

Abstract
Activation of innate immunity (natural killer cell/interferon-γ: NK cell/IFN-γ) has been shown to play an important role in anti-viral and anti-tumor defenses as well as anti-fibrogenesis. However, little is known about the regulation of innate immunity during chronic liver injury. Here, we compared the functions of NK cells in early and advanced liver fibrosis induced by a 2-week or a 10 week-carbon tetrachloride (CCl4) challenge, respectively. Injection of poly I:C or IFN-γ induced NK cell activation and NK cell killing of hepatic stellate cells (HSCs) in the 2-week CCl4 model. Such activation was diminished in the 10-week CCl4 model. Consistent with these findings, the inhibitory effect of poly I:C and IFN-γ on liver fibrosis was markedly reduced in the 10-week vs. the 2-week CCl4 model. In vitro co-culture experiments demonstrated that 4-day cultured (early-activated) HSCs induce NK cell activation via an NKG2D-retinoic acid-induced early gene 1 (RAE1)-dependent mechanism. Such activation was reduced when co-cultured with 8-day cultured (intermediately-activated) HSCs due to the production of transforming growth factor-β (TGF-β) by HSCs. Moreover, early-activated HSCs were sensitive, while intermediately-activated HSCs were resistant to IFN-γ mediated inhibition of cell proliferation, likely due to elevated expression of suppressor of cytokine signaling 1 (SOCS1). Disruption of the SOCS1 gene restored the IFN-γ inhibition of cell proliferation in intermediately-activated HSCs. Production of retinol metabolites by HSCs contributed to SOCS1 induction and subsequently inhibited IFN-γ signaling and functioning, while production of TGF-β by HSCs inhibited NK cell function and cytotoxicity against HSCs.
Conclusion
The anti-fibrogenic effects of NK cell/IFN-γ are suppressed during advanced liver injury, which is likely due to the increased production of TGF-β and expression of SOCS1 in intermediately-activated HSCs.
Keywords: hepatic stellate cell, STAT1, SOCS1, TGF-β, retinol
Liver lymphocytes are enriched in natural killer (NK) cells, which play a crucial role in innate immune responses against tumors and pathogenes.1, 2 Recent studies have demonstrated that NK cells also play an important role in suppressing liver fibrosis via killing activated hepatic stellate cells (HSCs) and producing interferon-γ (IFN-γ) in mice and humans.3-5 IFN-γ not only directly induces HSC apoptosis and cell cycle arrest 6 but also stimulates the cytotoxicity of NK cells against activated HSCs via increasing the number of NK cells and upregulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) on NK cells.4, 7 Recently, we have demonstrated that retinol metabolites plays an important role in enhancing the sensitivity of activated HSCs to NK cell killing.8 HSCs store large amounts of vitamin A (retinol) in their cytoplasm. Upon activation, they lose their retinol via either release or metabolism of retinol into retinoic acid, which has been implicated in the pathogenesis of liver fibrogenesis.8-10 Retinoic acid is elevated in the HSCs during activation and upregulates the expression of a variety of genes, including retinoic acid-induced early gene 1 (RAE1), an NK cell activating ligand. RAE1 then activates NK cells and increases the susceptibility of HSCs to NK cell killing.4, 8
Emerging evidence suggests that liver fibrosis can be reversed and prevented either via inhibiting HSC activation and proliferation or inducing HSC apoptosis in various immune cell and cytokines-dependent manners.2-7, 9, 10 Among these mechanisms, NK cells/IFN-γ have been suggested to be one of the most potent negative regulators of liver fibrosis. In vivo activation of NK cells by poly I:C or treatment with IFN-γ ameliorates liver fibrosis induced by carbon tetrachloride (CCl4) or dimethylnitrosamine in rodents.4, 6, 11, 12 In addition, clinical studies have shown that IFN-γ treatment attenuates liver fibrosis in some patients with chronic viral hepatitis B (HBV) and HCV infection.13, 14 However, other clinical trials reported that IFN-γ therapy had no beneficial effects in attenuating the severity of advanced fibrosis and cirrhosis in patients with chronic HCV infection.15 The reasons for these controversial reports are not clear. One of possible explanation may be due to the selection of patients with different degrees of liver diseases. In the present study, we compared the antifibrotic efficacy of NK cells/IFN-γ on early and advanced liver fibrosis in vivo and the effects of NK cells/IFN-γ on the different stages of activated HSCs in vitro.
Materials and Methods
Animals
C57BL/6J mice and IFN-γ−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). IFN-γ−/−SOCS1−/− mice were kindly provided by Dr. James Ihle, St. Jude Children’s Research Hospital (Memphis, TN). All mice used in the present study were housed in a specific pathogen–free facility and were cared for in accordance with National Institutes of Health (NIH) guidelines. All animal experiments were approved by the Institutional Animal Care and Use Committees of the NIAAA, NIH.
Liver fibrosis induction and co-treatment with CCl4 plus poly I:C or IFN-γ
Hepatic fibrosis in mice was induced experimentally by intraperitoneal injection (i.p) of CCl4 as previously described.6 Mice were injected with CCl4 for 2 weeks or 8 weeks and co-treated with or without poly I:C (2μg/g, 3 times a week, i.p) or IFN-γ (2000 IU/g, 7 times per week, SC) for an additional 2 or 4 weeks. Control mice received CCl4 plus saline. For acute poly I:C treatment, mice were injected (i.p) with poly I:C once.
Statistical analysis
Data are expressed as means ± SEM. To compare values obtained from two or more groups, the student t-test or one-way analysis of variance was performed. A value of P < 0.01 or 0.05 was considered significant.
Other methods
All other methods are described in the supporting documents.
Results
Poly I:C-mediated activation of NK cells is diminished in advanced liver fibrosis compared with early stage of liver fibrosis
To investigate functions of NK cells on early stage (2-week CCl4) or advanced liver fibrosis (10-week CCl4), mice were injected with CCl4 for 2 or 10 weeks without or with co-treatment of poly I:C (for the final 2 weeks). In 2-week CCl4 group, serum IFN-γ levels were significantly increased after poly I:C treatment, but such elevation was not observed in 10-week CCl4 (Fig. 1A). In addition, basal levels of serum IFN-γ from the 10-week CCl4 mice without poly I:C treatment were lower than those from 2-week CCl4 mice. Flow cytometry analyses showed the basal levels of liver NK cell population and NKG2D expression on these cells were significantly lower in 10-week CCl4 mice compared with those in 2-week CCl4 mice (Fig. 1B). Interestingly, poly I:C treatment resulted in about 2-fold induction of liver NK cell population and NKG2D expression in both 2-week and 10-week CCl4 mice (Fig. 1B). Furthermore, poly I:C induction of several NK cell-associated genes in the livers of 10-week CCl4 mice was diminished compared to those of 2-week CCl4 mice (Fig. 1C).
Fig. 1.
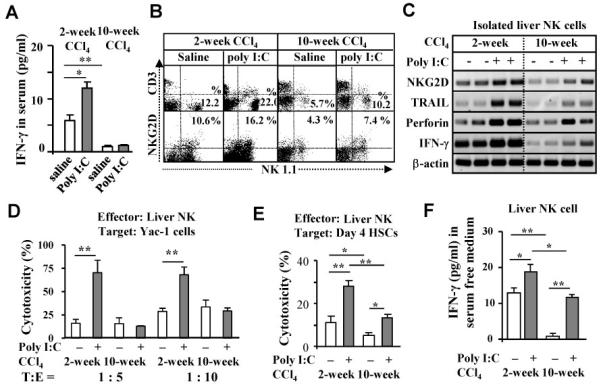
Poly I:C activation of liver NK cells is suppressed in 10-week CCl4 model compared with 2-week CCl4 model. Two-week or 10-week CCl4-treated mice were co-injected with poly I:C for the last 2 weeks. (A) Serum levels of IFN-γ. (B-C) Liver mononuclear cells (MNCs) and NK cells were isolated and subject to FACS analyses (B) and RT-PCR analyses (C). (D-E) Liver NK cells were used as effector cells for cytotoxicity assay against Yac-1 cells (D) or cultured day 4 HSCs (D4 HSCs) (E). (F) Liver NK cells from 2-week or 10-week CCl4-treated mouse with or without poly I:C were cultured in serum free medium for 16 h for measurement of IFN-γ levels. *P<0.05, **P<0.01.
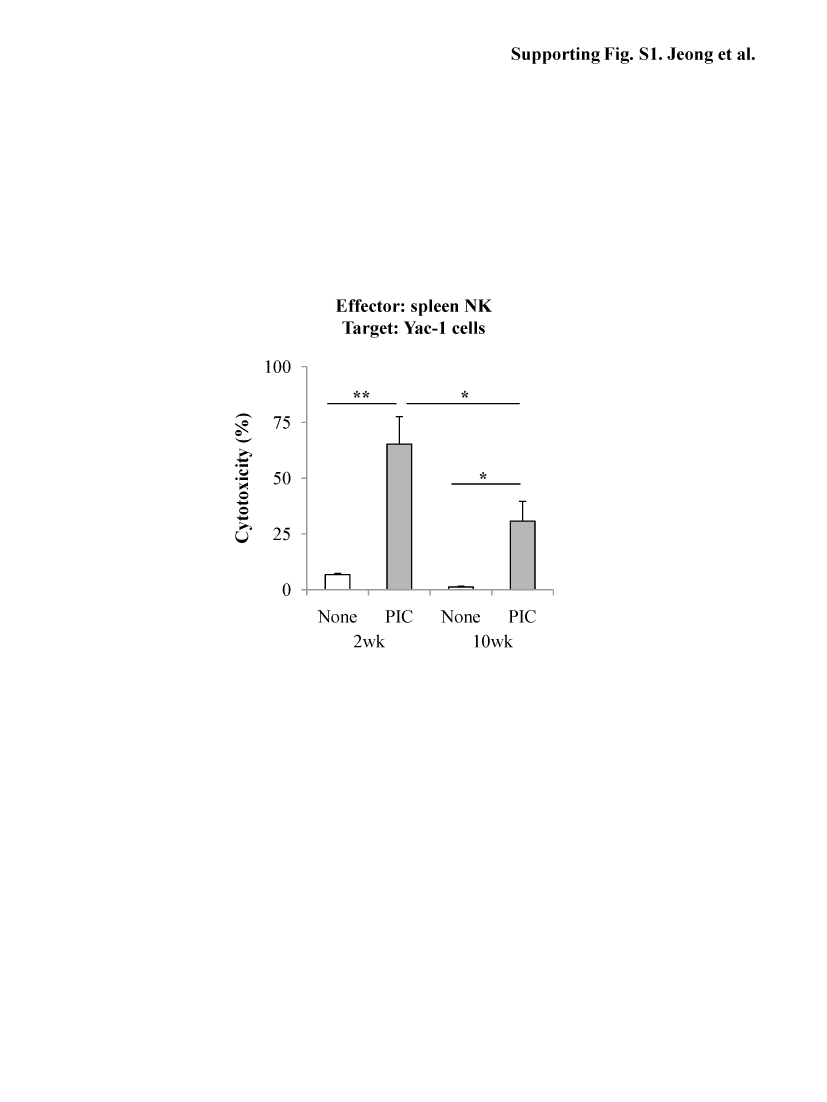
Cytotoxicity assay demonstrated that poly I:C treatment significantly increased cytotoxicity of NK cells isolated from 2-week CCl4 mice against Yac-1 cells (NK sensitive target cells) but not those of NK cells isolated from 10-week CCl4 mice (Fig. 1D). Cytotoxicity of spleen NK cells against Yac-1 cells was also diminished in 10-week CCl4 vs 2-week CCl4 mice (Supporting Fig. S1). In addition, NK cells isolated from poly I:C-treated or non-treated mice of 10-week CCl4 mice showed significant reductions in killing of early-activated HSCs (Fig. 1E) and IFN-γ production (Fig. 1F) compared to those of 2-week CCl4 mice.
IFN-γ-mediated activation of NK cells are diminished in advanced liver fibrosis compared to early stage of liver fibrosis
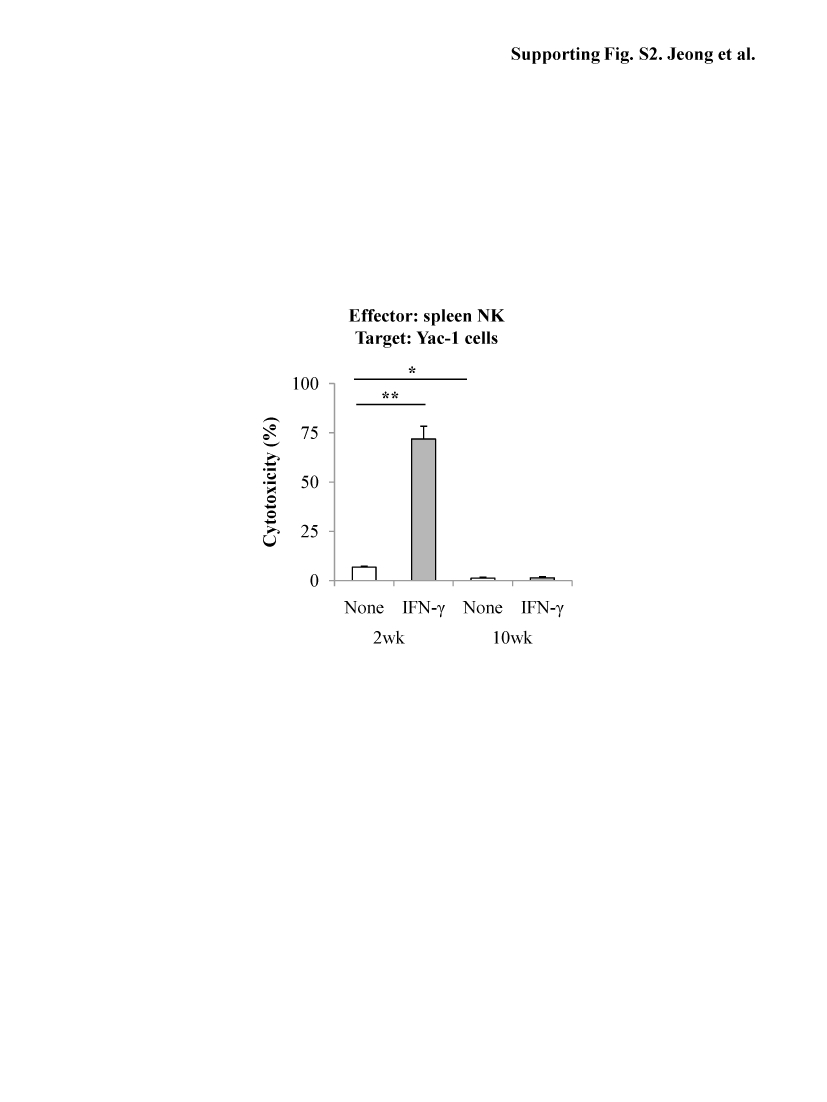
Previously, it has been demonstrated that IFN-γ enhances the cytotoxicity of NK cells against activated HSCs via increasing the number of NK cells and production of IFN-γ.4, 6, 7 Fig. 2A shows that the basal levels of liver NK cells and NKG2D expression were lower in 10-week CCl4 mice than those in 2-week CCl4 mice although IFN-γ treatment resulted in a similar fold induction of these parameters in both groups. RT-PCR analyses showed IFN-γ treatment markedly upregulated the expression of NKG2D, TRAIL, perforin and IFN-γ genes in liver NK cells from 2-week CCl4 mice but not from 10-week CCl4 mice (Fig. 2B). Cytotoxicity assay against Yac-1 cells showed IFN-γ treatment significantly increased cytotoxicity of NK cells isolated from 2-week CCl4 mice but not those from 10-week CCl4 mice (Fig. 2C). In the case of spleen NK cells, cytotoxicity against Yac-1 cells was also diminished in 10-week CCl4 mice compared to 2-week CCl4 mice (Supporting Fig. S2). Additionally, NK cells isolated from IFN-γ-treated or non-treated mice of 10-week CCl4 mice had lower killing activity against activated HSCs compared to those of 2-week CCl4 mice (Fig. 2D).
Fig. 2.
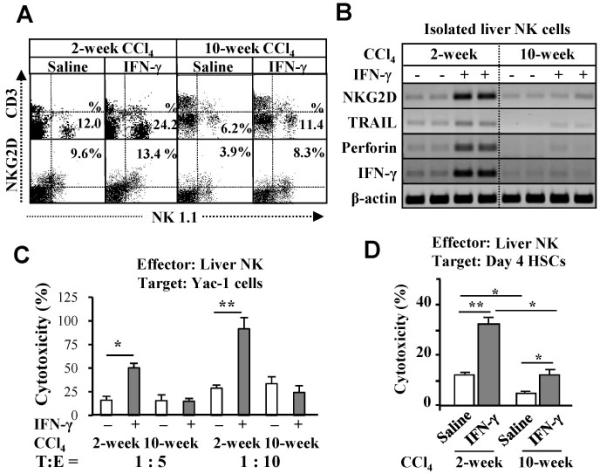
IFN-γ activation of liver NK cells is diminished in 10-week CCl4 model compared with 2-week CCl4 model. Two-week or 10-week CCl4-treated mice were injected with IFN-γ for the last 2 weeks. MNCs and NK cells were isolated and subjected to FACS analyses (A) and RT-PCR analyses (B). Liver NK cells were used as effector cells for cytotoxicity assay against Yac-1 cells (C) and D4 HSCs (D). *P<0.05, **P<0.01.
Poly I:C or IFN-γ treatment does not ameliorate advanced liver fibrosis
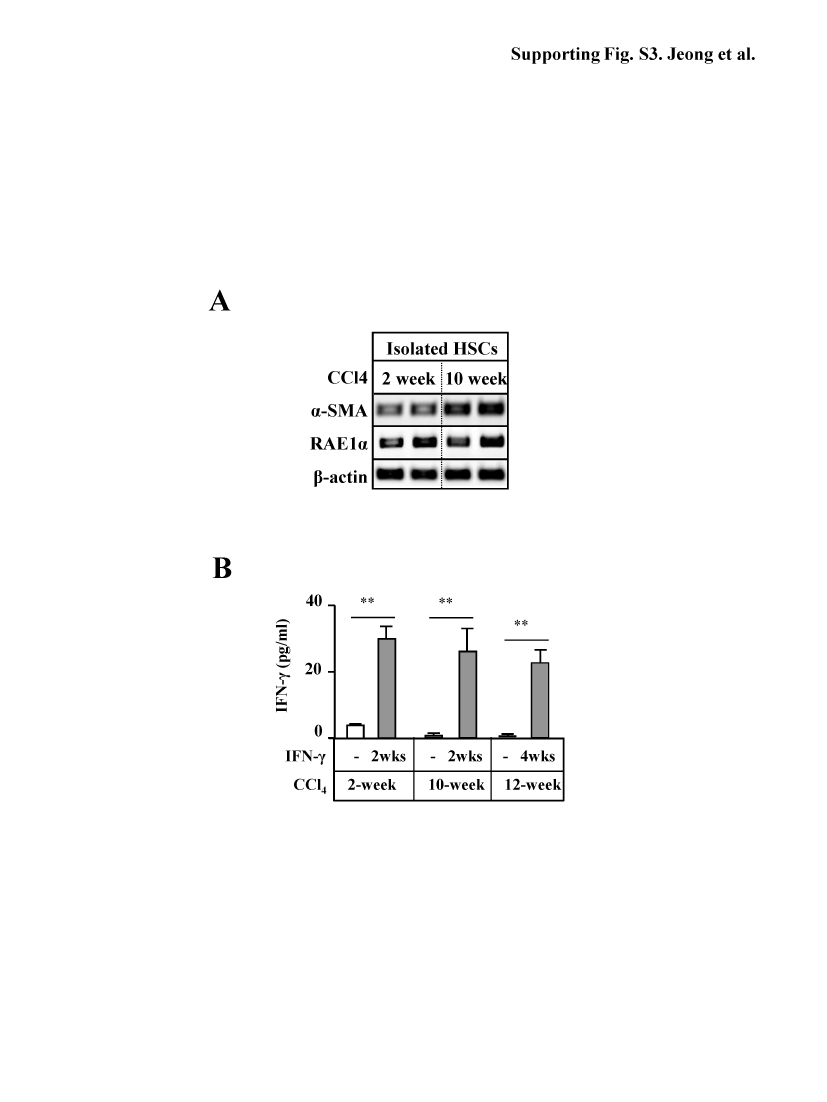
We and others have previously shown that poly I:C or IFN-γ treatment ameliorates early liver fibrosis in mice.4, 6, 11, 12 Here we show that treatment with poly I:C inhibited liver fibrosis induced by a 2-week CCl4 treatment but had no inhibitory effects on advanced liver fibrosis induced by a 10-week CCl4 challenge (Figs. 3A-B). Moreover, poly I:C treatment reduced expression of α-SMA and transforming growth factor-β1 (TGF-β1) in HSCs from 2-week CCl4 mice, while such inhibition was not observed in HSCs from 10-week CCl4 mice (Fig. 3C). HSCs from 10-week CCl4 mice had higher levels of α-SMA expression compared with those from 2-week CCl4 mice, while expression of RAE1, an NK cell activating ligand, was comparable in the HSCs from both groups (supporting Fig. S3A). Next, we examined the effects of IFN-γ on advanced liver fibrosis induced by 10- or 12- week CCl4 administration. Treatment with IFN-γ for 2 weeks inhibited liver fibrosis in 2-week CCl4 group; however, IFN-γ treatment for the final 2 weeks or 4 weeks did not affect 10- or 12-week CCl4-induced liver fibrosis as determined by α-SMA staining and hydroxyproline contents (Figs. 3D-E). After IFN-γ injection, serum IFN-γ levels increased in all groups (Supporting Fig. S3B).
Fig. 3.

Poly I:C or IFN-γ treatments inhibits liver fibrosis induced by 2-week CCl4 but not by 10-week CCl4 treatment. Two-week or 10-week CCl4-treated mice were co-injected with poly I:C for the last 2 weeks. (A-B) Liver tissues were collected for immunohistochemistry analyses with anti-α-SMA antibody (A), measurement of hepatic hydroxyproline content (B). (C) HSCs were isolated from the mouse liver and then subject to Western blotting. (D-E) Mice were injected with CCl4 plus IFN-γ for 2 weeks, or with CCl4 for 8 weeks and then co-treatment of CCl4 plus IFN-γ for an additional 2 or 4 weeks. Liver tissues were subsequently collected and subject to immunohistochemistry analyses with anti-α-SMA antibody (D) and measurement of hepatic hydroxyproline content (E). (F) Protein extracts from HSCs were subject to Western blotting. *P<0.05.
To understand the mechanism underlying the ineffectiveness of IFN-γ treatment on advanced liver fibrosis, IFN-γ activation of STAT1 was investigated in isolated HSCs from 2-week, 8-week, or 12-week CCl4 mice. As illustrated in Fig. 3F, IFN-γ treatment inhibited the expression of α-SMA and TGF-β1 in 2-week CCl4 mice but not in 10- or 12-week CCl4 mice. STAT1 was phosphorylated in isolated HSCs of the IFN-γ treated-2-week group, but not in HSCs of the IFN-γ treated 10- or 12-week group. Finally, expression of SOCS1 protein, a negative regulator of STAT1,16 in HSCs was upregulated in 2-week CCl4 mice after IFN-γ treatment. HSCs isolated from 10- or 12 week CCl4 mice had higher basal levels of SOCS1 protein than those from 2-week CCl4 mice, which were not further upregulated after IFN-γ treatment (Fig. 3F).
Early-activated D4 HSCs induce more NK cell activation than intermediately-activated D8 HSCs: Suppression by TGF-β1
To further understand the underlying mechanism of suppressed NK cell function observed in the advanced liver fibrosis, day 4 (early-activated) or day 8 (intermediately activated) cultured HSCs (D4 or D8 HSCs) were co-cultured with liver NK cells for 24 hours. After co-culturing with HSCs, IFN-γ production by NK cells was significantly increased in co-culturing with D4 HSCs or with D8 HSCs. Higher levels of IFN-γ were observed when co-cultured with D4 HSCs than those with D8 HSCs (Fig. 4A). Co-culture studies of IFN-γ-deficient cells suggest that the source of IFN-γ production is from NK cells (Fig. 4B). Furthermore, incubation with NKG2D neutralizing antibody diminished IFN-γ production in the co-culture experiments (Fig. 4C), suggesting that activated HSCs induce IFN-γ production by NK cells via an NKG2D-dependent mechanism.
Fig. 4.
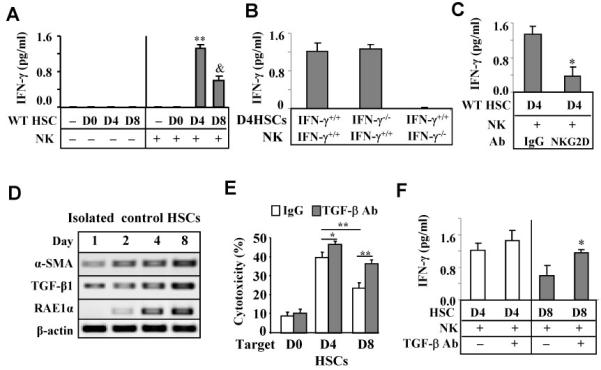
Early-activated D4 HSCs induce more NK cell activation (IFN-γ production) than intermediately-activated D8 HSCs: suppression by TGF-β. (A-C) IFN-γ levels from the supernatant of co-culture of liver NK cells and HSCs for 24 h. (A) Co-culture of NK with D0-D8 HSCs. (B) Co-culture of IFN-γ+/+ or IFN-γ−/− NK cells with IFN-γ+/+ or IFN-γ−/− D4 HSCs. (C) Co-culture of D4 HSCs with NK cells in the presence of anti-NKG2D antibody or IgG control. (D) Western blotting of cultured HSCs. (E) Activated liver NK cells from poly I:C-treated mice were used as effector cells in cytotoxicity assay against D0-D8 HSCs in the presence of anti-TGF-β antibody or IgG controls. (F) D8 HSCs were incubated with liver NK cells with or without anti-TGF-β antibody for 24 h. IFN-γ level in the supernatant was measured. *P<0.05, **P<0.01 in comparison with corresponding groups, &P<0.05 in comparison with D4 HSC.
Expression of TGF-β protein was significantly higher in D8 HSCs compared with D4 HSCs (Fig. 4D). Since TGF-β is a potent inhibitor for NK cells,7, 17 we hypothesized that TGF-β1 produced by co-cultured HSCs may inhibit IFN-γ production and cytotoxicity of NK cells. As illustrated in Fig. 4E, incubation with TGF-β neutralizing antibody markedly enhanced NK cell cytotoxicity against D8 HSCs as well as D4 HSCs (albeit to a lesser extent). In addition, TGF-β antibody treatment increased IFN-γ production by NK cells when co-cultured with D8 HSCs but did not affect IFN-γ production in co-culture experiment with D4 HSCs (Fig. 4F). Furthermore, the addition of TGF-β1 ligand suppressed the cytotoxicity of NK cells against D4 and D8 HSCs (Supporting Fig. S4).
Intermediately-activated HSCs are less responsive to IFN-γ stimulation than early-activated HSCs due to upregulation of SOCS1 expression
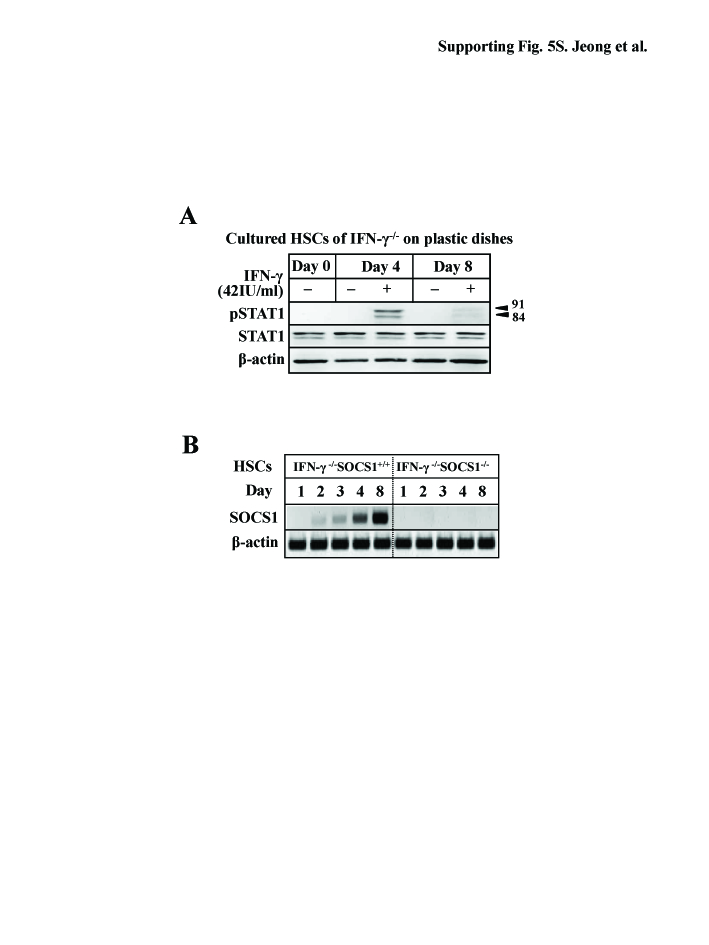
Although IFN-γ-mediated STAT1 activation has been well documented in HSCs,6, 11, 12, 18 the aforementioned experiments show that IFN-γ activation of STAT1 in HSCs from livers with advanced liver fibrosis appears to be disrupted (Fig. 3F). To study the underlying mechanisms responsible for the disruption, IFN-γ-mediated inhibitory cell proliferation and activation of STAT1 were compared on D4 and D8 HSCs. As shown in Fig. 5A, IFN-γ treatment suppressed cell proliferation of D4 HSCs, but not D8 HSCs. Western blotting showed that IFN-γ induced STAT1 activation (phosphorylated STAT1) in D4 HSCs, but this activation was markedly attenuated in D8 HSCs (Fig. 5B and Supporting Fig. S5A). Immunocytochemical analyses confirmed that IFN-γ treatment induced STAT1 activation in D4 HSCs, but not in D8 HSCs (Fig. 5C). Intriguingly, D8 HSCs expressed higher levels of SOCS1 mRNA compared to D4 HSCs, although expression of IFN-γR1 and R2 were similar in both types of cells (Fig. 5D). To determine whether induced SOCS1 protein is responsible for the insensitivity of D8 HSCs to IFN-γ, IFN-γ−/−SOCS1−/− and IFN-γ−/−SOCS1+/+ HSCs were isolated and cultured for 8 days. SOCS1−/− mice are not embryonic lethal but they die within 3 weeks after birth.19 Therefore, we used IFN-γ−/−SOCS1−/− mice that survive well to isolate SOCS1−/− HSCs. Lack of SOCS1 protein and mRNA expression in IFN-γ−/−SOCS1−/− HSCs was confirmed (Fig 5E and Supporting Fig. S5B). IFN-γ activation of STAT1 and inhibition of HSC proliferation were restored in D8 IFN-γ−/−SOCS1−/− HSCs (Figs. 5E-F).
Fig. 5.

D8 HSCs are resistant to IFN-γ stimulation due to induction of SOCS1. HSCs were cultured for 4 days (D4 HSCs) or 8 days (D8 HSCs). (A) D4 and D8 HSCs were treated with IFN-γ and [3H] thymidine uptake was determined. D4 and D8 HSCs was treated with IFN-γ for 30 min, followed by Western blot analysis (B) or immunocytochemistry analyses (C) with anti-pSTAT1, STAT1 or SOCS1 antibodies. In panel C, the black arrows indicate positive pSTAT1 staining in the nuclei. Open arrows indicate negative staining. (D) RT-PCR analyses of cultured HSCs. (E) Western blot analyses of D8 IFN-γ−/−SOCS1+/+ and IFN-γ−/−SOCS1−/− HSCs treated with IFN-γ for 30 min. (F) [3H] thymidine uptake analyses of D8 IFN-γ−/−SOCS1+/+ and IFN-γ−/−SOCS1−/− HSCs treated with IFN-γ. **P<0.01 compared with corresponding groups.
Inhibition of retinol metabolism by 4-methyl pyrazole (4-MP) blocks SOCS1 induction
Retinol metabolites, in particular retinoic acid (RA), have been reported to induce SOCS3 mRNA expression in primary cultured astrocytes and C6 cells.20 Furthermore, HSCs produce high levels of RA during activation.8 Thus, we hypothesized that induction of RA may also contribute to SOCS1 induction in HSCs during activation. To test this hypothesis, 4-MP, a selective inhibitor of alcohol dehydrogenase (ADH) enzymes, was used to inhibit retinol metabolism in HSCs. As illustrated in Fig. 6A, 4-MP treatment significantly reduced metabolism of retinol into retinaldehydes (Ralds) and RAs. Induction of SOCS1 and SOCS3 mRNAs on HSCs was remarkably reduced in the 4-MP-treated group (Fig. 6B). Western blotting showed that expression of SOCS1 protein was detected in vehicle-treated, but not in 4-MP-treated D8 HSCs (Fig. 6C). Finally, 4-MP treatment restored IFN-γ activation of STAT1 (Fig. 6C) and IFN-γ inhibition of cell proliferation in D8 HSCs (Fig. 6D).
Fig. 6.
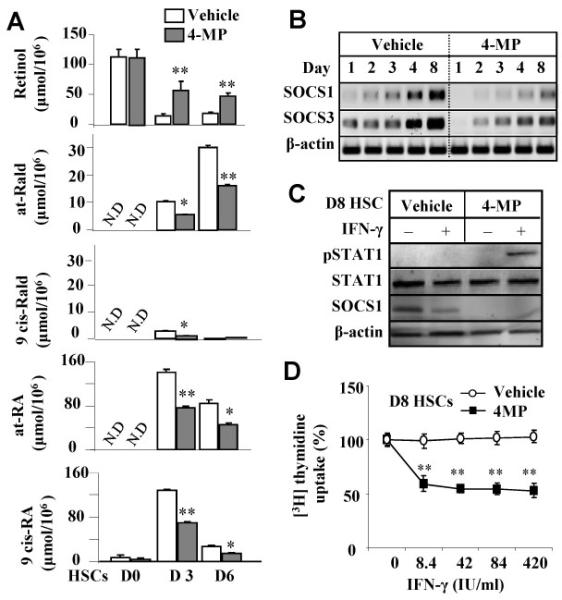
Inhibition of retinol metabolism by 4-MP blocks SOCS1 induction in intermediately-activated HSCs. (A) Intracellular concentrations of retinol and retinol metabolites from cultured HSCs treated with 4-MP. (B) RT-PCR analyses of cultured HSCs treated with 4-MP. (C) Western blot analyses of HSCs treated with 4 MP and/or IFN-γ. (D) HSCs were cultured with or without 4MP for 8 days, treated with IFN-γ and then [3H] thymidine uptake was determined. *P<0.05, **P<0.01 compared with corresponding vehicle-treated groups.
Retinol metabolites induce SOCS1 protein in HSCs
The above findings suggest that retinol metabolites may induce SOCS1 expression in HSCs. To test this notion, the effects of various retinol metabolites (retinaldehydes and retinoic acids) on SOCS1 expression in HSCs were investigated. Expression of SOCS1 gene was markedly induced in HSCs treated with 9-cis Rald and 9-cis RA, whereas all trans Rald and all trans RA treatments only resulted in slight induction (Fig. 7A). To investigate which types of retinoic acid receptors (RARs) and retinoid X receptors (RXRs) were involved in SOCS1 induction, HSCs were treated with RAR agonist (CD437) and a RXR agonist (methoprene acid, MA). After treatment, only CD437 RAR agonist induced SOCS1 expression in D2 HSCs (Fig. 7B). Additionally, the effects of 9-cis Rald, 9-cis RA and CD437 on IFN-γ signaling in HSCs were explored. As illustrated in Figs. 7C-D, IFN-γ treatment induced pSTAT1 activation and SOCS1 expression in vehicle-treated D4 HSCs. Such STAT1 activation was attenuated in 9-cis Rald, 9-cis RA and CD437-pretreated HSCs compared to control (vehicle) group. Furthermore, treatment with 9-cis Rald and CD437 but not 9-cis-RA upregulated SOCS1 protein in HSCs compared to vehicle group.
Fig. 7.
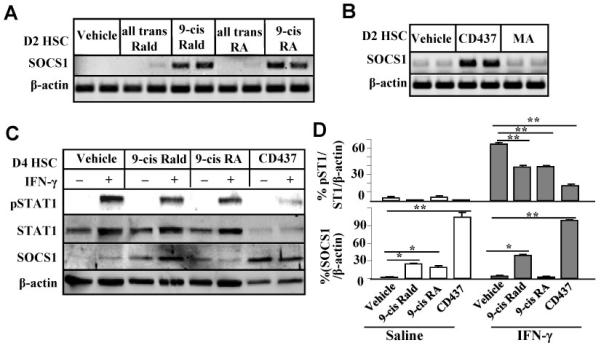
Retinol metabolites induce SOCS1 gene expression in HSCs, rendering HSC resistant to IFN-γ treatment. (A-B) RT-PCR analyses of SOCS1 mRNA expression from HSCs treated with retinol metabolites, RAR agonist CD437 or RXR agonist MA for 2 days. (C) HSCs were cultured with various retinol metabolits for 4 days and then cultured in serum-free medium for an additional 6 h, followed by treatment with IFN-γ for 30 min. Cell extracts were then prepared for Western blotting. (D) The densities of bands in panel C were quantified. *P<0.05, **P<0.01.
Discussion
Numerous studies have shown that treatment with poly I:C or IFN-γ, at the onset or early stages, prevents liver fibrosis in rodents via enhanced activation of NK cells/IFN-γ against HSCs.4-6, 11, 12 In this report, we demonstrated that the antifibrotic effects of poly I:C and IFN-γ were diminished in advanced liver fibrosis induced by a 10-week CCl4 treatment, and that retinol metabolites play an important role in inhibiting the anti-fibrotic effects of NK cell and IFN-γ via induction of TGF-β1 and SOCS1 protein, respectively. In addition, retinol metabolites may also enhance NK cell function via induction of NK cell activating ligand expression on HSCs. We have summarized our findings into a proposed model as shown in Fig. 8.
Fig. 8.
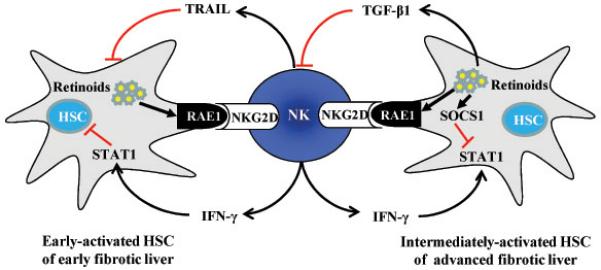
A double edged sword role of retinol metabolites in HSCs against NK cell activation. During HSC activation, retinols are metabolized into Rald or RA, which affect the interaction of NK and HSCs in 2 ways. (1) RA induces expression of NK cell activating ligand RAE1, which then activates NK cells through interaction with NKG2D. Activated NK cells can directly kill early-activated HSCs via releasing TRAIL or inhibit HSCs via releasing IFN-γ. IFN-γ induces cell cycle arrest and apoptosis of HSCs via activation of STAT1. (2) Retinoids (Rald and RA) can inhibit IFN-γ-mediated activation of STAT1 via induction of SOCS1, a key negative inhibitor of IFN-γ signaling, in intermediately-activated HSCs. Also, RA can activate latent TGF-β, which then inhibits NK cell activity. Therefore, retinol metabolism during HSC activation can promote NK cell activation via induction of RAE1, but can also downregulate NK cell activity via induction of TGF-β that inhibits NK cell activity, or induction of SOCS1 that inhibits IFN-γ signaling.
The antifibrotic effects of NK cells and IFN-γ have been previously documented in various models; however, the majority of these studies were conducted on the model of early stage of liver fibrosis.4-6, 11, 12, 18 Here we provided multiple lines of evidence suggesting that the anti-fibrotic functions of NK cells/IFN-γ are suppressed in advanced liver fibrosis. First, poly I:C and IFN-γ treatment did not ameliorate advanced liver fibrosis induced by a 10-week CCl4 treatment; Second, serum levels of IFN-γ were not increased by poly I:C treatment in the model of advanced liver fibrosis (Fig. 1A); Third, liver NK cells from 10-week CCl4 challenged mice display lower cytotoxicity against both Yac-1 cells and HSCs than those from 2-week CCl4 mice (Figs. 1-3), suggesting NK cell functions are impaired in advanced liver fibrosis; Fourth, the number of activated NK cells and expression of NK cells-associated genes were lower in advanced liver fibrosis vs. early stage of liver fibrosis (Figs. 1 and 2). Finally, IFN-γ activation of STAT1, a major mediator of IFN-γ signaling, was suppressed in HSCs from the advanced fibrotic liver (Fig. 3F) or in intermediately-activated D8 HSCs (Fig. 5B) despite expression of high levels of IFN-γRs on these HSCs (Supporting Fig. S6 and Fig. 5D). These data suggest that IFN-γ treatment is likely effective in treating early stages of liver fibrosis but not advanced liver fibrosis, and lack of effects of IFN-γ therapy on liver fibrosis observed in clinical trials may be due to the selection of patients with advanced liver fibrosis.15
The next question is what are the mechanisms underlying the decreased anti-fibrotic effects of NK cells/IFN-γ in advanced liver fibrosis. The studies from current paper suggest that TGF-β and retinoic acid contribute to inhibition of NK cell functions and IFN-γ signaling pathways, respectively, in advanced liver fibrosis. Recently, it has been suggested that the enhanced production of IFN-γ by NK cells could be derived from interaction with activating HSCs in liver diseases.1, 2, 4-8 In the current study, we provide several lines of evidence suggesting that early-activated HSCs express high levels of RAE1, an NK cell activating ligand, which can induce NK cell activation and production of IFN-γ, while intermediately-activated HSCs may inhibit NK cell activity via producing high levels of TGF-β. First, IFN-γ production by NK cells was significantly enhanced when co-cultured with early-activated D4 HSCs, yet compared to D4 HSCs, IFN-γ production was lower when co-cultured with intermediately-activated D8 HSCs (Fig. 4). Second, Western blotting and RT-PCR analyses showed that TGF-β1 mRNA and protein expression were highly induced in HSCs from advanced liver fibrosis (Figs. 3C-F) and intermediately-activated D8 HSCs (Fig. 4D). Third, blocking TGF-β with a neutralizing antibody increased NK cell killing and restored IFN-γ production of NK cells (Fig. 4E,F), whereas treatment with TGF-β decreased NK cell cytotoxicity (Supporting Fig. S4). Finally, TGF-β is known to inhibit NK cell-mediated cytotoxicity and cytokine production. 7, 17, 21 Taken together, TGF-β likely plays an important role in inhibiting the anti-fibrotic effect of NK cells and resistance of intermediately-activated HSCs to NK cell killing is likely mediated by the overproduction of TGF-β. In addition to HSCs that are known to be one of the major sources for TGF-β production in the fibrotic liver and its production is proportional to the grade of liver fibrosis,9, 10 Kupffer cells also play an important role in producing TGF-β during liver fibrogenesis.22 Future studies are required to determine whether Kupffer cells can also negatively regulate NK cell functions via production of TGF-β in the advanced liver fibrosis.
In addition to resistant to NK cell killing, HSCs isolated from advanced fibrotic liver or intermediately-activated D8 HSCs are also less responsive to IFN-γ stimulation (Fig. 3F and Supporting Figs. S3 and S6). The reduced responsiveness of these cells to IFN-γ stimulation is likely due to the increased expression of SOCS1 (Fig. 3F and Figs. 5B-D) as SOCS1 is known to be a key mediator in suppressing IFN-γ signaling.23 This conclusion was supported by the fact that IFN-γ inhibition of cell proliferation and activation of STAT1 were restored in D8 cultured IFN-γ−/−SOCS1−/− HSC compared to D8 IFN-γ−/−SOCS1+/+ HSCs (Figs. 5E-F). Further experiments suggest that upregulation of SOCS1 in D8 HSCs is due to RA production during HSC activation.
Quiescent HSCs store approximately 80% retinols of the body which are released or metabolized into Ralds by alcohol dehydrogenase (ADH) and subsequently RA by retinaldehyde dehydrogenases (Raldh) during HCS activation.8, 24, 25 An increase of RA and a decrease of retinol content have been reported in CCl4 and thioacetamide-induced fibrotic livers of rats, and in cultured HSCs.8, 26 In the current paper, we demonstrated that inhibition of retinol metabolism by 4-MP reduced expression of SOCS1 and subsequently increased the IFN-γ activation of STAT1 signaling in HSCs (Fig. 6), suggesting a role of retinol metabolites in the induction of SOCS1 in HSCs. The fact that treatment with 9-cis Rald, 9-cis RA or CD437 (an agonist of RAR-β/γ)8, 25, 27 induced SOCS1 expression and attenuated STAT1 phosphorylation of IFN-γ in HSCs (Fig. 7) clearly indicates that retinol metabolites inhibit IFN-γ signaling via induction of SOCS1 in HSCs.
In conclusion, the current study demonstrated that intermediately-activated HSCs displayed resistance to IFN-γ stimulation and NK cell killing via a RA-mediated SOCS1 and TGF-β dependent manner despite the enhanced expression of RAE1 (Fig. 8). These data potentially provide insight into the mechanisms underlying the resistance to NK cells/IFN-γ therapy in patients with advanced liver fibrosis. Therefore, retinol metabolites/SOCS1/TGF-β could be a potential therapeutic target for improving the efficacy of IFN-γ treatment and NK cell therapy in treating liver fibrosis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial Support
This study was supported in part by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A090292), and in part by the intramural program of NIAAA, NIH
List of Abbreviations
- ADH
alcohol dehydrogenase
- ALT
alanine aminotransferase
- CCl4
carbon tetrachloride
- HSC
hepatic stellate cell
- HYP
hydroxyproline
- IFN-γ
interferon-γ
- MA
methoprene acid
- MNC
mononuclear cells
- 4-MP
4-methylpyrazole
- NK
natural killer cell
- NKG2D
natural killer group 2 member D
- Poly I:C
polyinosinic-polycytidylic acid
- RA
retinoic acid
- Rald
retinaldehyde
- RAE
retinoic acid early inducible gene
- Raldh
retinaldehyde dehydrogenase
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- SOCS1
suppressor of cytokine signaling 1
- α-SMA
alpha-smooth muscle actin
- STAT1
signal transducers and activators of transcription 1
- TGF-β1
transforming growth factor-beta1
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
References
- 1.Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729–36. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 2.Notas G, Kisseleva T, Brenner D. NK and NKT cells in liver injury and fibrosis. Clin Immunol. 2009;130:16–26. doi: 10.1016/j.clim.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Muhanna N, Doron S, Amer J, Azzeh M, Mahamid M, Friedman S, Safadi R. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2010 doi: 10.1136/gut.2010.211136. TL. in press. [DOI] [PubMed] [Google Scholar]
- 4.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–52. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 5.Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 6.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–51. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 7.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–58. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–16. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 9.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, Benedetti A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–99. doi: 10.1002/hep.510230538. [DOI] [PubMed] [Google Scholar]
- 12.Rockey DC, Chung JJ. Interferon gamma inhibits lipocyte activation and extracellular matrix mRNA expression during experimental liver injury: implications for treatment of hepatic fibrosis. J Investig Med. 1994;42:660–70. [PubMed] [Google Scholar]
- 13.Weng HL, Wang BE, Jia JD, Wu WF, Xian JZ, Mertens PR, Cai WM, Dooley S. Effect of interferon-gamma on hepatic fibrosis in chronic hepatitis B virus infection: a randomized controlled study. Clin Gastroenterol Hepatol. 2005;3:819–28. doi: 10.1016/s1542-3565(05)00404-0. [DOI] [PubMed] [Google Scholar]
- 14.Muir AJ, Sylvestre PB, Rockey DC. Interferon gamma-1b for the treatment of fibrosis in chronic hepatitis C infection. J Viral Hepat. 2006;13:322–8. doi: 10.1111/j.1365-2893.2005.00689.x. [DOI] [PubMed] [Google Scholar]
- 15.Pockros PJ, Jeffers L, Afdhal N, Goodman ZD, Nelson D, Gish RG, Reddy KR, Reindollar R, Rodriguez-Torres M, Sullivan S, Blatt LM, Faris-Young S. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-gamma1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology. 2007;45:569–78. doi: 10.1002/hep.21561. [DOI] [PubMed] [Google Scholar]
- 16.Brysha M, Zhang JG, Bertolino P, Corbin JE, Alexander WS, Nicola NA, Hilton DJ, Starr R. Suppressor of cytokine signaling-1 attenuates the duration of interferon gamma signal transduction in vitro and in vivo. J Biol Chem. 2001;276:22086–9. doi: 10.1074/jbc.M102737200. [DOI] [PubMed] [Google Scholar]
- 17.Dasgupta S, Bhattacharya-Chatterjee M, O’Malley BW, Jr., Chatterjee SK. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. 2005;175:5541–50. doi: 10.4049/jimmunol.175.8.5541. [DOI] [PubMed] [Google Scholar]
- 18.Rockey DC, Maher JJ, Jarnagin WR, Gabbiani G, Friedman SL. Inhibition of rat hepatic lipocyte activation in culture by interferon-gamma. Hepatology. 1992;16:776–84. doi: 10.1002/hep.1840160325. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, Kishimoto T. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–87. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 20.Choi WH, Ji KA, Jeon SB, Yang MS, Kim H, Min KJ, Shong M, Jou I, Joe EH. Anti-inflammatory roles of retinoic acid in rat brain astrocytes: Suppression of interferon-gamma-induced JAK/STAT phosphorylation. Biochem Biophys Res Commun. 2005;329:125–31. doi: 10.1016/j.bbrc.2005.01.110. [DOI] [PubMed] [Google Scholar]
- 21.Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM, Jr., Mao H, Yokohama A, Bhatt D, Shen L, Davuluri R, Weinstein M, Marcucci G, Caligiuri MA. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. 2006;24:575–90. doi: 10.1016/j.immuni.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–48. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davey GM, Heath WR, Starr R. SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens. 2006;67:1–9. doi: 10.1111/j.1399-0039.2005.00532.x. [DOI] [PubMed] [Google Scholar]
- 24.Duester G, Mic FA, Molotkov A. Cytosolic retinoid dehydrogenases govern ubiquitous metabolism of retinol to retinaldehyde followed by tissue-specific metabolism to retinoic acid. Chem Biol Interact. 2003;143-144:201–10. doi: 10.1016/s0009-2797(02)00204-1. [DOI] [PubMed] [Google Scholar]
- 25.Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440:139–62. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 26.Natarajan SK, Thomas S, Ramachandran A, Pulimood AB, Balasubramanian KA. Retinoid metabolism during development of liver cirrhosis. Arch Biochem Biophys. 2005;443:93–100. doi: 10.1016/j.abb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Hellemans K, Verbuyst P, Quartier E, Schuit F, Rombouts K, Chandraratna RA, Schuppan D, Geerts A. Differential modulation of rat hepatic stellate phenotype by natural and synthetic retinoids. Hepatology. 2004;39:97–108. doi: 10.1002/hep.20015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.