

Abstract
Objective
We previously generated MRL/lpr mice deficient in the activation-induced deaminase (AID) who lack isotype switching and immunoglobulin hypermutation. These mice have high levels of unmutated (germline) autoreactive IgM yet experienced an increase in survival and an improvement in lupus nephritis that exceeded that of MRL/lpr mice lacking IgG. Herein, we test the hypothesis that high levels of germline autoreactive IgM in these mice confer protection against lupus nephritis.
Methods
Autoreactive IgM antibodies of various specificities including against dsDNA from AID-deficient-MRL/lpr mice were given to asymptomatic MRL/lpr mice and the levels of cytokines, proteinuria, immune complex deposition in the kidneys, and glomerulonephritis were examined. Novel AID-deficient MRL/lpr mice that lack any antibodies were generated to compare to AID-deficient-MRL/lpr mice that secrete only IgM.
Results
Anti-dsDNA IgM treatment resulted in a dramatic improvement in lupus nephritis. Other autoreactive IgM’s such as anti-phospholipid and anti-Smith antigen did not alter pathology. Secretion of pro-inflammatory cytokines by macrophages, and the levels of inflammatory cells and apoptotic debris in the kidneys were lower in mice receiving anti-dsDNA IgM. Protective IgM derived from AID-deficient-MRL/lpr mice, displayed a distinct B cell repertoire, with a bias towards members of the Vh7183 family.
Conclusions
Anti-dsDNA IgM protected MRL/lpr mice from lupus nephritis likely by stopping the inflammatory cascade leading to kidney damage. A distinct repertoire of Vh usage in anti-dsDNA IgM hybridomas from AID-deficient mice, suggests enrichment in these mice of a dedicated B cell population that secretes unmutated protective IgM.
Autoreactive B cells play a critical role in the pathogenesis of Systemic Lupus Erythematosus (SLE) (1–5). SLE is characterized by the circulation of autoantibodies and immune complex deposition in various tissues particularly the kidneys leading to glomerulonephritis (6). Hallmark SLE autoantibodies include antibodies against double-stranded DNA (dsDNA) and ribonuclear proteins (7). These pathogenic autoantibodies form immune complexes that trigger inflammatory responses leading to organ damage. Factors that have been implicated in SLE and in lupus-like syndromes in mice include defective apoptosis, toll receptor signaling, defects in B and T cell tolerance, complement activation, cytokine regulation, and defects in endothelial cell function (8–19).
Strong evidence suggests an antibody-independent contribution of B cells to autoimmunity (20). Lupus-prone mice with B cells unable to secrete antibodies developed a milder form of lupus nephritis, while mice without B cells did not. The evidence suggests that B cells contribute to autoimmunity by activating autoreactive T cells, likely as antigen-presenting cells (20).
MRL-Faslpr/lpr (MRL/lpr) mice develop an autoimmune syndrome similar to SLE (21–22). MRL/lpr mice display high levels of autoantibodies to dsDNA, and lupus nephritis development through immune complex deposition, glomerular disease, and tissue infiltration by inflammatory cells. Many loci contribute to autoimmunity in MRL/lpr mice leading to defects in cytokine regulation, lymphocyte tolerance, and apoptosis (8–17).
We generated MRL/lpr mice deficient in AID (23), a molecule required for isotype switching and immunoglobulin hypermutation (24). Heterozygotes experienced a delay in the onset of lupus nephritis that correlated with delayed accumulation of high affinity anti-dsDNA antibodies (25). Homozygous AID-deficient-MRL/lpr mice lack anti-dsDNA IgG but have high levels of autoreactive IgM. Despite this, AID-deficient-MRL/lpr mice failed to develop significant kidney disease and experienced survival levels exceeding that of mice with B cells but lacking secreted antibodies. This prompted us to speculate that, in addition to the lack of pathogenic IgG, another factor contributed to improved survival. Herein, we demonstrate the protective factor to be unmutated anti-dsDNA IgM.
METHODS AND MATERIALS
Mice
AID−/−MRL/lpr mice were developed by backcrossing C57BL6-AID-deficient onto the MRL/lpr background, using the MRL/MpJ-Faslpr/2J (stock number: 006825) for six generations. They were then backcrossed an additional nine generations into cryorecovered MRl/lpr mouse strain, MRL/MpJ-Faslpr/J, obtained from Jackson Laboratory (Stock Number: 000485, Bar Harbor, Maine). Mouse strain B6;129S4-Igh-6tm1Che/J with a mutation in the IgM secretory exon, from Jackson Laboratory was described previously (26). B6;129S4-Igh-6tm1Che/J mice were backcrossed with cryo-recovered MRl/lpr mice for four generations and with the 15th-generation backcrossed AID−/−MRL/lpr mice for an additional 4 generations to generate μS−/−AID−/−MRL/lpr. These are AID-deficient MRL/lpr mice lacking the secretory exon of IgM. Because B cells from these mice cannot secrete IgM, nor can undergo class switch recombination, they lack secreted antibodies. All mice were housed in specific pathogen-free facilities at NIEHS. All experiments included the same number of males and females among groups.
Production of anti-dsDNA monoclonal IgM
Anti-dsDNA IgM-producing hybridomas were generated following standard protocols (27). Spleens were collected from uninmunized AID+/+, AID+/−, and AID−/− MRL/lpr mice (10–12 weeks of age, 3–6 mice/group). Splenocytes were fused with murine myeloma NS1 (ratio of 5:1) in 50% polyethylene glycol (1500 PEG, Roche, Basel, Switzerland). Supernatants were screened for anti-dsDNA specificity by ELISA. Cell clones secreting anti-dsDNA IgM were injected (2×106 cells) into the peritoneal cavity of RAG-1 ko mice (Jackson Laboratory) pretreated with 0.2ml Pristane (Sigma, St. Louis, MO) to induce ascites. Ascites fluid was collected 2–3 weeks after injection and filtered through a small filter on the tip of the serum-containing syringe (Millex, Bedford, MA). IgM concentrations and specificity to dsDNA was confirmed by ELISA (Bethyl Laboratories, Montgomery, TX). A control non-autoreactive IgM-producing hybridoma clone was confirmed by anti-nuclear antibody assay. A mouse B-cell line secreting antiphospholipid IgM was a gift from Laurent Verkoczy (Duke University). An anti-Smith IgM-producing hybridoma was provided by Barbara Vilen (UNC at Chapel Hill). These lines were injected into the peritoneal cavity of RAG-1-deficient mice pretreated with Pristane. All monoclonal antibodies (mAb) were generated using the same strategy.
Passive transfer of serum or monoclonal antibodies
MRL/lpr mice of 8–10 weeks of age were treated with pooled sera collected from older MRL/lpr mice, sera from age-matched AID−/−MRL/lpr mice, or with PBS by i.p. 200ul/mouse, twice a week for 8 weeks. Wild-type sera contained both IgM and IgG autoantibodies, while AID-deficient sera contained high levels of IgM autoantibodies but no IgG. Urine samples were collected weekly for proteinuria testing. After 8 weeks, mice were euthanized. In antibody-transfer experiments, MRL/lpr mice were treated with anti-dsDNA IgM mAbs at 100ug, twice a week for 8–15 weeks depending on the experiment. For therapeutic experiments, treatment was not started until the mice had moderate proteinuria (50–60 mg/dl).
Detection of urine protein
Urine protein was tested with Multistix 10 SG (Siemens Healthcare Diagnostics Inc., Tarrytown, NY), and scored as described previously (23).
ELISA
IgM and IgG levels in cell culture supernatants, ascites fluid, and sera were determined with commercial ELISA kits (Bethyl Laboratories). Mouse anti-dsDNA IgM was measured as described previously (23). Reading at 3-fold over background was set as cut-off value.
Sequencing of Vh regions from hybridomas and splenic B cells
Anti-dsDNA monoclonal antibody-producing hybridomas were lysed with TRIzol (Invitrogen, Carlsbad, CA) for RNA. Two micrograms of RNA was used to synthesize cDNA by SuperScript III First-Strand Synthesis (Invitrogen). Five microliters of cDNA reactions was used to amplify V regions of immunoglobulins by AccuPrimeTM Pfx DNA polymerase (Invitrogen). Universal primers were designed as described previously (28): universal primer MVH-F(5′-AGGTSMARCTGCAGSAGTCWGG-3′) paired with MCμ-R(5′-CAGGGGGCTCTCGCAGGAGACGAGG-3′) for in VHDJH-Cμ amplification, while MVH-F paired with MCγ-R( 5′-GGACAGGGATCCAGAGTTCC-3′) for VHDJH-Cγ amplification. The PCR cycle conditions included: 95°C for 2 min; 35 cycles at 95°C for 15 sec, 58°C for 1 min, and 68°C for 1 min. The amplified VH DNA fragments were purified with QIAquick® Gel Extraction Kit (Qiagen, Valencia, CA). The purified VH fragments were sequenced using BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) plus MCμ-R or MCγ-R primer depending on VH fragments.
For repertoire analysis, spleens were collected from AID-WT and –deficient MRL/lpr mice (3 mice/group, 10–14 weeks of age). CD19+ B cells were purified with anti-CD19 MACS MicroBeads (Miltenyi Biotech GmbH, Bergisch Gladbach, Germany) and lysed with TRIzol. cDNA was synthesized and Vμ fragments were amplified and sequenced as above.
Immunofluorescence and histoimmunochemical staining
Frozen kidney samples were stained for C3 as reported previously (23). For each animal, 20 randomly selected glomeruli were evaluated to obtain an average score as follows: +, weak staining with limited localization in the glomerulus(<50%); ++, moderate stain intensity where localization in the glomerulus was more diffuse (50%–75%); and +++, intense stain with diffused and homogeneous stain covering most of the glomerulus (>75%).
For F4/80 staining, paraffin-fixed kidney slides were deparaffinized, hydrated, and treated with 3% hydrogen peroxide. Slides were placed in a Decloaking Chamber (Biocare Medical, Concord, CA). Slides were blocked with 5% rabbit serum (Jackson Immunoresearch Laboratories, Inc., West Grove, PA) and avidin block from the Avidin/Biotin Blocking Kit (Vector Laboratories, Inc., Burlingame, CA). Rat anti-F4/80 monoclonal antibody (BM8) (Santa Cruz Biotechnology Santa Cruz, CA) was applied at a 1:25 dilution. Biotinylated Rabbit Anti-Rat IgG (H+L) (Vector Laboratories, Inc. Burlingame, CA) was used at 1:200 dilution. Slides were incubated with DAB chromagen and counterstained with Harris Hematoxylin. F4/80 staining in glomeruli was scored on a scale of 0–3 in 20 randomly selected high-power microscopic fields (x400) per animal as follows: 0, No F4/80 positive cell; +, 1–9 F4/80 positive cells; ++, 10–20 F4/80 positive cells; +++, > 20 F4/80 positive cells.
Cleaved-caspase-3 staining was performed similarly to F4/80 staining except rabbit anti-cleaved Caspase-3 Antibody (Promega Corporation, Madison, WI) was used at 1:500 dilution and the biotinylated goat anti-rabbit IgG (Vector Laboratories, Inc. Burlingame, CA) was used at 1:1000 dilution. Cleaved-Caspase-3 positive cells were counted in 10 randomly selected views from High-Power Microscopic Field (x200) per animal. The presence of apoptotic cells was graded according to apoptotic index which equals Cleaved-Caspase-3 positive cell number/per HPMF: 0, No positive cell;+, 1–4 positive cells; ++, 5–10, positive cells; +++, > 10 positive cells.
Histology
Kidneys were prepared for hematoxylin and eosin stain and periodic acid-Schiff stain as previously described (23). Glomerulonephritis and lymphocyte infiltration in kidneys were examined by a pathologist and graded for each animal on a scale of 1–4 with 1 = minimal, 2= mild, 3 = moderate, and 4 = marked, as reported previously (23). For electron microscopy, tissues were prepared and examined as described previously (23).
Detection of cytokines
Serum cytokines levels (IFNγ, IL-1beta, IL-12, TNFα, IL-6, IL-4, IL-5, and IL-10) were determined with Bio-Plex Mouse Cytokine Assays (Bio-Rad Laboratories, Hercules, CA) and Mouse TH1/TH2 9-PlexUltra-Sensitive Kit (Meso Scale Discovery, Gaithersburg, Maryland). Splenic macrophages were purified with CD11b Micro-beads and MACS Separation Columns (Miltenyi Biotec, Auburn, CA), and incubated with LPS (1ug/ml). Mouse TH1/TH2 9-PlexUltra-Sensitive Kit (Meso Scale Discovery) was used to measure cytokine production.
Survival analysis
μS−/−AID−/− MRL/lpr mice (N = 22) (AID-deficient MRL/lpr mice without secreted antibodies; see “mice” section) and AID−/− MRL/lpr mice (N = 26) were followed for at least 56 weeks to examine mortality with or without secreted IgM.
Statistical analysis
Mann-Whitney Rank Sum Test was performed to analyze the data among different treatment groups. For survival analysis, Log-rank (Mantel-Cox) Test was used to compare survival curves. For repertoire analysis, the Likelihood Ratio test was used.
RESULTS
We hypothesized that the survival increase in AID-deficient-MRL/lpr mice, was not only due to the lack of IgG, but also to a protective factor in their serum. Given the high levels of autorective IgM, we speculated the factor to be anti-dsDNA IgM. To test this, we carried out passive transfer experiments using sera or monoclonal anti-dsDNA IgM antibodies from AID-deficient-MRL/lpr mice. The results are discussed below.
Mice receiving sera or anti-dsDNA IgM from AID-deficient-MRL/lpr mice experienced an improvement in lupus nephritis
To determine whether sera from AID deficient MRL/lpr mice contained a factor that may contribute to increased survival, we transferred sera from AID-deficient, AID-WT, and AID-heterozygous MRL/lpr into young asymptomatic MRL/lpr mice for at least eight weeks. These mice were 8–10 weeks of age and did not exhibit evidence of proteinuria, initially. There was a significant reduction in the levels of proteinuria in the mice receiving the sera from AID-deficient-MRL/lpr mice compared to PBS or WT sera (Figure 1a). Accordingly, MRL/lpr mice recipients of AID-deficient MRL/lpr sera, also experienced decreased levels of immune complex deposition in the kidneys, as measured by the amount of complement factor 3 immunofluorescence in glomeruli (C3) (Figure 1b). The group receiving serum from AID wild-type MRL/lpr mice exhibited a trend for increased kidney damage but it was not significantly different from PBS.
Figure 1.

Transfer of serum or anti-dsDNA IgM antibodies from AID-deficient-MRL/lpr mice into asymptomatic MRL/lpr mice conferred protection against kidney damage. A. Decreased levels of urine protein in mice receiving the AID-deficient derived serum (N = 6) but not the AID-WT serum (N = 6) or PBS control (N = 6). B. Decreased immune complex deposition in the glomeruli of kidneys from mice receiving the AID-deficient derived serum. Fluorescent antibodies against C3 complement factor were used to visualize immune complexes. The right panel depicts representative images for each score and were taken from an untreated MRL/lpr mouse. C&D. Decreased levels of proteinuria in mice receiving anti-dsDNA IgM (For C, all groups were N = 8; and for D, all groups were N = 11). Clone number indicated in parenthesis. NA indicate a non-autoreactive IgM. Mann-Whitney Rank Sum Test was used to test for significance. Error bars depict standard error.
To determine whether increased levels of autoreactive IgM in the serum of AID-deficient mice contributed to reduced lupus nephritis in mice receiving serum from AID-deficient-MRL/lpr mice, anti-dsDNA IgM secreting hybridomas were generated. There was a 7-fold increase in anti-dsDNA clone numbers in hybridomas from AID-deficient-MRL/lpr mice (Supplemental Figure 1). As expected, AID-deficient mice produced only IgM-secreting clones. Clones secreting anti-dsDNA IgM from AID-WT and AID-deficient MRL/lpr mice were used in passive transfer experiments. In addition, a non-autoreactive, IgM-secreting clone (as determined by anti-nuclear antibody assay, Supplemental Figure 2), was used as a control. Following treatment with anti-dsDNA IgM, asymptomatic MRL/lpr mice experienced a significant delay in the onset of lupus nephritis as measured by proteinuria (Figure 1c–d) and IgG-immune complex deposition (supplemental Figure 3). Accordingly, the kidneys of mice receiving anti-dsDNA IgM treatment were smaller than those from other groups in three out of four groups receiving anti-dsDNA IgM (Supplemental Figure 4). In most cases, mice receiving this treatment failed to develop significant kidney disease even after 10 weeks of treatment; a point where most mice were at least 6 months old and when all of the mice in the PBS or the non-autoreactive antibody-receiving group had developed moderate to severe kidney damage (Figure 1c–d).
To ask if other autoreactive IgM antibodies could also confer protection against lupus nephritis, we repeated these experiments, including an anti-phospholipid IgM and an anti-Smith antigen IgM. The anti-Smith antigen IgM has been previously described and it cross-reacts with single-stranded DNA but not with dsDNA (29). Mice receiving the anti-dsDNA IgM fared best compared to all groups including mice treated with anti-Smith and anti-phospholipid IgM which displayed little or no protection (Figure 2a–b). Histopathology analysis of these groups confirmed that the anti-dsDNA IgM-receiving groups experienced decreased kidney damage (Figure 2c–d). There was a trend for reduced mononuclear cell infiltration, albeit not statistically significant (Figure 2c), and a significant reduction in glomerulonephritis (Figure 2d).
Figure 2.

Not all autoreactive IgM protected against lupus nephritis. A&B. Decreased levels of urine protein and of immune complex deposition in mice receiving the AID-deficient derived anti-dsDNA IgM (AID−/−) compared to mice receiving an anti-phospholid IgM (AP), anti-Smith antigen IgM (AS), a non-autoreactive IgM (NA) and PBS. C&D. Mononuclear cell infiltration in the kidneys and glomerulonephritis was also reduced in mice receiving the AID-deficient derived anti-dsDNA IgM. Glomerulonephritis scores for each group were based from analysis of 20 randomly selected glomeruli from each group visualized with hematoxylin and eosin stain (H&E) and Periodic Acid Schiff stain (PAS). Only the anti-dsDNA IgM from AID−/− background was significant (other comparisons had p values > 0.10). Mann-Whitney Rank Sum Test was used to test for significance. Error bars depict standard error.
Testing various doses of anti-dsDNA IgM confirmed a positive correlation for better protection with increasing dose up to 100μg, twice a week, the dose used in our studies. There was little gain when using double that amount (Figure 3a). Using 100 μg, twice a week, we treated symptomatic mice (proteinuria of >60 mg/dl) and found that even in mice with significant proteinuria, anti-dsDNA IgM treatment resulted in reduction in kidney damage with (Figure 3b–c).
Figure 3.

Dose-dependent therapeutic effects of anti-dsDNA IgM in MRL/lpr mice with significant proteinuria. A. Increasing dose correlates with better protection up to a certain point (100mg). MRL/lpr mice, at 10–11 weeks of age, were divided into five groups, 9–10 mice/group. The mice were i.p. injected with anti-dsDNA IgM mAb (clone 12H5) derived from AID−/−MRL/lpr mice or PBS only, twice a week for 8 weeks, each time, at the indicated dose. B&C. Therapeutic effect of anti-dsDNA IgM in mice with significant proteinuira at start of treatment. B. Decreased levels of urine protein in mice receiving the anti-dsDNA IgM (N = 12 for all groups). C. Decreased immune complex deposition in the glomeruli of kidneys from symptomatic mice receiving the anti-dsDNA IgM compared to PBS group, as measured by C3 staining. Mann-Whitney Rank Sum Test was used to test for significance. Error bars depict standard error.
Anti-dsDNA IgM treatment was associated with reduced macrophage infiltration and a reduction in the levels of apoptotic debris in the kidneys
Electron microscopy analysis of glomeruli, revealed the following characteristics seen in most groups except mice receiving anti-dsDNA IgM from AID-deficient hybridomas: macrophage and lymphocytic infiltration, the presence of electron dense deposits in glomeruli, and the presence of apoptotic cells/debris in the mesangium (Supplemental Table 1). Next, kidney tissues were stained for immunohistochemistry with either F4/80 antibody (macrophage detection) or anti-cleaved Caspase-3 antibody (apoptosis detection). Mice receiving anti-dsDNA IgM treatment displayed lower levels of F4/80 and in the case of the 12H5 clone, for cleaved-Caspase-3, confirming the electron microscopy observations (Figure 4). As seen for proteinuria and C3 deposition, treatment with other autoreactive IgM, such as anti-phospholipid and anti-Smith, failed to reduce cleaved-Caspase-3 or F4/80 positive areas (Figure 4). Accordingly, LPS–activated splenic B cells and macrophages were examined for cytokine production. There was a trend for reduced secretion of TNF-alpha by cultured macrophages and for decreased levels of circulating IgG-containing immune complexes in mice receiving anti-dsDNA IgM, but it reached statistical significance for some but not all of the anti-dsDNA IgM antibodies tested (Supplemental Figure 5). There was no correlation with decreased activation of autoreactive T cells by B cells or by an increase in the proportion of regulatory T cells, with anti-dsDNA IgM treatment (Supplemental Figure 6), but, activated macrophages from mice receiving the protective IgM, secreted less interferon γ (Supplemental Figure 7) and there was a trend for mice receiving the anti-dsDNA IgM to have more of the anti-inflammatory cytokines IL4 and IL10 in the serum, albeit was not consistent across experiments (data not shown). Combined, these results suggest anti-dsDNA IgM blocks the inflammatory cascade leading to tissue damage and cell death. The variability among anti-dsDNA IgM antibodies in their impact on inflammatory correlates suggests that while all decrease proteinuria and immune complex deposition, there is variation among them, in terms of effectivity and mechanism, which may relate to their specificity or polyereactivity.
Figure 4.

Mice receiving anti-dsDNA IgM displayed evidence of decreased inflammation and of ongoing apoptosis in the kidneys. A. Staining of kidneys with anti-F4/80 antibody which recognizes a glycoprotein expressed by macrophages, is reduced in MRL/lpr mice receiving anti-dsDNA IgM compared to MRL/lpr mice receiving PBS control. B. Staining of kidneys with anti-cleaved-caspase-3 antibody, which binds to the large fragment of activated caspase-3 following cleavage during apoptosis is also reduced in MRL/lpr mice receiving anti-dsDNA IgM. Positive cells are stained brown. A normal C57BL6 control is included. Significant P values were obtained with the Mann-Whitney Rank Sum Test. Error bars depict standard error.
AID deficient MRL/lpr mice with secreted IgM outlived AID-deficient, μS-deficient MRL/lpr mice lacking secreted antibodies, despite both strains having minimal levels of proteinuria
μS−/−MRL/lpr mice were crossed to AID-deficient-MRL/lpr mice to generate MRL/lpr mice with no secreted antibodies; μS−/−MRL/lpr mice lack secreted IgM because of a mutation in the IgM secretory exon (26) and without AID, they are unable to switch to IgG or other isotypes. Comparing these mice to AID-deficient-MRL/lpr mice, secreting only IgM, enabled the examination of two nearly identical strains differing only on whether they secrete IgM. Minimal levels of proteinuria was observed in both strains after 52 weeks suggesting anti-dsDNA IgM protects against lupus nephritis through an IgG-mediated process (Figure 5a). This is in contrast to the parental strain, μS−/− MRL/lpr, which experienced accelerated lupus nephritis as they lacked secreted IgM but secreted all other isotypes (26). Surprisingly, AID-deficient-MRL/lpr mice with IgM experienced significantly lower mortality rates than AID-deficient-MRL/lpr mice without secreted antibodies which experienced ~ 70% mortality at 60 weeks (Figure 5b), despite both groups having little proteinuria. This suggests an additional mechanism of IgM-mediated protection.
Figure 5.
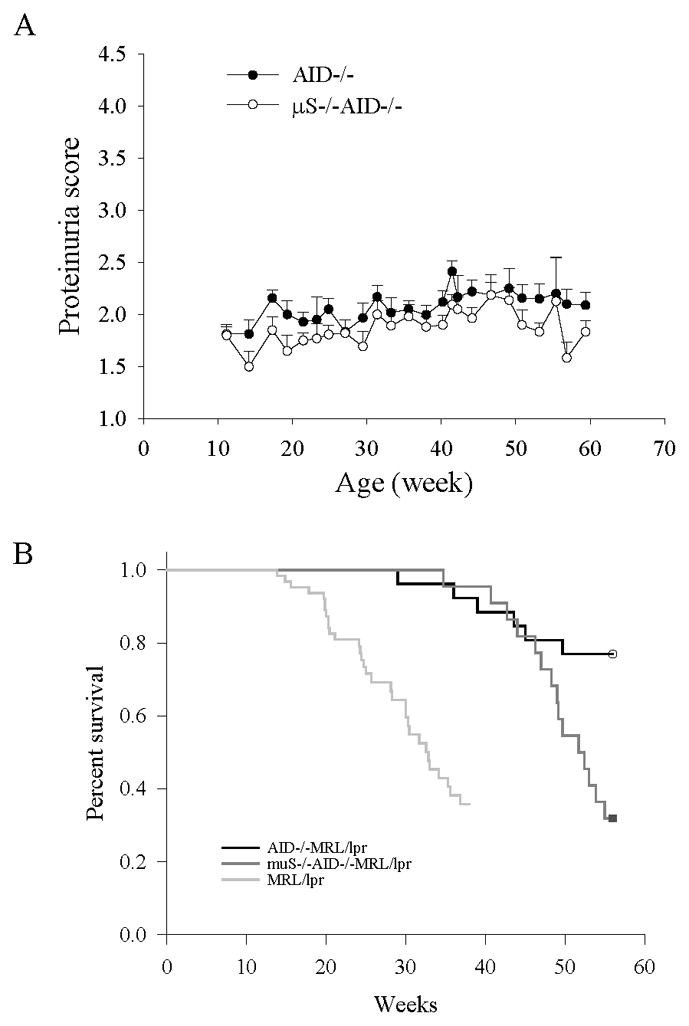
MRL/lpr mice lacking secreted antibodies had a similar phenotype in terms of kidney damage as mice secreting only IgM but exhibited increased mortality. A. Similar levels of proteinuria in AID-deficient MRL/lpr mice and μS−/−AID−/− MRL/lpr mice with no secreted IgG or other isotypes. By crossing μS−/− MRL/lpr mice to AID-deficient MRL/lpr mice, we generated mice unable to secrete antibodies of any kind. The resulting μS−/−AID−/− MRL/lpr mice (N = 22) were followed for at least 56 weeks and levels of proteinuria compared to those of AID-deficient MRL/lpr mice (N = 26), secreting only IgM. There was no difference between the two groups suggesting IgM protects from lupus nephritis through an IgG-mediated mechanism. Error bars depict standard error. B. AID-deficient MRL/lpr mice experienced increased survival levels compared to μS−/−AID−/− MRL/lpr despite both μS−/−AID−/− MRL/lpr and AID-deficient MRL/lpr mice having similar levels of proteinuria (p < 0.05). This suggests an additional protective role for IgM, besides lupus nephritis. The two survival curves were compared by using Log-rank (Mantel-Cox) Test. Survival data from AID-WT MRl/lpr mice (N = 63) was included.
The Vh usage repertoire of B cells secreting anti-dsDNA IgM from AID-deficient-MRL/lpr mice favors Vh7183 members, while that from AID-WT-MRL/lpr mice favors the J558 family
To examine the Vh genes used in anti-dsDNA IgM antibodies, we sequenced hybridomas from AID-deficient and WT MRL/lpr mice. Over half of the hybridomas from AID-deficient-MRL/lpr mice were Vh7183, while most derived from WT mice were VhJ558-related (Figure 6a). The rest of AID-deficient hybridomas were either J558 or J606. Among the antibodies used in the passive transfer studies, 4A5 (WT) originated from the VhJ558 family, 12H5 (AID−/− ) from the Vh7183 family and 13D2 (AID−/− ) from the VhJ606 family. These results cannot be explained by a biased B cell repertoire in AID-deficient-MRL/lpr mice (Figure 6b). IgM repertoire analysis of splenic B cells from AID-deficient and AID-WT MRL/lpr mice using the same primers did not reveal an increase in Vh7183 usage in AID-deficient mice (Figure 6b). To examine if the difference in repertoire and protection correlated with affinity to dsDNA, apparent affinities were measured for the antibodies used in the study, but it did not reveal a correlation. For example, anti-dsDNA IgM 4A5 had the highest apparent affinity value at 12.96 but did not protect better than clones 12H5 and 13D2, with apparent affinities of 0.76 and 0.21 respectively.
Figure 6.

Repertoire of anti-dsDNA IgM secreting hybridomas from AID-deficient MRL/lpr mice is different from the repertoire derived from AID WT counterparts. A. Sequencing of hybridomas revealed that at least half the clones of AID−/−MRL/lpr hybridomas (N = 11) used Vh7183 while AID WT hybridomas (N = 10) used mostly J558. This difference was significant (P < 0.05) using the Likelihood Ratio test. B. The repertoire differences in the hybridomas is not due to a splenic B cell repertoire shift with AID deficiency.
DISCUSSION
Despite having high levels of autoreactive IgM, AID-deficient-MRL/lpr mice experienced a dramatic increase in survival (23). Herein we hypothesized that autoreactive IgM is not only nonpathogenic in lupus nephritis, but may be protective. Treatment with anti-dsDNA IgM antibodies from AID-deficient and AID-WT MRL/lpr – derived hybridomas revealed an improvement in all measures of lupus nephritis, such as proteinuria, immune complex deposition, and glomerulonephritis in MRL/lpr mice. These results suggest that both factors, high anti-dsDNA IgM levels and absence of pathogenic IgG, contributed to increased survival of AID-deficient-MRL/lpr mice. In another study, B cells from MRL/lpr mice were found to express high AID levels and to secrete highly mutated autoantibodies, suggesting that high AID levels increase levels of autoreactive antibodies (30). Also, it was previously noted that peptides with specificity to dsDNA may be therapeutic in lupus nephritis (31) and autoimmune mice lacking secreted IgM but having secreted IgG experienced accelerated autoimmunity (26, 32).
The differential contribution of autoreactive IgM versus IgG to autoimmunity remains controversial. IgG autoreactive antibodies are thought to be pathogenic in autoimmune disease because of their isotype and affinity to self-antigen. Somatic hypermutation and class switch recombination tend to co-occur which means that much of the serum IgG has been fine-tuned to a specific antigen through affinity maturation. In SLE and in MRl/lpr mice, this could mean enhanced affinity to self-antigen (33–34). AID-heterozygous MRL/lpr mice experienced a delay in lupus nephritis onset that correlated with reduced levels of high affinity anti-dsDNA IgG and reduced hypermutation (25). IgG subclass is known to influence autoantibody pathogenicity (35–37), and FcR and FcRIII deficient mice experienced a reduction in kidney damage (38–39). So it is apparent that both, class switch and affinity maturation against self-antigens through somatic hypermutation contribute to pathogenic IgG formation. IgM’s role is less clear since IgM-antibodies are likely to be unmutated. However, the dramatic increase in survival experienced by AID-deficient-MRL/lpr mice, which secrete nothing but IgM, much of it autoreactive, casts serious doubts on the pathogenecity of autoreactive IgM in lupus nephritis (23). The data presented here points to anti-dsDNA IgM as a protective agent. Recently, it was shown that SLE patients in remission have high levels of autoreactive naive B cells (40), which in light of our findings, suggests these are cells secreting protective IgM. Also, a positive correlation was found between a high antidsDNA IgG to IgM ratio in SLE and lupus nephritis (41). It will be important to determine if the predictability of the IgG/IgM ratio for lupus nephritis can be improved by refining the IgM value to reflect protective IgM.
Not all autoreactive IgM is protective. Anti-phospholipid and anti-Smith IgM did not protect significantly. Furthermore, some autoreactive IgM may contribute to autoimmune disease. Natural IgM antibodies are associated with exacerbation of ischemia/reperfusion injury (42) and Durandy and colleagues demonstrated that there is an increased incidence of autoimmune cytopenias and other autoimmune disorders with AID deficiency in humans (43) that is almost certainly associated with specific autoreactive natural IgM. However, this is observed not just with AID-deficiency but with most immunodeficiencies suggesting that the immune deregulation associated with immunodeficiency results in increased activation of residual autoreactive lymphocyte populations (44). A recent study, using AID-deficient lpr mice in the C57BL6 strain showed a modest increase in proteinuria with AID deficiency (45). There was also increased proliferation of B cells, consistent with a known component of the AID-deficiency phenotype (24). Because in an autoimmune environment, the available population to experience hyperproliferation is polyreactive, one might expect that to contribute to autoimmunity. This implies that AID deficiency can enhance autoreactivity by increasing the population of autoreactive B cells, known to have an antibody-independent role in enhancing autoimmunity (20), but ameliorates it by pathogenic IgG-deficiency and elevated levels of protective IgM. It is possible that when lupus nephritis is mild in the parental strain like in C57Bl6 lpr mice (46), AID-deficiency-mediated increase in polyreactive B cells enhances autoimmunity. However, when the parental strain displays antibody-dependent severe lupus nephritis, the benefits of AID–deficiency such as high levels of protective IgM and absence of pathogenic IgG, outweigh the effects from an increase in polyreactive B cells. It will be interesting to see if mortality was increased in the lpr mice in the C57BL6 background with AID deficiency; in the MRL/lpr background, AID deficiency resulted in a dramatic improvement in survival (23).
The mechanism by which anti-dsDNA IgM antibodies protect against lupus nephritis is unknown. There was a trend for macrophages from mice receiving anti-dsDNA IgM to secrete less pro-inflammatory cytokines such as TNF-alpha and interferon γ, consistent with decreased macrophages in the kidneys. A correlation between anti-dsDNA IgG antibodies and activation of macrophages to secrete pro-inflammatory cytokines has been made previously (47); perhaps IgM has the opposite effect. If true, it suggests that protective IgM helps create an environment less prone to inflammation-induced tissue injury, perhaps by quickly clearing apoptotic debris as suggested previously (48). However, while all anti-dsDNA IgM tested decreased proteinuria and immune complex deposition, not all correlated with a reduction in inflammatory markers or apoptotic debris in the kidneys, suggesting variation in mechanism. In a previous study, we found similar levels of IgM in the glomeruli of AID-deficient vs AID-WT MRL/lpr mice, suggesting that despite its size, pentameric IgM is able to penetrate glomeruli, (23). Thus, it is possible IgM also works by blocking the formation of IgG-immune complexes locally. Indeed, AID-deficient MRL/lpr mice with a defect in secreted IgM (therefore lacking secreted antibodies) experienced a reduction in proteinuria, similar to AID-deficient-MRL/lpr mice with IgM, suggesting that anti-dsDNA IgM protects against nephritis through an IgG-mediated process.
Of note, while antibody-deficient mice had no signs of lupus nephritis, their AID-deficient but IgM-secretion proficient littermates, experienced significantly higher survival levels. Indeed, close to 70% of the antibody-deficient mice had died by 62 weeks while less than 25% of the IgM-secreting mice had died by that time. Both had survival rates that were better than conventional MRL/lrp mice, which tend to die earlier from lupus nephritis. Given that proteinuria was very low in both groups, these results suggest additional protective roles for IgM. This finding is currently under investigation.
What defines a protective antibody in lupus nephritis? Our data suggests that the IgM isotype and anti-dsDNA specificity are important. In contrast to pathogenic IgG and pathogenicity, high affinity to dsDNA did not enhance protection by anti-dsDNA IgM. This suggests that dsDNA specificity is a correlate to other self-antigen important for protection or that high affinity correlates with reduced polyreactivity and it is the polyreactivity that is important. Intriguingly, other autoreactive IgM such as against phospholipids and against the Smith antigen (a ribonuclear protein) did not confer protection against lupus nephritis. We are currently investigating the role of polyreactivity vs anti-dsDNA specificity in IgM-mediated protection against lupus nephritis by examining a large panel of anti-dsDNA IgM antibodies. Given that B cells from AID-deficient-MRL/lpr mice only secrete unmutated IgM, this novel strain represents a new tool to better define the role of unmutated autoreactive IgM and natural antibodies in autoimmunity.
AID-deficient-MRL/lpr mice-derived hybridomas displayed distinct Vh usage from AID-WT MRL/lpr mice-derived hybridomas. Most AID-deficient hybridomas used Vh7183 or J606, genes typically seen in the neonatal repertoire (49). None of the hybridomas from AID-WT mice used Vh7183 or J606; most used J558. This difference was not the result of a change in the overall B cell repertoire with AID-deficiency. Previous reports detected anti-dsDNA IgM using Vh7183 from MRL/lpr mice but at a lower frequency (50), suggesting Vh7183 IgM-bearing B cells with anti-dsDNA specificity are present regardless of AID status but are enriched in AID-deficient MRL/lpr mice because of a B cell repertoire rich in polyreactive IgM. The Vh usage in anti-dsDNA IgM hybridomas from AID-deficient MRL/lpr mice suggests a B cell population specialized in secreting protective antibodies. If true, it may be possible that this population could be specifically targeted for activation in SLE.
Supplementary Material
Acknowledgments
This work was supported, in whole or in part by National Institutes of Health Grant (Intramural Research Program, NIEHS) 1ZIAES101603-07
We are grateful to Silvia Bolland, Michael Fessler, and Laurent Verkoczy for helpful comments. We are indebted to Ron Herbert, Greg Travlos, and Natasha Clayton for invaluable assistance with histology and cytokine analysis. Special thanks to Connie Cummings for electron microscopy analysis. This work was supported by the National Institutes of Health Grant (Intramural Research Program, NIEHS) 1ZIAES101603-07.
References
- 1.van Es JH, Gmelig Meyling FH, van de Akker WR, Aanstoot H, Derksen RH, Logtenberg T. Somatic mutations in the variable regions of a human IgG anti-double-stranded DNA autoantibody suggest a role for antigen in the induction of systemic lupus erythematosus. J Exp Med. 1991;173:461–470. doi: 10.1084/jem.173.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winkler TH, Fehr H, Kalden JR. Analysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomas. Eur J Immunol. 1992;22:1719–1728. doi: 10.1002/eji.1830220709. [DOI] [PubMed] [Google Scholar]
- 3.Kasaian MT, Casali P. B-1 cellular origin and VH segment structure of IgG, IgA, and IgM anti-DNA autoantibodies in patients with systemic lupus erythematosus. Ann N Y Acad Sci. 1995;764:410–423. doi: 10.1111/j.1749-6632.1995.tb55856.x. [DOI] [PubMed] [Google Scholar]
- 4.Manheimer-Lory AJ, Zandman-Goddard G, Davidson A, Aranow C, Diamond B. Lupus-specific antibodies reveal an altered pattern of somatic mutation. J Clin Invest. 1997;100:2538–2546. doi: 10.1172/JCI119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenks SA, Sanz I. Altered B cell receptor signaling in human systemic lupus erythematosus. Autoimmun Rev. 2009;8:209–213. doi: 10.1016/j.autrev.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oates JC, Gilkeson GS. Mediators of injury in lupus nephritis. Curr Opin Rheumatol. 2002;14:498–503. doi: 10.1097/00002281-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Yung S, Chan TM. Anti-DNA antibodies in the pathogenesis of lupus nephritis--the emerging mechanisms. Autoimmun Rev. 2008;7:317–321. doi: 10.1016/j.autrev.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Peng SL, Craft J. T cells in murine lupus: propagation and regulation of disease. Mol Biol Rep. 1996;23:247–251. doi: 10.1007/BF00351176. [DOI] [PubMed] [Google Scholar]
- 9.Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Exp Med. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theofilopoulos AN, Lawson BR. Tumour necrosis factor and other cytokines in murine lupus. Ann Rheum Dis. 1999;58(Suppl 1):I49–55. doi: 10.1136/ard.58.2008.i49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786–794. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196:1543–1552. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 15.Pascual V, Banchereau J, Palucka AK. The central role of dendritic cells and interferon-alpha in SLE. Curr Opin Rheumatol. 2003;15:548–556. doi: 10.1097/00002281-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Culton DA, O’Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ, Clarke SH. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176:790–802. doi: 10.4049/jimmunol.176.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert MR, Carnathan DG, Cogswell PC, Lin L, Baldwin AS, Jr, Vilen BJ. Dendritic cells from lupus-prone mice are defective in repressing immunoglobulin secretion. J Immunol. 2007;178:4803–4810. doi: 10.4049/jimmunol.178.8.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Martin D, Diaz-Zamudio M, Crispin JC, Alcocer-Varela J. Interleukin 2 and systemic lupus erythematosus: beyond the transcriptional regulatory net abnormalities. Autoimmun Rev. 2009;9:34–39. doi: 10.1016/j.autrev.2009.02.035. [DOI] [PubMed] [Google Scholar]
- 19.Casali P. Polyclonal B cell activation and antigen-driven antibody response as mechanisms of autoantibody production in SLE. Autoimmunity. 2000;5:147–150. doi: 10.3109/08916939009002973. [DOI] [PubMed] [Google Scholar]
- 20.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999a;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 23.Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, Diaz M. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 25.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci U S A. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scearce RM, Eisenbarth GS. Production of monoclonal antibodies reacting with the cytoplasm and surface of differentiated cells. Methods Enzymol. 1983;103:459–469. doi: 10.1016/s0076-6879(83)03032-3. [DOI] [PubMed] [Google Scholar]
- 28.Liang Z, Xie C, Chen C, Kreska D, Hsu K, Li L, Zhou XJ, Mohan C. Pathogenic profiles and molecular signatures of antinuclear autoantibodies rescued from NZM2410 lupus mice. J Exp Med. 2004;199:381–398. doi: 10.1084/jem.20030132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloom DD, Davignon JL, Retter MW, Shlomchik MJ, Pisetsky DS, Cohen PL, Eisenberg RA, Clarke SH. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr mice. J Immunol. 1993;150:1591–1610. [PubMed] [Google Scholar]
- 30.Zan H, Zhang J, Ardeshna S, Xu Z, Park SR, Casali P. Lupus-prone MRL/faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity. 2009;42:89–103. doi: 10.1080/08916930802629554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iikuni N, Hahn BH, La Cava A. Potential for anti-DNA immunoglobulin peptide therapy in systemic lupus erythematosus. Expert Opin Biol Ther. 2009;9:201–206. doi: 10.1517/14712590802681636. [DOI] [PubMed] [Google Scholar]
- 32.Melamed D, Miri E, Leider N, Nemazee D. Unexpected autoantibody production in membrane Ig-mu-deficient/lpr mice. J Immunol. 2000;165:4353–4358. doi: 10.4049/jimmunol.165.8.4353. [DOI] [PubMed] [Google Scholar]
- 33.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, Gonzalez JB, Pascual V, Stichweh D, Wardemann H, Nussenzweig MC. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A. 2008;105:9727–9732. doi: 10.1073/pnas.0803644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi S, Nose M, Sasaki J, Yamamoto T, Kyogoku M. IgG3 production in MRL/lpr mice is responsible for development of lupus nephritis. J Immunol. 1991;147:515–519. [PubMed] [Google Scholar]
- 36.Berney T, Fulpius T, Shibata T, Reininger L, Van Snick J, Shan H, Weigert M, Marshak-Rothstein A, Izui S. Selective pathogenicity of murine rheumatoid factors of the cryoprecipitable IgG3 subclass. Int Immunol. 1992;4:93–99. doi: 10.1093/intimm/4.1.93. [DOI] [PubMed] [Google Scholar]
- 37.Panka DJ, Salant DJ, Jacobson BA, Minto AW, Marshak-Rothstein A. The effect of VH residues 6 and 23 on IgG3 cryoprecipitation and glomerular deposition. Eur J Immunol. 1995;25:279–284. doi: 10.1002/eji.1830250146. [DOI] [PubMed] [Google Scholar]
- 38.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, van de Winkel JG, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 39.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 40.Yurasov S, Tiller T, Tsuiji M, Velinzon K, Pascual V, Wardemann H, Nussenzweig MC. Persistent expression of autoantibodies in SLE patients in remission. J Exp Med. 2006;203:2255–2261. doi: 10.1084/jem.20061446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus. 2004;13:36. doi: 10.1191/0961203304lu485oa. [DOI] [PubMed] [Google Scholar]
- 42.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006;177:4727–4734. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 43.Quartier P, Bustamante J, Sanal O, Plebani A, Debre M, Deville A, Litzman J, Levy J, Fermand JP, Lane P, Horneff G, Aksu G, Yalcin I, Davies G, Tezcan I, Ersoy F, Catalan N, Imai K, Fischer A, Durandy A. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clin Immunol. 2004;110:22–29. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 44.Haymore BR, Mikita CP, Tsokos GC. Common variable immune deficiency (CVID) presenting as an autoimmune disease: role of memory B cells. Autoimmun Rev. 2008;7:309–312. doi: 10.1016/j.autrev.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, Guo L, Tian J, Zheng B, Han S. Deficiency in activation-induced cytidine deaminase promotes systemic autoimmunity in lpr mice on a C57BL/6 background. Clin Exp Immunol. 2010;159:169–175. doi: 10.1111/j.1365-2249.2009.04058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vidal S, Kono DH, Theofilopoulos AN. Loci predisposing to autoimmunity in MRL-Fas lpr and C57BL/6-Faslpr mice. J Clin Invest. 1998;101:696–702. doi: 10.1172/JCI1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang EJ, Nahm DH, Jang YJ. Mouse monoclonal autoantibodies penetrate mouse macrophage cells and stimulate NF-kappaB activation and TNF-alpha release. Immunol Lett. 2009;124:70–76. doi: 10.1016/j.imlet.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Werwitzke S, Trick D, Kamino K, Matthias T, Kniesch K, Schlegelberger B, Schmidt RE, Witte T. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB × NZW)F1 mouse. Arthritis Rheum. 2005;52:3629–3638. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 49.Kearney JF, Bartels J, Hamilton AM, Lehuen A, Solvason N, Vakil M. Development and function of the early B cell repertoire. Int Rev Immunol. 1992;8:247–257. doi: 10.3109/08830189209055577. [DOI] [PubMed] [Google Scholar]
- 50.Radic MZ, Mascelli MA, Erikson J, Shan H, Shlomchik M, Weigert M. Structural patterns in anti-DNA antibodies from MRL/lpr mice. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 2):933–946. doi: 10.1101/sqb.1989.054.01.108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.