|---|---|
| Entry | R | k (M-1 s-1)a, b, c | Yield (%)d |
| 1 | C6H5 (6a) | 3.44 ± 0.03 | 55 |
| 2 | 2-Me-C6H4 (6g) | 3.20 ± 0.03 | 51 |
| 3 | C6H5-CH2CH2 (6h) | 1.38 ± 0.01 | 90 |
Rate constant was determined from isolated oximes;
Second order rate constants were determined from pseudo first order rate constants at various concentration of nitrile oxides at 25 ± 0.1°C.
Pseudo first order kinetics were determined using UV-Vis spectroscopy following the decay of the absorbance of 2 at 305 nm; [2] = 6.0 × 10-5 M; for details on the concentrations of nitrile oxides, see supporting information.
Isolated yields of combined isomers.
Rate constants were measured for the tandem sequence of oxidation of oximes to nitrile oxides followed by 1,3-dipolar cycloaddition with 2 establishing that the cycloaddition is the rate-limiting step and highlighting that oxidation with BAIB is exceptionally fast. For example, when benzaldehyde oxime was employed, the rate constant of the reaction was 3.44 M-1.s-1, which is almost the same to the value obtained when benzaldehyde imidoyl chloride was employed (3.38 M-1.s-1). Furthermore, the kinetic data for compounds 6g and 6h demonstrate further that the nature of the substituent has only a small effect on the rate of the reactions.
Convenient bioorthogonal reactions require that transformations are modular, have a high tolerance for the presence of functional groups, and proceed at ambient temperature using benign solvents and reagents. To determine whether SPANOC complies with these requirements, we examined the tagging of a carbohydrate with a biotin probe. Complex carbohydrates are involved in a wide variety of biological processes28 and fluorescent, biotin, multivalent and immobilized saccharide derivatives are important tools to study the intriguing properties of this class of biomolecules.29 It was expected that such derivatives can easily be prepared by reaction of sugar oximes by a sequential reaction of an aldose with hydroxylamine to give an oxime which can then be functionalized by reaction with DIBO derivatives in the presence of BAIB. The attraction of such an approach is that it allows functionalization of the reducing end of complex carbohydrates with various probes using low equivalents of expensive reagents. Thus, reaction of the readily available oxime 1430 with 2 or biotin-modified DIBO 15 (equimolar amounts) in the presence of BAIB for 10 min gave the sugar derivatives 16 and 17, respectively. It is interesting to note that the use of BAIB did not oxidize primary hydroxyls of lactose or sulfur of biotin.31 Compounds such as 16 that are modified with a biotin tag can, for example, be employed for immobilization to a surface coated with Streptavidine.
We envisaged SPANOC can also be used for the installation of tags into sialic acid containing glycoproteins by mild treatment with NaIO4 to form a C-7 aldehyde, which upon treatment with hydroxylamine will give an oxime that can be oxidized to a nitrile oxide for reaction with derivatives of DIBO. The attraction of such a strategy is that tags can be installed into glycoproteins by stable isoxazoles linkages.32 To examine the usefulness of such a strategy, the glycoprotein fetuin was treated with a 1 mM solution of NaIO4 for 5 min after which the excess of oxidizing reagent was removed by spin filtration. The resulting aldehyde containing glycoprotein was treated with hydroxyl amine to install an oxime, which was immediately oxidized to a nitrile oxide by short treatment with BAIB and then reacted with 15 for 15 min to give a biotin containing sialic acid. As a control, BSA, which does not contain sugar moieties, was subjected to the same sequence of reactions. The presence of biotin was examined by Western blotting using anti-biotin antibody conjugated to HRP. As can be seen in Figure 3, fetuin showed strong reaction when subjected to the sequential three-step procedure whereas BSA was not detected. Furthermore, exclusion of one of the reaction step abolished detection confirming the selectivity of the procedure. Quantitative protein and biotin determination indicated that two biotin moieties were installed in each fetuin molecule.
Figure 3.

Labeling and detection of sialic acids on the glycoprotein fetuin using SPANOC. Fetuin (samples in lanes 1-4) and BSA (samples in lanes 5-8) were subjected to periodate oxidation using NaIO4 (samples in lanes 3, 4, 7 and 8). Next, the generated C-7 aldehyde (on sialic acid) was reacted with HONH2.HCl to form an oxime, which was oxidized by reacting with BAIB to produce nitrile oxide that was reacted with DIBO 15 (samples in lanes 2, 4, 6 and 8). Incorporated biotin was then detected by Western blot using an anti-biotin antibody conjugated to HRP. Total protein loading was confirmed by Coomassie staining.
The large difference in reactivity of the cycloaddition of DIBO with the various 1,3-dipoles should make it possible to perform sequential click reactions, which may provide opportunities to prepare multifunctional compounds or materials by a simple synthetic procedure. In particular, it was expected that a highly reactive nitrile oxide can selectively undergo a cycloaddition in the presence of an azide. Furthermore, we envisaged that oximes can function as latent 1,3-dipoles, and therefore, a cyclooctyne should react with an azide without affecting an oxime. However, in the presence of BAIB, an oxime is rapidly converted into a nitrile oxide, which can then be reacted with another functionalized cyclooctyne. Thus, by careful selection of appropriate reagents, it should be possible to selectively modify a bifunctional linker (or complex compound) containing an azide and oxime moiety.
As expected, the addition of monosaccharide-modified DIBO 19 to bifunctional azido-oxime linker 18 in methanol resulted in selective cycloaddition at the azide moiety to provide the triazole 20 in high yield (Scheme 3). However, when linker 18 was treated with DIBO derivative 19 in the presence of BAIB, the oxime moiety was rapidly oxidized to a highly reactive nitrile oxide, which underwent a fast SPANOC resulting in the selective formation of isoxazole 21.
Scheme 3.

Selective cycloadditions between galactoside-modified DIBO 19 with either the azide or oxime moiety of linker 18.
Having established the orthogonality of azides and oximes/nitrile oxides, we examined sequential SPAAC-SPANOC click reactions of bifunctional linker 18 with a biotin (15) or a fluorescent probe (22) and a cluster of glycosides (25)10b modified with DIBO. Thus, treatment of azido-oxime linker 18 with DIBO modified-biotin (15) or DIBO modified–coumarin (22) in methanol or THF, respectively, at ambient temperature for 2 h led to clean formation of monofuntionalized triazoles 23 and 24, respectively. Next, triazoles 23 and 24 were exposed to a mixture of BAIB to convert the oxime moiety into a highly reactive nitrile oxide, and reaction with DIBO-modified saccharide cluster 25, lead to a fast SPANOC to give bifunctional compounds 26 and 27, displaying a cluster of galactoses conjugated to biotin or a fluorescent tag, respectively. It is of interest to note that neither oxidation of biotin moiety by BAIB, nor cycloaddition of the in-situ generated nitrile oxide at the carbon double bond of coumarin,33 was observed, highlighting that SPANOC is perfectly suitable for the conjugation of sensitive compounds.
Conclusion
Although metal free cycloadditions between cyclooctynes and azides to give stable 1,2,3-triazoles have found wide utility in chemical biology1 and material sciences,8 there is an urgent need for faster and more versatile bioorthogonal reactions. We have found that 1,3-dipolar cycloadditions of cyclooctynes with nitrile oxides exhibit much faster kinetics than similar reactions with azides. The nitrile oxides could easily be prepared by direct oxidation of the corresponding oximes with BAIB and these reaction conditions made it possible for oxime formation, oxidation and cycloaddition to be performed as a one-pot procedure. The transformations have a high tolerance for the presence of functional groups, proceed at ambient temperature using benign solvents and reagents and make it possible to modify compounds by a modular approach. Furthermore, the results presented here demonstrate that oximes and azides provide an orthogonal pair of functional groups for sequential metal free click reactions. In this respect, sequential click reactions have been reported by Cu(I)-catalyzed alkyne azide cycloaddition34 (CuAAC) using terminal- and silyl-protected alkynes35 and by exploiting the differential reactivity of CuAAC with SPAAC and thiol-ene click reactions.36 The usefulness of these approaches has been demonstrated by the controlled modification of oligonucleotides,37 proteins38 and fullerenes39 with two or more tags. The results reported here demonstrate, for the first time, that strain promoted click reactions can be performed in a sequential manner by tuning the reactivity of 1,3-dipoles or by using a latent 1,3-dipole. The attractiveness of the new approach is that it offers chemical flexibility, avoids toxic metal catalysts and makes it possible to multi-functionalize compounds by simple chemical manipulations.
A variety of methods have been reported for convenient installment of aldehydes in biomolecules,40 which can easily be converted into oximes. Thus, it is to be expected that a variety of biomolecules can be modified by SPANOC. Metal-free click reactions have found entry into materials science,8 and it is to be expected that SPANOC will provide an additional tool for the preparation of increasingly complex materials by simple and flexible chemical manipulations. Finally, we anticipate that SPANOC will offer an attractive alternative to the well-established oxime ligation41 because the synthesis of oximes is simple, the isoxazole products are stable, and a combined use with SPAAC will make it possible to introduce two different functional groups.
Experimental Procedures
Kinetic Measurements
The rate measurements of cycloadditions of dibenzocyclooctynol 2 with various dipoles were conducted by using Cary 50 and Cary 100 UV-Vis spectrophotometer at 25.0 ± 0.1°C. A calculated amount of 0.1 M solutions of a dipole (5a-b,d-f, 7, 9, 11, 13a-g,h, 14) required to achieve desired dipole concentration (2.5 × 10−4 - 2.7 × 10−2 M) was added to a thermally equilibrated solution of dibenzocyclooctynol 2 (3.0 × 10−5 − 6.0 × 10−5 M) in MeOH. In the case of nitrile oxide derivatives of 5a-b,d-f, the imidoyl chlorides 5a,b-d-f in methanol (6.0 × 10-4 - 1.5 × 10-2 M) were treated with triethylamine and then added to a thermally equilibrated solution of 2. Whereas nitrile oxide derivatives of 13a-g,h were generated by the oxidation of oximes 13a-g,h using [bis(acetoxy)iodo]benzene. Reactions were monitored by following the decay of the characteristic absorbance of dibenzocyclooctynol 2 at 305 nm. Observed rate constants of the cycloaddition reactions at various concentrations of dipoles are summarized in Tables S1-S14.26
In the case of the cycloaddition of dibenzocyclooctynol 2 with the nitrile oxide 5c, 0.1 M solutions of 2 was required to achieve the desired concentration of 2 (7.0 × 10−4 - 1.75 × 10−3 M) and triethylamine (concentration of triethylamine in the reaction mixture was 1.2 × 10−4 M) were added to a thermally equilibrated solution of 5c (6.0 × 10−5 M) in methanol. Reaction kinetics of nitrone 9 were monitored by following the second order growth of the product at 330 nm decay in the equimolar mixture of reagents. Second order rate constants were determined by fitting the curves with the following equation.
y = observed absorbances at given time “t”; A0 = Initial concentration of the starting materials in molarity; ESM = sum of extinction coefficients of starting materials; Ep = extinction coefficients of the product; k = second order rate constants in M-1s-1. The obtained second order rate constants were summarized in Table S15.
1-Benzyl-8,9-dihydro-1H-dibenzocycloocta[1,2,3]triazol-8-ol (8)
Benzyl azide (7) (13 μL, 0.1 mmol) was added dropwise to a solution of 4-dibenzocyclooctynol (2) (22 mg, 0.1 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature for 30 min. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using a mixture of hexane and ethyl acetate to give pure triazole 8 (34 mg, 97%): 1H NMR (300 MHz, CDCl3) δ 2.90-3.65 (m, 2H, CH2CH), 4.55-5.10 (m, 1H, CHOH), 5.40-5.85 (m, 2H, CH2N), 6.90-7.70 (m, 13H, aromH); HRMS (MALDI-ToF) 354.1295 (C23H20N3O (M+H+) requires 354.1601).
2-Methyl-3-phenyl-2,3,8,9-tetrahydrodibenzo[3,4:7,8]cycloocta-isoxazol-9-ol (10)
Phenyl nitrone 9 (14 mg, 0.1 mmol) was added to a solution of 4-dibenzocyclooctynol (2) (22 mg, 0.1 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature for 30 min. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using a mixture of hexane and ethyl acetate to give pure N-methyl dihydroisoxazole 10 (33 mg, 92%): 1H NMR (300 MHz, CDCl3) δ 1.50-1.95 (m, 1H, OH), 2.95-3.65 (m, 5H, CH3, CH2CH), 4.90-5.26 (m, 2H, CHN, CHOH), 6.75-7.65 (m, 13H, aromH); HRMS (MALDI-ToF) 356.1299 (C24H22NO2 (M+H+) requires 356.1645).
N-Benzyl-9-hydroxy-8,9-dihydro-3H-dibenzocyclooctapyrazole-3-carboxamide (12)
Diazo benzylamide 11 (18 mg, 0.1 mmol) was added to a solution of 4-dibenzocyclooctynol (2) (22 mg, 0.1 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature overnight. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using a mixture of hexane and ethyl acetate to give pure pyrazole 12 (36 mg, 92%): 1H NMR (300 MHz, CDCl3) δ 2.75-3.60 (m, 2H, CH2CH), 4.00-4.60 (m, 2H, CH2NH), 4.70-5.15 (m, 1H, CHOH), 6.65-7.90 (m, 15H, aromH, CHN, NH); HRMS (MALDI-ToF) 396.1426 (C25H22N3O2 (M+H+) requires 396.1707).
General Procedure for the Formation of Dibenzocyclooctyl-isoxazoles 6a-f from Imidoyl Chlorides 5a-f
Imidoyl chloride 5a-f (0.11 mmol) was added to a solution of 4-dibenzocyclooctynol (22 mg, 0.1 mmol) and triethylamine (16 μL, 0.11 mmol) in methanol (10 mL). The reaction mixture was stirred at room temperature for 10 min. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using an appropriate mixture of hexane and ethyl acetate to give pure dibenzocyclooctyl-isoxazoles 6a-f.
3-Phenyl-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6a)
1H NMR (300 MHz, CDCl3) δ 2.09 (s, 1H, OH), 2.75-3.25 (m, 2H, CH2), 4.66-5.05 (m, 1H, CHOH), 6.55-7.55 (m, 13H, aromH); HRMS (MALDI-ToF) 340.1075 (C23H18NO2 (M+H+) requires 340.1332).
3-(4’-Methoxyphenyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6b)
1H NMR (300 MHz, CDCl3) δ 208 (s, 1H, OH), 3.10-3.50 (m, 2H, CH2), 3.70-3.76 (m, 3H, OMe), 5.00-5.65 (m, 1H, CHOH), 6.75-7.55 (m, 12H, aromH); HRMS (MALDI-ToF) 370.1027 (C24H20NO3 (M+H+) requires 370.1438).
3-(4’-Nitrophenyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6c)
1H NMR (300 MHz, CDCl3) δ 2.08 (brs, 1H, OH), 3.15-3.75 (m, 2H, CH2), 5.00-5.75 (m, 1H, CHOH), 6.70-7.80 (m, 10H, aromH), 8.00-8.20 (m, 2H, aromH); HRMS (MALDI-ToF) 385.0961 (C23H17N2O4 (M+H+) requires 385.1183).
3-(4’-Fluorophenyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6d)
1H NMR (300 MHz, CDCl3) δ 1.97 (brs, 1H, OH), 3.15-3.75 (m, 2H, CH2), 5.10-5.70 (m, 1H, CHOH), 6.75-7.55 (m, 12H, aromH); HRMS (MALDI-ToF) 358.1016 (C23H17FNO2 (M+H+) requires 358.1238).
3-(4’-Chlorophenyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6e)
1H NMR (300 MHz, CDCl3) δ 2.08 (brs, 1H, OH), 3.15-3.75 (m, 2H, CH2), 5.00-5.70 (m, 1H, CHOH), 6.75-7.65 (m, 12H, aromH); HRMS (MALDI-ToF) 374.0579 (C23H1735ClNO2 (M+H+) requires 374.0942).
3-(4’-Bromophenyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6f)
1H NMR (300 MHz, CDCl3) δ 2.07 (brs, 1H, OH), 3.15-3.75 (m, 2H, CH2), 5.00-5.60 (m, 1H, CHOH), 6.70-7.85 (m, 12H, aromH); HRMS (MALDI-ToF) 417.9794 (C23H1779BrNO2 (M+H+) requires 418.0437).
General Procedure for the One-Pot Formation of Dibenzocyclooctyl-isoxazoles 6a,g-h from the Corresponding Aldehydes 13a,g-h
Hydroxylamine hydrochloride (10.4 mg, 0.15 mmol) was added to a solution of aldehyde 13a,g-h (1.0 mmol) and sodium hydroxide (6 mg, 0.15 mmol) in methanol (5 mL). The reaction mixture was stirred at room temperature for 2 h. (monitored by TLC). [Bis(acetoxy)iodo]benzene (BAIB) (64 mg, 0.20 mmol) was then added and the reaction mixture was stirred for 5 min at room temperature. 4-Dibenzocyclooctynol (22 mg, 0.1 mmol) was then added and the reaction mixture was stirred for an additional 10 min at room temperature. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using an appropriate mixture of hexane and ethyl acetate to give pure dibenzocyclooctyl-isoxazole 6a,g-h.
3-(2’-Toluyl)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (6g)
1H NMR (300 MHz, CDCl3) δ 1.88-2.20 (m, 4H, CH3, OH), 3.20-3.65 (m, 2H, CH2), 5.12-5.48 (m, 1H, CHOH), 6.60-7.60 (m, 12H, aromH); HRMS (MALDI-ToF) 354.1031 (C24H20NO2 (M+H+) requires 354.1489).
3-(2-Phenylethyl)-8,9-dihydrodibenzocyclooctanyl-isoxazol-9-ol (6h)
1H NMR (300 MHz, CDCl3) δ 1.50-1.90 (m, 1H, OH), 2.65-3.65 (m, 6H, 3×CH2), 4.90-5.10 (m, 1H, CHOH), 6.90-7.70 (m, 13H, aromH); HRMS (MALDI-ToF) 368.1210 (C25H22NO2 (M+H+) requires 368.1645).
General Procedure for the Formation of Lactose Derivatives
4-Dibenzocyclooctyl-derivative 2 or 15 (0.1 mmol) was added to a solution of [bis(acetoxy)iodo]benzene (35mg, 0.11 mmol) and lactose oxime 14 (40 mg, 0.11 mmol) in methanol (4 mL), premixed for 1 min. The reaction mixture was then stirred at room temperature for 10 min (TLC monitoring). The solution was concentrated in vacuo and the residue was purified either by Iatrobeads using a mixture of 10% of water in acetonitrile (for 16) or by RP-HPLC (0-2min 0.1% TFA/H2O, v/v; 2-5 min gradient of 0-20% 0.1% TFA/CH3CN, v/v; 5-30 min gradient of 20%-60% 0.1% TFA/CH3CN v/v, 30-35 min gradient of 60-100% 0.1% TFA/CH3CN, v/v, 35-45 min gradient of 100-0% 0.1% TFA/CH3CN, v/v; t=21.8 and 23.9 min. Appropriate fractions were combined and lyophilized to give pure dibenzocyclooctyl-isoxazole 16 and 17, respectively.
3-(Lactose)-8,9-dihydro-dibenzocyclooctanyl-isoxazol-9-ol (16)
1H NMR (600 MHz, CDCl3) δ 3.00-3.85 (m, 11H, 2×CH2OH, 4×CHgal, 3×CHglu), 3.95-4.30 (m, 2H, CH2Ar), 4.35-5.45 (m, 3H, ArCHOH, OCHO, CHC=N), 7.20-7.80 (m, 8H, aromH); HRMS (MALDI-ToF) 598.1693 (C28H33NO12Na (M+Na+) requires 598.1895).
Lactose-biotin isoxazole 17
1H NMR (600 MHz, D2O) δ 1.00-1.45 (m, 6H, 3×CH2biotin), 2.00-2.10 (m, 2H, CH2biotin), 2.40-4.30 (m, 32H, 6×CH2PEG, CH2biotin, 3×CHbiotin, CH2Ar, 2×CH2OH, 5×CHgal, 4×CHglu), 5.90-6.00 (m, 1H, ArCHOCO), 7.00-7.48 (m, 8H, aromH); HRMS (MALDI-ToF) 998.2939 (C45H61N5O17SNa (M+Na+) requires 998.3681).
Labeling of Sialic Acid Residues on Glycoproteins
Fetuin (sialylated) and BSA (non-sialylated) as control were subjected to periodate oxidation (1 mM NaIO4) for 5 min at 4°C. The protein solution was spin filtered at 14,000 × g for 15 min to remove excess reagent. Next, the generated C-7 aldehyde (on sialic acid) was reacted with HONH2.HCl (100 μM in DPBS, pH 6.7) for 1 h at RT. The generated oxime was oxidized by reacting with BAIB for 5 min at RT to produce nitrile oxide. After removal of excess reagent by centrifugation at 14,000 × g for 15 min, the nitrile oxide was reacted with DIBO 15 by a copper-free cycloaddition reaction for 30 min at RT. The samples (25 μg of protein per lane) were resolved on a 4–20% SDS-PAGE gel (Bio-Rad) and transferred to a nitrocellulose membrane. Next, the membrane was blocked in blocking buffer (nonfat dry milk (5%; Bio-Rad) in PBST (PBS containing 0.1% Tween-20 and 0.1% Triton X-100)) for 2 h at RT. The blocked membrane was then incubated for 1 h at RT with an anti-biotin antibody conjugated to horseradish peroxidase (HRP) (1:100000; Jackson ImmunoResearch Lab, Inc.) in blocking buffer and washed with PBST (4 × 10 min). Final detection of HRP activity was performed using ECL Plus chemiluminescent substrate (Amersham), exposure to film (Kodak) and development using a digital X-ray imaging machine (Kodak). Coomassie Brilliant blue staining was used to confirm total protein loading.
Biotin Quantitation
Incorporation of biotin into the protein was quantified using the Fluorescence Biotin Quantitation Kit (Thermo Scientific) according to the manufacturer’s protocol. Briefly, the biotinylated protein was dissolved in PBS, and DyLight Reporter (a premix of fluorescent avidin and 4′-hydroxyazobenzene-2-carboxylic acid (HABA)) was added to the biotinylated samples and a range of biocytin standards. The avidin in this reporter fluoresces when the weakly interacting HABA is displaced by the biotin. A calibration curve of the biocytin standards was used for calculations. The extent of biotinylation is expressed as mol biotin/mol protein.
Triazole 20
Azide 18 (10 mg, 0.03 mmol) was added to a solution of galactose-DIBO derivative 19 (14.3 mg, 0.03 mmol) in methanol (2 mL). The reaction mixture was stirred at room temperature for 2 h. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using a mixture of 10% methanol in CH2Cl2 to give pure triazole 20 (23 mg, 93%): 1H NMR (500 MHz, CD3OD) δ 1.74 (m, 2H, CH2), 2.90-3.28 (m, 4H, 2×CH2), 3.35-4.24 (m, 23H, 8×CH2, CHCH2, CH2gal, 3×CHgal), 4.50-4.62 (m, 2H, 2×CHgal), 5.85-6.20 (m, 2H, CH2CHO, NH), 6.80-7.70 (m, 12H, aromH), 8.01 (s, 1H, CH=N); HRMS (MALDI-ToF) 844.3492 (C41H51N5O13Na (M+Na+) requires 844.3376).
Isoxazole 21
A methanolic solution (1 mL) of galactose-DIBO derivative 19 (14.3 mg, 0.03 mmol) was added dropwise to a solution of oxime 18 (12.2 mg, 0.036 mmol) and BAIB (11.6 mg, 0.036 mmol) in methanol (1 mL). The reaction mixture was stirred at room temperature for 10 min. The solution was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel using a mixture of 8% methanol in CH2Cl2 to give pure isozaxole 21 (14.6 mg, 61%): 1H NMR (500 MHz, CD3OD) δ 1.70-1.84 (n, 2H, CH2), 3.30-4.30 (m, 29H, 10×CH2, CH2CHOH, CH2gal, 5×CHgal), 6.10-6.40 (m, 1H, CH2CHOH), 6.70-7.70 (m, 13H, aromH, NH); HRMS (MALDI-ToF) 842.2192 (C41H49N5O13Na (M+Na+) requires 842.3219).
General Procedure for SPAAC with Bifunctional Linker 18
Bifunctional linker 18 (0.03 mmol, 10.1 mg) and corresponding DIBO derivative 15 or 22 (0.03 mmol) were dissolved in MeOH or THF (in case of coumarin-DIBO derivative 22) (2 mL). The reaction mixture was stirred for 3 h and the solution was concentrated in vacuo. The residue was purified by column chromatography on silica gel.
Triazole 23
Purification by silica gel column chromatography (5 then 10% MeOH in CH2Cl2) gave 23 as a colorless oil (25.1 mg, 87%): 1H NMR (300 MHz, CD3OD) δ 1.34-1.45 (m, 2H, CHCH2CH2), 1.52-1.76 (m, 4H, CHCH2CH2CH2), 2.15-2.21 (m, 2H, CH2C=O), 2.64-2.69 (m, 1H, CHHS), 2.85-3.74 (m, 26H, CHHS, 9×CH2O, 2×CH2NH, CH2CHO, CHS), 3.83-4.06 (m, 4H, 2×CH2O), 4.21-4.28 (m, 1H, CHNH), 4.41-4.47 (m, 1H, CHNH), 4.55-4.61 (m, 2H, CH2-triazole), 5.89-6.17 (m, 1H, CH2CHO), 6.83-6.88 (m, 2H, aromH), 7.15-7.65 (m, 10H, aromH), 8.01 (s, 1H, CH=N); MS (MALDI-ToF) 981.4092 (C46H62N8O11SNa (M+Na+) requires 981.4157).
Triazole 24
Purification by silica gel column chromatography (3% MeOH in CH2Cl2) gave 24 as a yellow amorphous solid (22 mg, 73%): 1H NMR (300 MHz, CDCl3) δ 1.80-2.02 (m, 4H, 2×NCH2CH2CH2), 2.66-2.86 (m, 4H, 2×NCH2CH2CH2), 3.01-4.12 (m, 32H, 2×NCH2CH2CH2, 11×CH2O, 2×CH2NH, CH2CHO), 4.37-4.61 (m, 2H, CH2-triazole), 5.34-6.49 (m, 2H, CH2CHO, NH), 6.73-6.82 (m, 2H, aromH), 6.93-7.60 (m, 11H, aromH), 7.92-8.10 (m, 1H, NH), 8.56-8.68 (m, 1H, CH=N), 9.01-9.25 (m, 1H, CH-vinyl); MS (MALDI-ToF) 1022.4133 (C54H61N7O12Na (M+Na+) requires 1022.4270).
General Procedure for SPANOC between Triazoles 23 or 24 and Glycodendrimer 25
To a stirred solution of DIBO-glycodendrimer 25 (20.5 mg, 5.2 μmol) and oxime 23 or 24 (5.2 μmol) in MeOH/CH2Cl2 (3/1, v/v, 1.2 ml) was added a solution of BAIB (1.8 mg, 5.7 μmol) in MeOH (0.18 mL) and the reaction mixture was stirred for 30 min. The solvent was evaporated and the residue was purified by RP-HPLC. Appropriate fractions were combined and lyophilized.
Glycodendrimer-Biotin Conjugate 26
After RP-HPLC purification (0-5min 0% B, 5-40 min gradient of 0-100% B, t=29.4 min) and lyophilization, 26 was obtained as a white powder (14.0 mg, 55%): 1H NMR (500 MHz, D2O) δ 0.88-1.22 (m, 23H, 7×CH3, CHCH2CH2), 1.32-1.63 (m, 4H, CHCH2CH2CH2), 1.95-2.22 (m, 18H, 8×CH2CH2CH2-triazole, CH2C=O), 2.48-2.80 (m, 17H, CHHS, 8×CH2CH2-triazole), 2.80-3.02 (m, 17H, CHHS, 8×CH2CH2-triazole), 3.08-3.95 (m, 107H, 2×CH2CHO, 4×CH2NH, 15×CH2O, 8×CH-2gal, 8×CH-3gal, 8×CH-5gal, 8×CH2-6gal, 8×CH-4gal, 8×CH2CH2CH2-triazole, CHS), 3.99-4.55 (m, 56H, 9×CH2-triazole, 14×OCH2, 2×CHNH, 8×CH-1gal), 5.55-6.15 (m, 2H, 2×CH2CHO), 6.33-7.60 (m, 20H, aromH), 7.87 (s, 8H, 8×CHtriazole); MS (MALDI-ToF) 4933.4 (C218H310N34O92SNa (M+Na+) requires 4933.0).
Glycodendrimer-Coumarin Conjugate 27
After RP-HPLC purification (0-5min 0% B, 5-10 min gradient of 0-40% B, 10-30 min gradient of 40-60% B, t=25.3 min) and lyophilization, 27 was obtained as a yellow powder (15.1 mg, 61%): 1H NMR (500 MHz, D2O:CD3CN, 1:1, v/v) δ 0.99-1.20 (m, 21H, 7×CH3), 1.65-1.81 (m, 4H, 2×NCH2CH2CH2), 1.99-2.08 (m, 16H, 8×CH2CH2CH2-triazole), 2.59-2.62 (m, 20H, 2×NCH2CH2CH2, 8×CH2CH2-triazole), 2.84 (t, J = 7.3 Hz, 16H, 8×CH2CH2-triazole), 3.10-3.94 (m, 110H, 2×CH2CHO, 4×CH2NH, 15×CH2O, 2×NCH2CH2CH2, 8×CH-2gal, 8×CH-3gal, 8×CH-5gal, 8×CH2-6gal, 8×CH-4gal, 8×CH2CH2CH2-triazole), 3.94-4.25 (m, 36H, 14×OCH2, 8×CH-1gal), 4.25-4.45 (m, 18H, 9×CH2-triazole), 5.41-6.19 (m, 2H, 2×CH2CHO), 6.58-7.51 (m, 21H, aromH), 7.64 (s, 8H, 8×CHtriazole), 8.36-9.12 (m, 1H, CH-vinyl); MS (MALDI-ToF) 4972.8 (C224H309N33O93Na (M+Na+) requires 4974.0).
Supplementary Material
Figure 1.

Cyclooctynes for metal free click reactions.
Scheme 2.
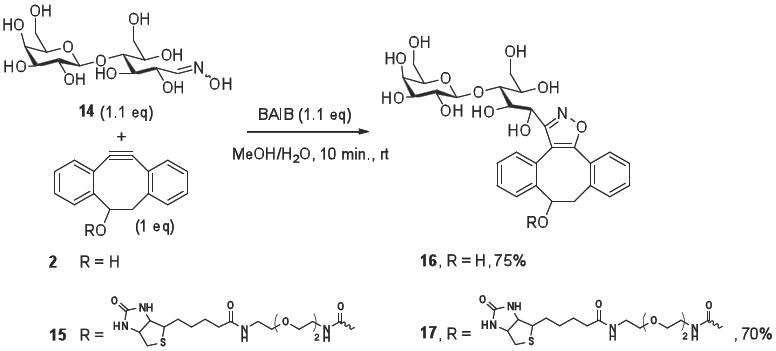
Modification of the reducing end of lactose by SPANOC employing oxime 14.
Scheme 4.
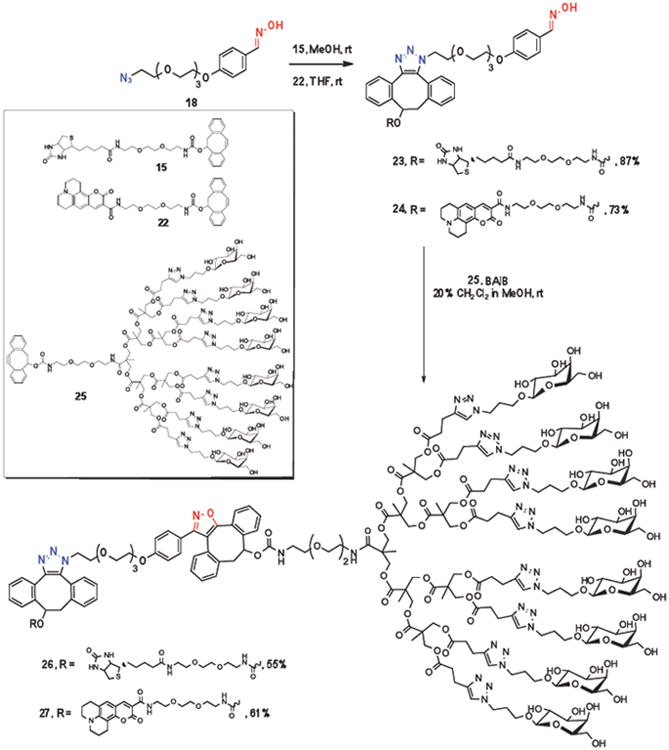
Preparation of a bifunctional compound by a sequential SPAAC and SPANOC
Acknowledgments
This research was supported by the National Cancer Institute of the US National Institutes of Health (R01 CA88986, G.-J.B.), the National Science Foundation Plant Genome Program (IOS-0923992, G.-J.B.), National Science Foundation (CHE-0449478, V.V.P.), and Georgia Cancer Coalition (V.V.P.).
Footnotes
Supporting Information Available. Synthesis of oximes 14a-f, imidoyl chlorides 5a-f, azido-oxime linker 18, DIBO-galactose 19, amino-PEG-coumarin 33 and DIBO-glycodendron 25. Spectral data of new compounds, and detailed reaction profiles in kinetic studies.
References
- 1.(a) Wittig G, Krebs A. Chem Ber. 1961;94:3260–3275. [Google Scholar]; (b) Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Debets MF, van der Doelen CWJ, Rutjes FPJT, van Delft FL. ChemBioChem. 2010;11:1168–1184. doi: 10.1002/cbic.201000064. [DOI] [PubMed] [Google Scholar]; (d) Becer CR, Hoogenboom R, Schubert U. Angew Chem Int Ed. 2009;48:4900–4908. doi: 10.1002/anie.200900755. [DOI] [PubMed] [Google Scholar]
- 2.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 3.(a) Link AJ, Vink MKS, Agard NJ, Prescher JA, Bertozzi CR, Tirrell DA. Proc Natl Acad Sci USA. 2006;103:10180–10185. doi: 10.1073/pnas.0601167103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tanrikulu IC, Schmitt E, Mechulam Y, Goddard WA, III, Tirrell DA. Proc Natl Acad Sci USA. 2009;106:15285–15290. doi: 10.1073/pnas.0905735106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Fernández-Suárez M, Baruah H, Martínez-Hernández L, Xie KT, Baskin JM, Bertozzi CR, Ting AY. Nat Biotechnol. 2007;25:1483–1487. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neef AB, Schultz C. Angew Chem Int Ed. 2009;48:1498–1500. doi: 10.1002/anie.200805507. [DOI] [PubMed] [Google Scholar]
- 5.Nessen MA, Kramer G, Back JW, Baskin JM, Smeenk LEJ, de Koning LJ, van Maarseveen JH, de Jong L, Bertozzi CR, Hiemstra H, de Koster CG. J Proteome Res. 2009;8:3702–3711. doi: 10.1021/pr900257z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kele P, Mezö G, Achats F, Wolfbeis OS. Angew Chem Int Ed. 2009;48:344–347. doi: 10.1002/anie.200804514. [DOI] [PubMed] [Google Scholar]
- 7.a) Jayaprakash KN, Pend CG, Butler D, Varghese JP, Maier MA, Rajeev KG, Manoharan M. Non-Nucleoside Building Blocks for Copper-Assisted and Copper-Free Click Chemistry for the Efficient Synthesis of RNA Conjugates. Org Lett. 2010 doi: 10.1021/ol102205j. In Press. [DOI] [PubMed] [Google Scholar]; b) van Delft P, Meeuwenoord NJ, Hoogendoorn S, Dinkelaar J, Overkleeft HS, van der Marel GA, Filippov DV. Synthesis of Oligoribonucleic Acid Conjugates Using a Cyclooctyne Phosphoramidite. Org Lett. 2010 doi: 10.1021/ol102357u. In Press. [DOI] [PubMed] [Google Scholar]
- 8.Wilson JT, Krishnamurthy VR, Cui W, Qu Z, Chaikof EL. J Am Chem Soc. 2009;131:18228–18229. doi: 10.1021/ja908887v. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JA, Baskin JM, Bertozzi CR, Koberstein JT, Turro NJ. Chem Commun. 2008:3064–3066. doi: 10.1039/b803043j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Ornelas C, Broichhagen J, Weck M. J Am Chem Soc. 2010;132:3923–3931. doi: 10.1021/ja910581d. [DOI] [PubMed] [Google Scholar]; (b) Ledin PA, Friscourt F, Guo J, Boons G-J. Convergent Assembly and Surface Modification of Multifunctional Dendrimers by Three Consecutive Click Reactions. Chem Eur J. 2010 doi: 10.1002/chem.201002052. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lallana E, Fernandez-Megia E, Riguera R. J Am Chem Soc. 2009;131:5748–5750. doi: 10.1021/ja8100243. [DOI] [PubMed] [Google Scholar]
- 12.Orski SV, Poloukhtine AA, Arumugam S, Mao L, Popik VV, Locklin J. J Am Chem Soc. 2010;132:11024–11026. doi: 10.1021/ja105066t. [DOI] [PubMed] [Google Scholar]
- 13.(a) Ess DH, Jones GO, Houk KN. Org Lett. 2008;10:1633–1636. doi: 10.1021/ol8003657. [DOI] [PubMed] [Google Scholar]; (b) Bach RD. J Am Chem Soc. 2009;131:5233–5243. doi: 10.1021/ja8094137. [DOI] [PubMed] [Google Scholar]; (c) Schoenebeck F, Ess DH, Jones GO, Houk KN. J Am Chem Soc. 2009;131:8121–8133. doi: 10.1021/ja9003624. [DOI] [PubMed] [Google Scholar]; (d) Chenoweth K, Chenoweth D, Goddard WA., III Org Biomol Chem. 2009;7:5255–5258. doi: 10.1039/b911482c. [DOI] [PubMed] [Google Scholar]
- 14.(a) Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ning X, Guo J, Wolfert MA, Boons G-J. Angew Chem Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, Popik VV. J Am Chem Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]; (c) Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Ning X, Temming RP, Dommerholt J, Guo J, Ania DB, Debets MF, Wolfert MA, Boons G-J, van Delft FL. Angew Chem Int Ed. 2010;49:3065–3068. doi: 10.1002/anie.201000408. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKay CS, Moran J, Pezacki JP. Chem Commun. 2010;46:931–933. doi: 10.1039/b921630h. [DOI] [PubMed] [Google Scholar]
- 18.Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Vol. 1. Wiley; New York: 1984. pp. 1–176. [Google Scholar]
- 19.Grünanger P, Vita-Finzi P. In: The Chemistry of Heterocyclic Compounds: Isoxazoles. Part I. Taylor EC, Weissberger A, editors. Vol. 49. Wiley- Interscience; New York: 1991. pp. 1–416. [Google Scholar]
- 20.Nair V, Suja TD. Tetrahedron. 2007;63:12247–12275. [Google Scholar]
- 21.König P, Zountsas J, Bleckmann K, Meier H. Chem Ber. 1983;116:3580–3590. For example using benzyne, see: Crossley JA, Browne DL. Tetrahedron Lett. 2010;51:2271–2273. Dubrovskiy AV, Larock RC. Org Lett. 2010;12:1180–1183. doi: 10.1021/ol902921s. Spiteri C, Mason C, Zhang F, Ritson DJ, Sharma P, Keeling S, Moses JE. Org Biomol Chem. 2010;8:2537–2542. doi: 10.1039/b927235f. Spiteri C, Sharma P, Zhang F, Macdonald SJF, Keeling S, Moses JE. Chem Commun. 2010;46:1272–1274. doi: 10.1039/b922489k. For example using norbornene, see: Gutsmiedl K, Wirges CT, Ehmke V, Carell T. Org Lett. 2009;11:2405–2408. doi: 10.1021/ol9005322.
- 22.Hansen TV, Wu P, Fokin VV. J Org Chem. 2005;70:7761–7764. doi: 10.1021/jo050163b. [DOI] [PubMed] [Google Scholar]
- 23.(a) Wittig von G, Hutchison JJ. Liebigs Ann Chem. 1970;741:79–88. [Google Scholar]; (b) Qi X, Ready JM. Angew Chem Int Ed. 2007;46:3242–3244. doi: 10.1002/anie.200700069. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He S, Chen L, Niu Y-N, Wu L-Y, Liang Y-M. Tetrahedron Lett. 2009;50:2443–2445. [Google Scholar]; (d) Jin T, Yamamoto Y. Angew Chem Int Ed. 2007;46:3323–3325. doi: 10.1002/anie.200700101. [DOI] [PubMed] [Google Scholar]; (e) Liu Z, Shi F, Martinez PDG, Raminelli C, Larock RC. J Org Chem. 2008;73:219–226. doi: 10.1021/jo702062n. [DOI] [PubMed] [Google Scholar]
- 24.Ramón RS, Bosson J, Díez-González S, Marion N, Nolan SP. J Org Chem. 2010;75:1197–1202. doi: 10.1021/jo902461a. [DOI] [PubMed] [Google Scholar]
- 25.(a) Grundmann C, Datta SK. J Org Chem. 1969;34:2016–2018. [Google Scholar]; (b) Liu K-C, Shelton BR, Howe RK. J Org Chem. 1980;45:3916–3918. [Google Scholar]
- 26.See Supporting Information
- 27.(a) Mendelsohn BA, Lee S, Kim S, Teyssier F, Aulakh VS, Ciufolini MA. Org Lett. 2009;11:1539–1542. doi: 10.1021/ol900194v. [DOI] [PubMed] [Google Scholar]; (b) Das B, Holla H, Mahender G, Banerjee J, Reddy MR. Tetrahedon Lett. 2004;45:7347–7350. [Google Scholar]
- 28.Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 29.(a) Paulson JC, Blixt O, Collins BE. Nat Chem Biol. 2006;2:238–248. doi: 10.1038/nchembio785. [DOI] [PubMed] [Google Scholar]; (b) Kiessling LL, Splain RA. Annual Rev Biochem. 2010;79:619–653. doi: 10.1146/annurev.biochem.77.070606.100917. [DOI] [PubMed] [Google Scholar]; (c) Laurent N, Voglmeir J, Flitsh SL. Chem Commun. 2008;37:4400–4412. doi: 10.1039/b806983m. [DOI] [PubMed] [Google Scholar]
- 30.Brand J, Huln T, Groth U, Jochims JC. Chem Eur J. 2006;12:499–509. doi: 10.1002/chem.200500325. [DOI] [PubMed] [Google Scholar]
- 31.In methanol, oxidation of biotin was only observed after 15 h. In water, trace amounts of sulfoxide byproduct were observed by MS after 10 min, therefore monitoring of reactions or premixing of BAIB with oximes was required.
- 32.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson J. Nature Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cycloaddition of nitrile oxide at C=C double bond of coumarin was reported after 72 h., see: Baldoli C, Gioffreda F, Zecchi G. J Heterocyclic Chem. 1994;31:251–253.
- 34.For original publications, see: Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;41:2596–2599. Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. For a detailed review, see: Meldal M, Tornøe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479.
- 35.Valverde IE, Delmas AF, Aucagne V. Tetrahedron. 2009;65:7597–7602. [Google Scholar]
- 36.Nurmi L, Lindqvist J, Randev R, Syrett J, Haddleton DM. Chem Commun. 2009:2727–2729. doi: 10.1039/b904249k. [DOI] [PubMed] [Google Scholar]
- 37.Isobe H, Fujino T, Yamazaki N, Guillot-Nieckowski M, Nakamura E. Org Lett. 2008;10:3729–3732. doi: 10.1021/ol801230k. [DOI] [PubMed] [Google Scholar]
- 38.(a) Aucagne V, Leigh DA. Org Lett. 2006;8:4505–4507. doi: 10.1021/ol061657d. [DOI] [PubMed] [Google Scholar]; (b) Kuijpers BHM, Groothuys S, Hawner C, ten Dam J, Quaedflieg PJLM, Schoemaker HE, van Delft FL, Rutjes FPJT. Org Proc Res Dev. 2008;12:503–511. [Google Scholar]
- 39.Iehl J, Nierengarten J-F. Chem Commun. 2010;46:4160–4162. doi: 10.1039/c0cc00252f. [DOI] [PubMed] [Google Scholar]
- 40.(a) Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem Int Ed. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]; (b) Carrico IS, Carlson BL, Bertozzi CR. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]; (c) Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Nat Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ebisu K, Tateno H, Kuroiwa H, Kawakami K, Ikeuchi M, Hirabayashi J, Sisido M, Taki M. ChemBioChem. 2009;10:2460–2464. doi: 10.1002/cbic.200900430. [DOI] [PubMed] [Google Scholar]
- 41.(a) Dawson PE, Kent SBH. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]; (b) Borgia JA, Fields GB. Trends Biotechnol. 2002;18:243–251. doi: 10.1016/s0167-7799(00)01445-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.