

Abstract
In the biological sciences the use of core-shell quantum dots (QDs) has gained wide usage, but analytical challenges still exist for characterizing the QD structure. The application of energy-dispersive x-ray spectroscopy and x-ray photoelectron spectroscopy (XPS) to bulk materials is relatively straightforward, however, for meaningful applications of surface science techniques to multilayer nanoparticles requires novel modifications and analysis methods. To experimentally characterize the elemental composition and distribution in CdSe/CdS/ZnS QDs, we first develop a XPS signal subtraction technique capable of separating the overlapped selenium 3s (core) and sulfur 2s (shell) peaks (both peaks have binding energies near 230eV) with higher precision than is typically reported in the nanoparticle literature. This method is valid for any nanoparticle containing selenium and sulfur. Then we apply a correction formula to the XPS data and determine that the 2 nm stoichiometric CdSe core is surrounded by 2 CdS layers and a stoichimetric ZnS monolayer. These findings and the multi-approach methodology represent a significant advancement in the detailed surface science study of multi-layer nanoparticles. In agreement with recent surprising findings, the time-of-flight secondary mass spectrometry measurements suggest that the surface sites of the QDs used in this study are primarily covered with a mixture of octadecylphosphonic acid and trioctylphophine oxide.
Keywords: Quantum Dots, X-ray Photoelectron Spectroscopy
Introduction
The synthetic history of semiconductor quantum dots (QDs) has been intimately tied to the passivation of the photoluminescent core and confinement of photo-generated excitons by higher bandgap shell overlayers (such as the ZnS shell1). QD core-shell structures with increased resistance to photo-oxidation and higher quantum yields (QY) have been used in numerous applications over the last decade as bright luminescent labels for molecular diagnostics, ultrasensitive in vitro assays and tumor imaging2–4. The notable optical properties of QDs include size-tunable, near-Gaussian emission profiles, broad excitation spectra, resistance to photo-bleaching and high QYs5–9. Similar in size to biological molecules such as proteins, the small size (<10 nm in diameter5), high QY (80–90%)10 and narrow emission line-widths (20–30 nm) of CdSe/ZnS core-shell structures have enabled their broad application in the biomedical sciences. The CdSe core is responsible for their fluorescent properties and the surrounding ZnS shell with a higher bandgap energy serves to enhance photoluminescent properties by attenuating photo-oxidation and surface defect effects6.
Although the development of a ZnS shell represents a significant advance, the lattice mismatch between CdSe and ZnS (~12%) causes strain inside the nanocrystal that can be reduced by introducing a transition layer with an intermediate degree of lattice mismatch and bandgap strength. Transition layers of CdS10 have been successfully developed and synthetically scaled-up for this purpose via the successive ion layer adsorption and reduction (SILAR)10, 11 method to produce CdSe/CdS/ZnS QDs. Recently the SILAR synthesis route was modified to synthesize multishell QDs with the aim of producing a new class of non-blinking QDs 12.
The high level of interest in these important multilayer nanostructures calls for rigorous analytical characterization to gain better insight into the chemical composition and elemental distribution13, 14. Basic analyses of CdSe/CdS/ZnS with transmission electron microscopy (TEM) and X-ray diffraction have been performed in the literature to measure QD dimensions and to verify the existence of the different phases (i.e., CdSe, CdS and ZnS). However, chemical information about the elemental distribution has not been achieved yet15, 16. The recent development of multishell QDs12 makes it even more critical to accurately quantify their elemental distribution.
Within the last year, breakthrough discoveries have reversed two long-standing views regarding the passivating surface ligand on the QD surface and of the reaction mechanism responsible for QD nucleation17. The organometallic synthetic procedure leaves the QD surface passivated with a monolayer of hydrophobic surfactant ligands8. Despite the assumption that the QDs are mainly coated with trioctylphosphine oxide (TOPO)10, 11, 18–21, recent studies have elegantly demonstrated that key impurities in the precursor solutions are actually the major surface ligand for thoroughly purified samples22,23.
Considering the nanometer-scale dimensions of QDs, determining the elemental distribution represents a significant challenge that requires novel analytical approaches. In this study, we characterize the elemental composition and distribution of CdSe/CdS/ZnS QDs with energy-dispersive x-ray spectroscopy (EDAX), x-ray photoelectron spectroscopy (XPS) and time-of-flight secondary mass spectrometry (ToF-SIMS). The complementary approach provides an estimate of the radius of the stoichiometric CdSe core as well as the thicknesses of the CdS layer and ZnS shell. For multilayer nanomaterials accurate delineation of individual layers solely by TEM has been achieved, but this is an extremely challenging experiment due to lack of contrast at boundaries of different semiconductor layers since Cd and Zn are relatively close in atomic number15, 16. Our method only requires TEM to determine the overall shape and size of the QDs, a much more straightforward measurement. A barrier facing XPS analysis of QDs is the overlap between the selenium and sulfur signals11, 19. Thus, previous XPS experiments have not attempted to accurately quantify the selenium and sulfur concentrations. To overcome this limitation we have developed a simple subtraction method, which can be used to determine the XPS elemental composition of any samples that contain selenium and sulfur. To investigate the organic surfactant layer covering the QDs, an area with exciting recent findings, we employ time-of-flight secondary mass spectrometry (ToF-SIMS). The ToF-SIMS results agree with recent literature findings22, 23 suggesting that the QD surface sites are mainly occupied with a mixture of phosphorous containing ligands such as trioctylphophine oxide (TOPO) and phosphonic acids. Our rigorous complementary approach represents an advance in the analytical characterization of multi-layer nanoparticles, which, as mentioned above, have gained increased attention for their reduced- and non-“blinking” fluorescence.
Materials and Methods
Reagents
Chloroform, hexane and methanol were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Selenium powder was purchased from Alfa Aesar. Powder samples of CdSe/CdS/ZnS (622 nm emission) QDs synthesized by the SILAR method 10, 11 and purified using standard hexane/methanol extraction11, 23 were obtained as a gift from Ocean Nanotech (Springdale, AR).
Optical analysis
A UV-2450 spectrophotometer (Shimadzu, Columbia, MD) and a Fluoromax4 fluorometer (Horiba Jobin Yvon, Edison, NJ) were used to characterize the absorption and emission spectra of CdSe/CdS/ZnS.
Sample preparation for electron microscopy and surface analysis
For TEM, XPS and ToF-SIMS analyses CdSe/CdS/ZnS QDs were dissolved in hexane. For TEM analysis 10–20 μl was drop cast onto a TEM grid. For XPS and ToF-SIMS analysis 20 drops (10 μl each) were dropped onto a clean silicon wafer substrate and dried under ambient conditions. The samples were stored in a desiccator prior the surface analysis experiments. For XPS analysis selenium powder was attached onto a silicon wafer using carbon double side tape.
Transmission Electron Microscopy (TEM)
TEM and EDAX analyses were carried out at the University of Washington Center for Nanotechnology using a FEI Tecnai G2 F20 TWIN 200kV TEM equipped with an EDAX detector. A dilute solution of QDs in hexane or water was dropped onto a Formvar-coated copper grid and allowed to dry under ambient conditions. TEM images were obtained with Gatan Digital Micrograph software and EDAX elemental analysis was analyzed with FEI TEM Image & Analysis software. Spectral acquisition data was averaged over at least 5 spots and 2 samples.
X-ray Photoelectron spectroscopy (XPS)
XPS data were acquired with a Surface Science Instruments S-probe spectrometer. This instrument has a monochromatized Al Kα x-ray source, hemispherical analyzer, multichannel detector and low-energy electron flood gun for charge neutralization. The x-ray spot size used for these experiments was approximately 800 μm × 800 μm. Pressure in the analytical chamber during spectral acquisition was less than 5 × 10−9 Torr. Spectra for elemental composition determination were acquired at an analyzer pass energy of 150 eV. The high-resolution spectra were acquired at an analyzer pass energy of 50 eV. The take-off angle (the angle between the substrate normal and the axis of the analyzer lens) was 55° for all XPS experiments. This take-off angle is close to the ‘magic’ angle (45°) which, depending on the particle size, minimizes the effects of surface curvature and roughness24, 25. Three spots on two replicates were analyzed for each sample type. The ESCA Analysis A program was used to determine peak areas.
Time-of-flight Secondary Ion Mass Spectrometry (ToF-SIMS)
ToF-SIMS data were acquired on an IONTOF 5–100 instrument. The Bi3+ primary ion source was operated in the high current bunched mode (i.e., the high mass resolution mode) and scanned over a 100 μm × 100 μm area. Five positive secondary ion and five negative secondary spectra were acquired for each sample type over a mass range from m/z = 0 to 850. The primary ion dose for each spectrum was below the static SIMS limit of 1 × 1012 ions/cm2. A low-energy electron beam was used for charge compensation. Positive secondary ion spectra were mass calibrated using the CH3+, NH4+, C2H3+, C2H5+, C3H5+ and C5H9+ peaks and negative secondary ion spectra were mass calibrated using the CH−, O−, OH−, C2−, C2H− and CNO− peaks. Data analysis was performed using the IonSpec data reduction software. Peak intensities were normalized to the most intense peak in each spectrum (C2H5+ and C2H−).
Results and Discussion
Photoluminescence (PL) excitation and absorbance measurements as well as TEM are the most common techniques used to characterize QDs. Figure 1 shows the absorbance peak position and the PL spectra for the CdSe/CdS/ZnS QDs. The TEM image in Figure 1 show nearly spherical crystalline particles with a narrow size distribution and diameter of ~5 nm. The QDs elemental composition measured with EDAX (Table 1) exhibit Cd/Se and S/Zn atomic ratios of approximately 3 and 8, respectively. Since the x-rays detected in the EDAX experiments are not significantly attenuated as they are emitted from the QDs (i.e., their sampling depth is orders of magnitude larger than the QD particle size), the EDAX measured elemental composition represents the bulk composition of the QDs. If the QDs consisted only of a two component CdSe core/ZnS shell structure then the Cd/Se and S/Zn ratios determined by EDAX should be unity. Because of the presence of the CdS intermediate layer these ratios are both greater than unity.
Figure 1.
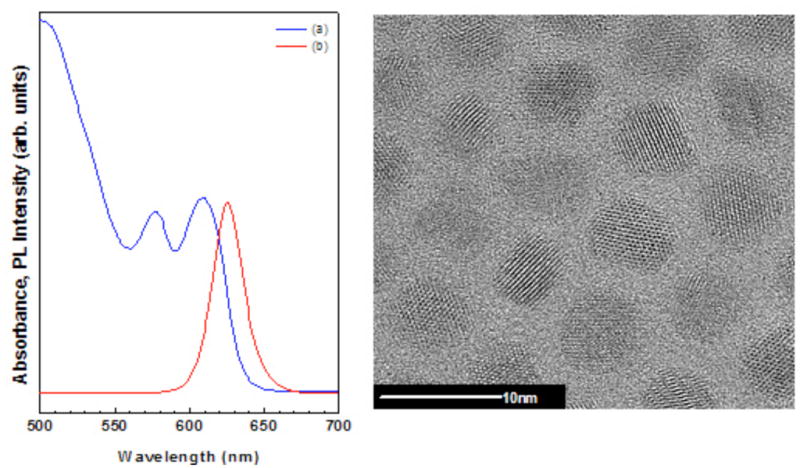
Characterization of QDs with standard methods. Absorbance (a, left panel) and photoluminescence (b, left panel) spectra of CdSe/CdS/ZnS QDs used in this study. High-resolution transmission electron microscopy images of CdSe/CdS/ZnS QDs show nearly spherical crystalline particles with a narrow size distribution and a diameter of ~5 nm (right panel).
Table 1.
EDAX determined bulk elemental composition of CdSe/CdS/ZnS QDs.
| Cd | Se | S | Zn | Cd/Se | S/Zn |
|---|---|---|---|---|---|
| 50.9±5.7 | 17.6±3.6 | 28.2±6.1 | 3.3±1.7 | 2.9 | 8.5 |
XPS is a state-of-the-art technique that is well known as a quantification tool for determining surface elemental compositions26. However, to date for nanoparticles, and QDs in particular, XPS has mainly been used to identify elements rather than determining their atomic composition. One challenge is the fact that the sulfur peaks (both 2s and 2p) from the QD shell overlap with the selenium peaks from the QD core11 (Figure 2i). For example, the S2s and the Se3s signals in Figure 2i cannot be distinguished with standard high-resolution XPS peak fitting due to uncertainty regarding the precise peak positions. Therefore, we have developed a new approach in which pure selenium was used as a reference material to measure the relative areas of the different selenium peaks (examples are shown in Figure 2ii a and b). These peak areas are related by the photoionization cross-section and their relative ratios are not affected by the chemical environment the Se atoms. Then, the ratios between the Se3d, which is the only selenium peak that does not overlap with peaks from other elements present in the QDs (Figure 2i), and the other Se peaks were calculated. This approach is also applicable to the P2p region that contains the P2p3/2 and P2p1/2 peaks as well as the Se(L2M4,5M4,5) Auger peak (Figure 2ii d). The ratios between the Se3d and Se(L2M4,5M4,5) or Se3s peaks determined from the reference spectra were Se3s/Se3d=0.62 and Se(L2M4,5M4,5)/Se3d=1.92. As stated above, these ratios are constant for all selenium containing samples. Also, since the kinetic energies of these photo- and Auger electrons are similar (ranging from ~1255 eV for Se3s and to ~1430 eV for Se3d), the sampling depth only varies by a few percent for these peaks. To calculate the QD elemental composition the areas of the Se3s photoemission peak and the Se(L2M4,5M4,5) Auger peak were determined by measuring the area of the Se3d peak and applying the known Se3s/Se3d and Se(L2M4,5M4,5)/Se3d peak area ratios. Then the estimated contributions of the Se peaks were subtracted from the total areas of Se3s/S2s or Se(L2M4,5M4,5)/P2p regions to determine the appropriate S2s or P2p peak areas (Figure 2ii c and d). Finally, the determined XPS measured surface atomic percentages for the CdSe/CdS/ZnS QDs in Table 2 were calculated by substituting the corrected intensities of S2s and P2p peaks into equation (1) which is used to determine XPS surface elemental composition26. In this equation, I is the peak area of element i, σ is the photoionization cross-section of peak i and K depends on an instrumental constant and the measurement parameters.
Figure 2.
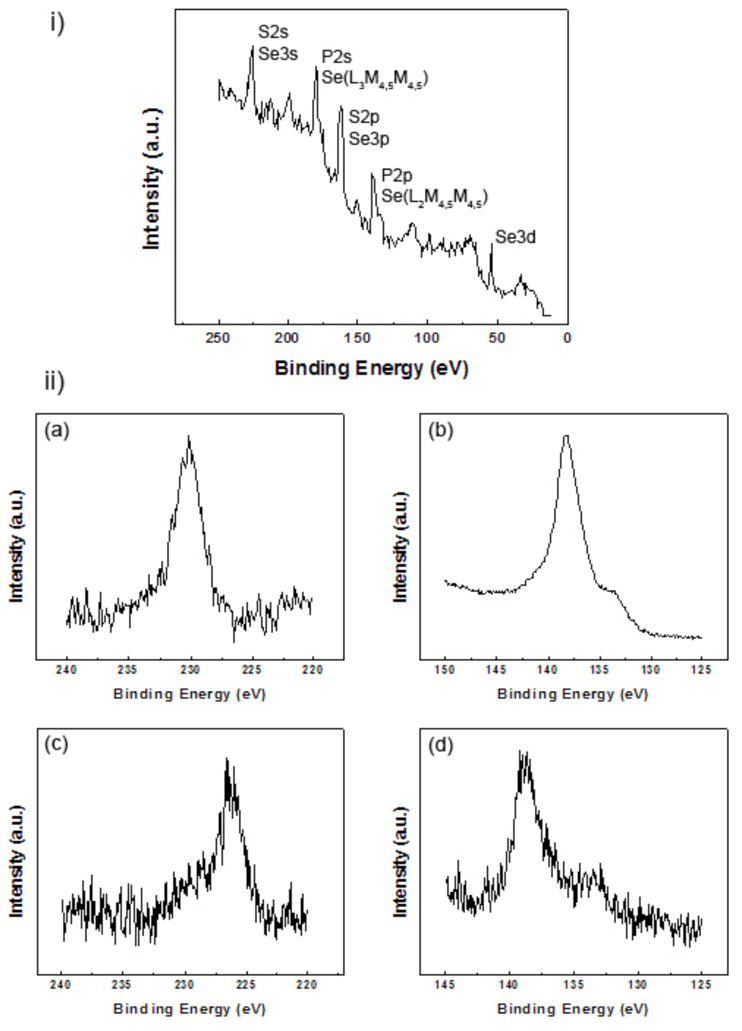
XPS analysis of QDs. (i) Survey spectrum of CdSe/CdS/ZnS QDs in the binding energy range between 0 and 250 eV showing the overlap between the selenium peaks with the sulfur and phosphorous peaks. (ii) High resolution spectra of Se3s (a) and Se(L2M4,5M4,5) (b) measured from pure Se powder and S2s (c) and P2p (d) regions measured from CdSe/CdS/ZnS QDs.
Table 2.
Initial XPS determined surface elemental compositions and corrected XPS atomic percentages of CdSe/CdS/ZnS indicate the presence of an intermediate CdS layer between the stoichiometric CdSe cores and the ZnS shell.
| at% | ||||
|---|---|---|---|---|
| Initial XPS surface atomic percentages using the peak area subtraction method | Corrected XPS atomic percentages Based on equation (3) | |||
| Element | All elements | Core elements | Homogenous CdSe core and ZnS shell | Three layer model: CdSe/CdS/ZnS |
| C 1s | 83.4±2.1 | |||
| O 1s | 5.7±0.9 | |||
| Cd 3d | 5.6±0.7 | 72.0 | 48.0 | 54.7 |
| Se 3d | 0.9±0.1 | 11.6 | 7.1 | 13.5 |
| P 2p | 0.6±0.1 | |||
| Zn 2p3 | 0.08±0.02 | 1.0 | 2.2 | 4.4 |
| S 2s | 1.2±0.4 | 15.5 | 42.8 | 27.4 |
| F 1s | 2.5±1.6 | |||
| Cd/Se | 6.2 | 6.2 | 6.8 | 4.1 |
| S/Zn | 15.5 | 15.5 | 19.5 | 6.2 |
| (1) |
In agreement with the EDAX measurements, XPS detected Cd, Se, Zn and S signals from the CdSe/CdS/ZnS QDs. In agreement with the EDAX data, the XPS Cd/Se and the S/Zn atomic ratios are greater than 1 (6 and 15, respectively). However, the XPS surface atomic ratios were significantly larger than the EDAX bulk atomic ratios because of the core-shell structure of the QDs (i.e., the surface composition is significantly different from the bulk composition). Since the XPS sampling depth is similar to the QD particle size, the XPS signals that originate below the outer surface of the QDs are attenuated (the XPS intensities are attenuated exponentially with distance below the outer surface). XPS also detected phosphorous, which is a unique marker for surface organic ligands such as TOPO and phosphonic acids23, 27–32. However, the XPS C/P and O/P atomic ratios are higher than the expected stoichiometric ratios for these compounds. The expected stoichiometric ratios are 24 C/P for TOPO and 3 O/P for alkyl phosphonic acids. This observed difference could to be due to the presence of organic residues from the solvents or adsorption of adventitious carbon33. Organic residues might also be the source of the fluorine that was detected. Recent work has shown that the phosphonic acid impurities contained in technical grade TOPO are retained on the QD surface22, 23.
An additional challenge with analysis of QDs with XPS is related to their size since it is comparable to the XPS photoelectron inelastic mean free path and therefore equation (1) is not valid. Equation 1 assumes the sample has a homogenous elemental composition (i.e., the surface composition is the same as the bulk composition). This is not the case for core-shell QDs, so the measured XPS surface elemental composition must be corrected by inserting the result from integral (2) into equation (3) to obtain the actual bulk elemental composition25:
| (2) |
| (3) |
The numeric solution to equation (2) includes the distribution of each element within the analyzed specimen. Thus, different models can be tested by applying equation (3) and comparing the corrected XPS elemental composition to the EDAX determined bulk elemental composition. When the corrected XPS elemental composition matches the EDAX determined bulk composition, that indicates the proposed core-shell model of the QD is consistent with the experimental measured compositions. As an initial model, the QDs were assumed to have a simple core-shell structure containing an CdSe inner core with a radius of ~2.4 nm surrounded by a shell of a ZnS monolayer. Applying equation (3) to the one shell model with a homogenous CdSe core and homogenous ZnS shell gave corrected Cd/Se and S/Zn XPS atomic ratios that were similar to the initial XPS composition and significantly higher than the EDAX values (see Table 2). Thus, the simple CdSe/ZnS core-shell model is not consistent with the measured elemental compositions. However, if a three-layer model is used then the corrected XPS Cd/Se and S/Zn atomic ratios are noticeably closer to the EDAX values. The three-layer model used for these calculations assumed a homogenous CdSe core with a 2 nm radius surrounded by 2 layers of CdS and covered by a shell with one monolayer of stoichiometric ZnS. Similar models with a core radiuses of 1.9–2.2 nm, 2–3 layers of CdS and 1–2 ZnS monolayer were also examined and also gave reasonable, but slightly worse, agreement between the corrected XPS elemental composition and the EDAX bulk elemental composition. The size of each QD (~5 nm diameter) is significantly smaller than the XPS analysis area (~800 × 800 μm2), so the XPS data is averaged over large numbers of QDs. Thus, it is reasonable to assume that the thicknesses of the CdSe core as well as the CdS intermediate layer and the ZnS shell vary within these ranges. However, over this range of values the three-layer model proposed above shows the best agreement with the EDAX data. A schematic of this three-layer model is shown in Figure 3. This three-layer QD structure is consistent with the SILAR synthesis route employed by Ocean Nanotech10, 11, 34. As stated above, the difference in sampling depths of the two techniques requires correction of the measured XPS surface elemental composition to obtain consistency with the EDAX measured bulk elemental composition. The EDAX sampling region is much larger than the QD diameter, whereas the XPS sampling depth is similar to the QD diameter. Since the outgoing photoelectrons in XPS interact more strongly with matter than the outgoing x-rays in EDAX, the significantly smaller XPS sampling depth is more sensitive to changes in the QD surface composition.
Figure 3.
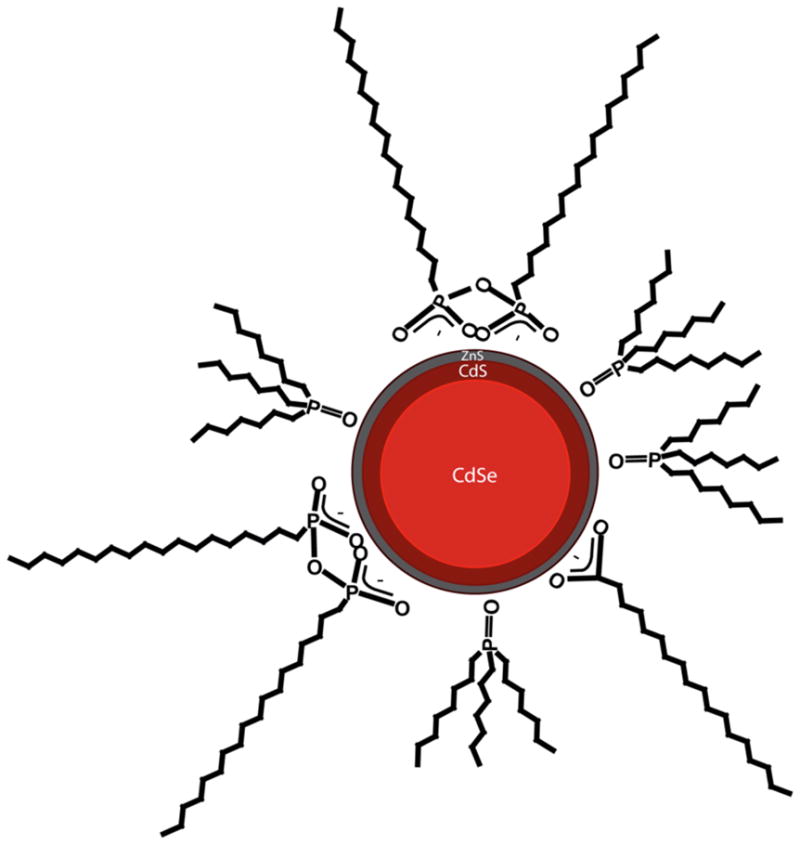
A schematic drawing of the three layer model based on the measured XPS surface and EDAX bulk elemental compositions: a homogenous CdSe core 4 nm in diameter surrounded by 2 layers of CdS and a shell that consists one monolayer of stoichiometric ZnS. This three-layer structure is primarily covered with a mixture of ODPA and TOPO and trace amounts of stearate.
ToF-SIMS was used to gain further understanding regarding the CdSe/CdS/ZnS structure. The main fragments detected from the QDs by ToF-SIMS contain Cdx+, Zn+, ZnCdx+, ZnSCdx+, CdSe+, Sex−, Sx−, SeS−, CdSx− and CdSe− (see Tables 3 and 4). These secondary ions are consistent with the elemental compositions observed by both EDAX and XPS. In particular, the relatively strong CdSx− fragments are consistent with the presence of the CdS intermediate layer (Table 3). ToF-SIMS also detected several phosphorous containig fragments. CxHyP− and CxHyPO− fragments are consistant with the presence of TOPO ligands on the QD surface. Phosphorous containing fragments with more than one oxygen (PO2 and PO3) were also detected and are associated with the presence of phosphonate ligands on the QDs surface23, 28, 29. Furthermore, fragments that can be assigned to octadecylphosphonic acid (ODPA) were found in the secondary ion spectra. In contrast, fragments that contain the entire TOPO molecule (i.e. (C8H17)3PO) or its derivatives were not detected. This difference could be the result of a different fragmentation pathway for TOPO and ODPA molecules, but more likely it is because the surface ODPA concentration on the QDs is significantly higher than the surface TOPO concentration. Both ODPA and TOPO molecules are present in solution during QDs synthesis34. Morris-Cohen et. al. showed that the existance of phosphonate ligands on QDs surfaces is responsible for preserving the QD QY23. As our QDs exhibit bright fluorescence and a high QY (Figure 1) this further supports the presence of phosphonic acid molecules on the QD surface. An additional impurity that arises from the synthetic reaction mixture is stearate23. The presence of C18HxO2− fragments in the secondary ion mass spectra was consistent with the presence of sterate. A schematic of the mixture of the surrounding mixture of ligands is shown in Figure 3.
Table 3.
ToF-SIMS negative secondary ion fragments from CdSe/CdS/ZnS QDs. The observed fragments are consistent with the QD elemental compositions. The Cd and Se fragments are associated with the CdSe QD core; the CdSx fragments are associated with the CdS intermediate layer; the phosphrous containing fragments are associated with ODPA and TOPO ligands.
| Negative secondary ion mass | Molecular Species (−1) | Assignment | Relative Intensity % | |
|---|---|---|---|---|
| 25.007 | C2H | Most intense peak | 100.00 | |
| 31.972 | S | CdSe/CdS/ZnS | 11.8 | |
| 63.947 | S2 | 4.6 | ||
| 79.923 | Se | 3.4 | ||
| 145.884 | CdS | 4.5 | ||
| 159.842 | Se2 | 0.6 | ||
| 177.863 | CdS2 | 3.8 | ||
| 191.814 | CdSe | 0.7 | ||
| 209.846 | CdS3 | 1.6 | ||
| 45.997 | CH3P | Organic Ligands | TOPO/ODPA | 8.5 |
| 61.993 | CH3PO | 21.4 | ||
| 74.995 | C2H4PO | 2.1 | ||
| 237.087 | C16H14P | 1.7 | ||
| 255.267 | C16H32P | 3.1 | ||
| 62.964 | PO2 | ODPA | 14.6 | |
| 78.972 | PO3 | 42.9 | ||
| 79.963 | PO3H | 17.4 | ||
| 80.973 | PH2O3 | 4.8 | ||
| 90.994 | C2H4PO2 | 0.4 | ||
| 93.980 | CH3PO3 | 0.6 | ||
| 106.983 | C2H4PO3 | 0.5 | ||
| 333.249 | C18H37PO3H (M−H) | 0.9 | ||
| 334.265 | C18H37PO3H2 | 0.6 | ||
| 348.2623 | C18H37PO4 (M−2H+O) | 3.7 | ||
| 349.260 | C18H37PO4 H (M−H+O) | 1.1 | ||
| 281.288 | C18H33O2 (M−4H) | Stearate23 | 7.0 | |
| 283.301 | C18H35O2 (M−2H) | 4.2 | ||
| 284.270 | C18H36O2 (M−H) | 0.9 | ||
| 285.265 | C18H37O2 | 0.7 | ||
Table 4.
ToF-SIMS positive secondary ion fragments from CdSe/CdS/ZnS QDs. The observed fragments are consistent with the QD elemental compositions. The Cd fragments are associated with the CdSe QD core and CdS intermediate layer; the zinc containing fragments are associated with the ZnS shell.
| Positive secondary ion mass | Molecular Species (+1) | Assignment | Relative Intensity % |
|---|---|---|---|
| 29.039 | C2H5 | Most intense peak | 100.00 |
| 63.927 | Zn | CdSe/CdS/ZnS | 0.2 |
| 113.904 | Cd | 11.7 | |
| 177.84 | ZnCd | 0.2 | |
| 209.812 | ZnSCd | 0.1 | |
| 225.794 | Cd2 | 2.0 | |
| 289.741 | ZnCd2 | 0.3 | |
| 321.711 | ZnSCd2 | 0.2 | |
| 403.633 | ZnCd3 | 0.2 | |
| 435.607 | ZnSCd3 | 0.2 | |
| 191.814 | CdSe | 0.2 | |
| 515.510 | ZnCd4 | 0.5 |
Conclusions
The complementary, multi-technique results from this study provide a detailed understanding of the elemental composition of the CdSe/CdS/ZnS QDs. A model that is consistent with the measured XPS surface elemental compositions is a three-layered structure with a ~2 nm stoichiometric CdSe core covered with 2 CdS intermediate layers and a monolayer ZnS outer shell. ToF-SIMS probes the composition of the outermost organic capping layer and provides evidence, consistent with recent findings, that the QD surfaces are largely covered with phosphonic acids. This work represents a significant advance in the application of XPS as a quantitative tool to better elucidate the elemental composition and distribution in multilayer nanoparticles. The methodology developed in this study for 3-layers QDs can be extended to QDs will higher number of shells.
Acknowledgments
This research was funded by the National ESCA and Surface Analysis Center for Biomedical Problems (NIH grant EB-002027) and NIH grant GM-074511. S.R.D. acknowledges the NSF for generous fellowship support. The authors would like to thank Pavel Zrazhevskiy for help with QD preparation and purification, as well as OceanNanotech for their generous gift of the QD samples. Part of this work was conducted at the University of Washington NanoTech User Facility, a member of the NSF National Nanotechnology Infrastructure Network (NNIN).
References
- 1.Hines MA, Guyot-Sionnest P. Journal of physical chemistry. 1996;100:468–471. [Google Scholar]
- 2.Alivisatos P. Nature Biotechnology. 2004;22:47–52. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 3.Gao X, Cui Y, RML, Chung LWK, Nie S. Nature Biotechnology. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 4.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, AMW, Gambhir SS, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith AM, Nie SM. Analyst. 2004;129:672–677. doi: 10.1039/b404498n. [DOI] [PubMed] [Google Scholar]
- 6.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nature Materials. 2005;4:435–466. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 7.Parak WJ, Garison D, Pellegrino T, Zanchet D. Nanotechnology. 2003;14:R15–R27. [Google Scholar]
- 8.Smith AM, Dave S, Nie S, True L, Gao X. Expert Review of Molecular Diagnostics. 2006;6:231–244. doi: 10.1586/14737159.6.2.231. [DOI] [PubMed] [Google Scholar]
- 9.Zrazhevskiy P, Sena M, Gao X. Chemical Society Reviews. 2010 doi: 10.1039/B915139G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talapin DV, Mekis I, Gotzinger S, Kornowski A, Benson O, Weller H. Journal of Physical Chemistry B. 2004;108:18826–18831. [Google Scholar]
- 11.Li JJ, Wang A, Guo W, Keay JC, Mishima TD, Johnson MB, Peng X. Journal of American Chemical Society. 2003;125:12567–12575. doi: 10.1021/ja0363563. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Vela J, Htoon H, Casson JL, Werder DJ, Bussian DA, Klimov VI, Hollingsworth JA. Journal of American Chemical Society. 2008;130:5026–5027. doi: 10.1021/ja711379k. [DOI] [PubMed] [Google Scholar]
- 13.Baer DR, Gaspar DJ, Nachimuthu P, Techane SD, Castner DG. Analytical and Bioanalytical Chemistry. 2010;396:983–1002. doi: 10.1007/s00216-009-3360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grainger DW, Castner DG. Advanced Materials. 2008;20:867–877. [Google Scholar]
- 15.Wang X, Ren X, Kahen K, Hahn MA, Rajeswaran M, Maccagnano-Zacher S, Silox J, Cragg GE, Efros AL, Krauss TD. Nature. 2009;459:686–689. doi: 10.1038/nature08072. [DOI] [PubMed] [Google Scholar]
- 16.Yu Z, Guo L, Krauss T, Silox J. Nano letters. 2005;5:565–570. doi: 10.1021/nl048245n. [DOI] [PubMed] [Google Scholar]
- 17.Evans CM, Evans ME, Krauss TD. Journal of American Chemical Society. 2010;132:10973–10975. doi: 10.1021/ja103805s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alivatos AP. Journal of Physical Chemistry. 1996;100:13226–13239. [Google Scholar]
- 19.Peng X, Schlamp MC, Kadavanich AV, Alivatos AP. Journal of American Chemical Society. 1997;119:7019–7029. [Google Scholar]
- 20.Yu WW, Qu L, Guo W, Peng X. Chemistry of Materials. 2003;15:2854–2860. [Google Scholar]
- 21.Dabbousi BO, Rodriguez-Viejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. Journal of Physical Chemistry. 1997;101:9463–9475. [Google Scholar]
- 22.Kopping JT, Patten TE. Journal of American Chemical Society. 2008;130:5689–5698. doi: 10.1021/ja077414d. [DOI] [PubMed] [Google Scholar]
- 23.Morris-Cohen AJ, Donakowski MD, Knowles KE, Weiss EA. Journal of Physical Chemistry C. 2010;114:897–906. doi: 10.1021/jp102101f. [DOI] [PubMed] [Google Scholar]
- 24.Gunter PLJ, Gijzeman OLJ, Niemantsverdriet JW. Applied Surfece Science. 1997;115:342–346. [Google Scholar]
- 25.Frydman A, Castner DG, Schmal M, Campbell CT. Journal of Catalysis. 1995;157:133–144. [Google Scholar]
- 26.Ratner BD, Castner DG. Electron Spectroscopy for Chemical Analysis. In: Vickerman JC, Gilmore IS, editors. Surface Analysis The Principle Techniques. 2. Wiley; New York: 2009. p. 47. [Google Scholar]
- 27.Adden N, Gamble LJ, Castner DG, Hoffman A, Gross G, Menzel H. Langmuir. 2006;22:8197–8204. doi: 10.1021/la060754c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubey M, Weidner T, Gamble LJ, Castner DG. Langmuir. 2010;26:14747–14754. doi: 10.1021/la1021438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Textor M, Ruiz L, Hofer R, Rossi A, Feldman K, Hahner G, Spencer ND. Langmuir. 2000;16:3257–3271. [Google Scholar]
- 30.Tosatti S, Michel R, Textor M, Spencer ND. Langmuir. 2002;18:3537–3548. [Google Scholar]
- 31.Zorn G, Adadi R, Brener R, Yakovlev VA, Gotman I, Gutmanas EY, Sukenik CN. Chemistry of Materials. 2008;20:5368–5374. [Google Scholar]
- 32.Zorn G, Gotman I, Gutmanas EY, Adadi R, Salitra G, Sukenik CN. Chemistry of Materials. 2005;17:4218–1226. [Google Scholar]
- 33.Min H, Kim Y, Yu H, Moon DW, Lim SJ, Yoon HJ, Lee TG, Shin SK. Chemistry: A European Journal. 2008;14:8461–8464. doi: 10.1002/chem.200801301. [DOI] [PubMed] [Google Scholar]
- 34.Ocean Nanotech protocol - personal communication