

Abstract
The most deadly phase in cancer progression is attributed to the inappropriate acquisition of molecular machinery leading to metastatic transformation and spread of disease to distant organs. While it is appreciated that metastasis involves epithelial-mesenchymal interplay, the underlying mechanism defining this process is poorly understood. Specifically, how cancer cells evade immune surveillance and gain the ability to navigate the circulatory system remains a focus. One possible mechanism underlying metastatic conversion is fusion between blood-derived immune cells and cancer cells. While this notion is a century old, in vivo evidence that cell fusion occurs within tumors and imparts genetic or physiologic changes remains controversial. We have previously demonstrated in vivo cell fusion between blood cells and intestinal epithelial cells in an injury setting. Here, we hypothesize that immune cells, such as macrophages, fuse with tumor cells imparting metastatic capabilities by transferring their cellular identity. We used parabiosis to introduce fluorescent-labeled bone marrow-derived cells to mice with intestinal tumors, finding that fusion between circulating blood-derived cells and tumor epithelium occurs during the natural course of tumorigenesis. Moreover, we identify the macrophage as a key cellular partner for this process. Interestingly, cell fusion hybrids retain a transcriptome identity characteristic of both parental derivatives, while also expressing a unique subset of transcripts. Our data supports the novel possibility that tumorigenic cell fusion may impart physical behavior attributed to migratory macrophages, including navigation of circulation and immune evasion. As such, cell fusion may represent a promising novel mechanism underlying the metastatic conversion of cancer cells.
Introduction
Metastatic disease accounts for the majority of cancer fatalities, and is, unfortunately the least understood phase of tumor progression. The underlying mechanism by which cancer cells acquire the ability to escape the primary tumor site, evade immune system eradication, migrate to a distant location and re-establish aggressive tumorigenesis is not completely known. Clearly, it is a multi-faceted process involving changes in the tumor epithelia in conjunction with local influences emanating from the surrounding tumor microenvironment. It has long been speculated that fusion between mesenchymal cells and tumor cells can lead to phenotypic diversity in tumors, which is thought to be an important factor in tumor progression(1, 2). Moreover, we have previously shown that circulating bone marrow-derived cells (BMDCs) readily fuse with the intestinal epithelium upon tissue injury(3, 4), and that this process is augmented by an inflammatory and hyperproliferative microenvironment(3) characteristic of a tumor setting.
Importantly, cell fusion between blood leukocytes and tumor cells has been shown to occur in vitro(6); however, its physiologic consequence on tumorigenesis remains unknown. It has been proposed that cell fusion imparts migratory blood cell properties to tumor cells as a potential contribution to drive metastatic disease(1, 5), however no definitive in vivo evidence for acquisition of these properties has been demonstrated. Evidence for cell fusion in cancer progression has the potential to revolutionize our current understanding of the biology of metastatic disease.
We hypothesized that fusogenic immune cell populations, such as the macrophage, would facilitate the ability of tumor cells to acquire metastatic capabilities by transferring distinct cellular capabilities during a physical fusion event with cancer cells. Using parabiosis, the surgical joining of two mice to facilitate a shared blood supply, we demonstrate in vivo fusion between circulating blood-derived cells and tumor epithelia. Further, we identify the macrophage as a blood-derived mesenchymal cell fusion partner in this process. Moreover, we show that hybrid cells resulting from cell fusion possess a transcriptome identity similar to both macrophages and epithelial cells, as well as a set of unique transcripts, distinguishing them from their two parental lineages. Our data suggest that tumorigenic cell fusion has the potential to impart aggressive tumor behavior that has been attributed to epithelial-mesenchymal transition, namely the acquisition of macrophage-like properties such as migration and immune system evasion, and implicate cell fusion as a promising novel mechanism for the metastatic conversion of cancer cells.
Materials & Methods
Mice
Mice were housed in a specific pathogen-free environment under strictly controlled light cycle conditions, fed a standard rodent Lab Chow (#5001 PMI Nutrition International), and provided water ad libitum. All procedures, including bone marrow transplantation and parabiosis, were performed in accordance to the OHSU Animal Care and Use Committee as previously reported(3, 4). The C57Bl/6, ROSA26(6), Rag1-/-(7), and ApcMin/+(8) mice were obtained from The Jackson Laboratory. OsbYO1 (GFP)(9) mice were bred in-house.
FACS-isolation of blood-derived cells
Blood cell progenitors were isolated by FACS for subsequent limited-lineage transplantation. Bone marrow from GFP mice was isolated as described(4), and stained with the appropriate combination of antibodies against cell-surface antigens for the desired populations. For separate macrophage, B cell, and T cell isolation followed by transplantation, whole bone marrow (WBM) cells were first combined with peripheral blood before FACS isolation. Peripheral blood was obtained by retro-orbital bleeding of GFP mice, sedimented and red blood cells lysed. Antibody concentrations are listed in Supplementary Table 1. All antibody staining was performed for 30 minutes at 4°C. Cells were washed twice in HBSS and resuspended in HBSS+ [3% Fetal Bovine Serum (FBS), 5mg/ml propidium iodide (PI)]. Cell populations were isolated using a Becton Dickinson FACSVantage with DiVa (Digital Vantage) option, and a 70μm nozzle. Cell doublets were eliminated on the basis of pulse width. Purity of sorted populations described below, then only >99% pure populations were used for subsequent assays. Isolated pure populations were individually transplanted into lethally irradiated mice as described below.
Bone Marrow Transplantation
WBM and lineage limited bone marrow transplantation was performed as described with some modifications(4). Briefly, WBM was harvested from 5- to 12-week-old donor GFP-expressing mice(4), filtered for single-cell suspension then resuspended in Hank's balanced salt solution (HBSS). Sorted or whole bone marrow cells were injected into the retro-orbital sinus of recipient mice. Approximately 5×106 GFP-expressing whole bone marrow cells or 8×104-3×105 lineage-limited populations supplemented with 2×105 unlabeled carrier whole bone marrow, was injected. Six-week-old recipient male WT, ApcMin/+, ROSA26, or ROSA26/ApcMin/+ mice received whole-body γ-IR (12 Gy split dose 4 hours apart) prior to transplant. To confirm hematopoietic engraftment or to check for contamination in FACS-isolated populations, peripheral blood leukocytes were isolated from recipient mice 2 weeks post-transplantation as previously reported(10) and analyzed for lineage using a Becton Dickinson FACSCalibur.
Parabiosis
Surgery was performed between GFP and ROSA26/ApcMin/+ mice (n=8 pair) as described previously(3). Briefly, pairs of 6- to 12-week age-, gender- and weight-matched mice were surgically joined from the elbow to knee. Mice were separated 7 weeks after surgery, their blood analyzed to assess percent chimerism and intestinal tissue analyzed.
Cell culture and live cell microscopy
MC-38H2BmRFP colorectal cancer cells were generated by retroviral transduction of the mouse adenocarcinoma cell line MC-38 (a kind gift from J. Schlom, National Cancer Institute, Bethesda, MD) to express monomeric red fluorescent protein (mRFP) fused to histone 2B (H2B); GFP-macrophages were derived from the bone marrow of GFP transgenic mice(9). MC-38H2BmRFP cancer cells and GFP-macrophages were co-seeded onto 35-mm glass-bottom dishes (MatTek), grown in standard conditions and incubated in a humidified incubator with 5% CO2 at 37°C. Fluorescence imaging was conducted in a temperature and CO2 controlled stage enclosure, using a 40X oil objective on a DeltaVision-modified inverted microscope (IX71; Olympus) and SoftWorx software (Applied Precision, LLC). Z-series images were acquired using a camera (Nikon CoolPix HQ, Nikon) at 15 minute intervals. Z-series stacks were compiled by maximum intensity projection for presentation. The movie is compiled at 6 frames per second.
Intestinal analysis of transplanted and parabiotic mice
Cell fusion was identified using immunohistochemical analysis and co-localization of donor, GFP and recipient, β-galactosidase expression. Small intestine and colon from experimental and control animals was dissected en bloc, processed for wholemount imaging, frozen in OCT and sectioned as previously described(11). Five μm (or 50μm for confocal analysis) tissue sections were stained with hematoxylin and eosin or incubated with antibodies for β-galactosidase (1:500, ICL, Inc.) and GFP (1:500; Molecular Probes) followed by detection with fluorescent secondary antibodies (1:500, Alexa488, Molecular Probes; 1:250, Cy5, Jackson Immuno Research ) and confocal microscopy performed, as we have reported previously(3, 4). For transplantation studies, mice were analyzed at 4 or 8 weeks post-transplantation for all studies (n=3-10/experimental paradigm), with the exception of the early time course analysis, where the mice were analyzed every 24 hours post-transplantation for 7 days (n>4 mice/time point). Analysis of parabiotic pairs took place at time of separation (4-9 weeks; n=4). Blood cell antigens were detected with antibodies to CD45 (1:500; BD Pharmingen), B220 (1:500; BD Pharmingen) CD4 & CD8 (1:500; BD Pharmingen), and F4/80 (1:500; eBioscience). Epithelial cells were detected with anti-E-cadherin antibodies (1:1000; Zymed), and the basement membrane detected with anti-laminin antibodies (1:1000; Chemicon). Nuclei were counterstained with Hoechst (33258; Sigma; 0.1μg/ml). Tissue sections were analyzed with a Leica DMR microscope, digital images captured with a DC500 digital camera and IM50 Image Manager Software (Leica Microsystems). Confocal images were acquired using an IX81 Inverted Microscope equipped with Fluoview FV1000-Spinning Disc Confocal (Olympus) scan head and FV10 ASW 1.7 software (Olympus).
Statistics
Cell fusion was quantified by reporting the percentage of crypt/villus units harboring at least one GFP-positive cell. A “unit” was defined as one villus and its adjacent crypt. For each animal, tissue sections at least 125 μm apart were quantified and >1500 units were examined (Supplementary Fig.1m). We do not quantify on a per cell basis because this would overestimate the extent of cell fusion due to proliferative expansion of the initial fusion event. Statistical significance between experimental populations was determined using a Student's two-tailed, unpaired t-test as appropriate for each experimental scenario. P values <0.01 were considered statistically significant. Statistical analysis was performed using GraphPad Prism for Windows (GraphPad Software). All data are presented as the mean ± s.e.m.
Epithelial FACS
Epithelium was isolated for FACS and subsequent gene expression analysis. The intestinal epithelium was isolated using a modified Weiser preparation(12). Cells were then incubated in Type III Collagenase (30min; 15units/ml; Sigma), dispase (30min; 0.3units/ml; Invitrogen), then filtered through a 12×75mm filter (BD Falcon) for single-cell suspension. Cells were stained with anti-CD45-APC (1:100; BD Pharmingen) for 30 minutes at 4°C, washed twice in HBSS and resuspended in HBSS+1% Bovine Serum Albumin and PI. To exclude the intra-epithelial lymphocyte population and blood cell contamination from the epithelial cells, GFP+;CD45-;PI- epithelial cells were isolated with an InFlux flow cytometer (Cytopeia) using a 150 μm nozzle. Doublets were eliminated using pulse width. The purity of sorted populations was determined and only >99% pure populations were used in subsequent experiments.
It is possible that acquisition of macrophage gene expression could be attributed to contamination during the isolation procedure; therefore, to demonstrate that epithelial isolation results in an uncontaminated population, isolated epithelial cells were spun onto slides and analyzed for CD45+ and F4/80+ cells (Supplementary Fig.10). In unsorted epithelial samples, a small fraction of CD45+ cells were detected, but no F4/80 expressing cells were observed. However, in FACS-isolated populations, such as those used in the transcriptome analysis, neither CD45+ nor F4/80+ cells were present. Further, using qRT-PCR, F4/80 gene expression was only detected at a 5% contamination threshold in a limiting dilution of macrophage into intestinal epithelial RNA (Supplementary Fig.10), indicating that our isolated epithelial cell samples are devoid of detectible contaminating macrophage transcripts.
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
qRT-PCR was used to validate the results obtained from deep sequencing. FACS-isolated epithelial cells and macrophages from transplanted or untransplanted GFP-expressing mice were harvested as described above. Total RNA was isolated from sorted cells using RNeasy Mini kit (Qiagen). RNA quality was assessed using Agilent's PicoChip on the 2100 Bioanalyzer, then 20 ng RNA was amplified and cDNA synthesized using the NuGEN Ovation RNA Amplification System v2 protocol (NuGEN). qRT-PCR was performed using a SYBR Green-based assay, and analyzed using an Applied Biosystems 7900 HT Sequence Detector according to established protocols(12). Each cDNA sample was analyzed in triplicate, along with triplicate samples of the endogenous reference gene, Glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Epithelial and macrophage samples were harvested from n=3-4 animals. For analysis of cell fusion hybrids, multiple animals were pooled (n=3) or multiple qRT-PCR assays were conducted. Data is presented as fold change ± standard error. Primers are listed in Supplementary Table 2.
RNA-Seq and Bioinformatic Analyses
Intestinal epithelium was isolated from GFP-expressing mice (n=3) and WBM transplanted mice (n=4), as well as macrophages from GFP-expressing mice (n=3) by FACS sorting, as described above. Total RNA (0.1-1μg) was isolated using the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. Poly(A) RNA was purified using oligotex-dT30 latex beads (Qiagen). First-strand cDNA synthesis was conducted using Superscript III and OligodT-20 according to the manufacturer's instructions (Invitrogen). Second strand synthesis was generated according to standard methods, followed by double stranded cDNA fragmentation (100-700bp) with 65 watt pulse on a Misonix sonicator. cDNA was polished with the DNA terminator repair kit (Lucigen) and a single A base was added with Klenow exo-(3' to 5' exo minus) prior to ligation of genomic DNA adapters (Illumina Solexa Genomic 1G) at 22°C. Amplification of the library using 10 cycles of limited PCR using Phusion HF DNA polymerase (NEB) and genomic PCR primer (Illumina; Solexa Genomic primers 1.1 and 2.2) was conducted.
Double stranded cDNA libraries were sequenced on a Solexa G1 Genome Analyzer and image analysis and base-calling were conducted with the standard Illumina Analysis Pipeline 1.0 (Firescrest-Bustard). 36 bp sequence tags were mapped to the mouse genome (NCBI Build 37) by calling the Eland algorithm (Illumina Analysis pipeline Gerald module) with Perl scripts. A C++ program was used to count the number of uniquely mapped reads within exons of Ref-Seq genes (UCSC Genome Browser mm9 annotation). All statistical analyses were performed in the R statistical programming environment. RNA-Seq tag counts in Ref-Seq genes were mean-scaled and pair wise comparisons were performed using the χ2 statistic. The Storey Q-test was used to correct for multiple comparisons(13). Differentially regulated RefSeq genes with a p<0.01 were considered significant. Relational comparisons between data sets called an R annotation script and SQL data base queries.
To identify transcripts that were differentially regulated between at least two cell populations, RefSeq genes with a p<0.01 from comparisons between each pair of cell populations were compiled and represented 19,696 transcripts. Within this differentially regulated population of RefSeq genes, we identified genes in which cell fusion hybrids a) shared gene expression profiles with wild-type epithelium, b) shared gene expression profiles with macrophages, or c) expressed unique gene expression profiles. For category a) we selected genes that were differentially regulated between macrophages and cell fusion hybrid epithelium (p<0.01) but not between cell fusion hybrid epithelium and wild-type epithelium comparisons (p>0.05). For category b) we selected genes that were differentially regulated between cell fusion hybrid epithelium and macrophages (p<0.01) but not between wild-type epithelium and macrophages (p>0.05). For category c) we selected genes that were differentially regulated genes between wild-type epithelium and cell fusion hybrid epithelium (p<0.01) and wild-type epithelium and macrophages (p<0.01). Heatmaps for each of these categories were generated in the R programming environment and depict mean- and log-scaled total RefSeq gene tag counts mapped to an 8-bit color scale.
Results & Discussion
In vivo cell fusion in tumors
The observation that inflammation and epithelial proliferation, two key components of a tumor microenvironment(14), are strong mediators of cell fusion(3) provides evidence that cell fusion readily occurs within tumors in an in vivo context. To show that this is indeed the case, we used parabiosis (the surgical joining of two mice that facilitates shared blood supply) to introduce green fluorescent protein (GFP)-expressing circulating blood(9, 15) into an intestinal tumor-bearing ApcMin/+;ROSA26 mouse(6,8) (Fig.1a). Tumor analysis displayed epithelium that co-expressed the “donor” marker (GFP) from newly introduced circulating blood cells, and the “recipient” marker (β-galactosidase; β-gal), as determined by confocal microscopy (n=4; Fig.1b-c). We have previously documented the occurrence of cell fusion between BMDCs and the intestinal epithelium using alternative dual marker systems: GFP/β-gal, GFP/Y-chromosome, and a genetic approach using Cre recombinase-activated reporter gene expression to mark generation of cell fusion hybrids in vivo(3,4). In addition, as controls, we show here that endogenous GFP expression can be recognized by direct fluorescence as well as with antibodies to GFP with high fidelity (Supplementary Fig.1a-c) and that cell fusion in the epithelial compartment is not mistaken as GFP-expressing intraepithelial lymphocytes or tissue auto-fluorescence (Supplementary Fig.1d-l, Supplementary Fig.2). Therefore, the detection of tumor epithelia co-expressing GFP and β-gal in parabiotic mice strongly illustrates that cell fusion occurs in the natural context of tumorigenesis.
Figure 1. Intestinal cell fusion in tumorigenesis.
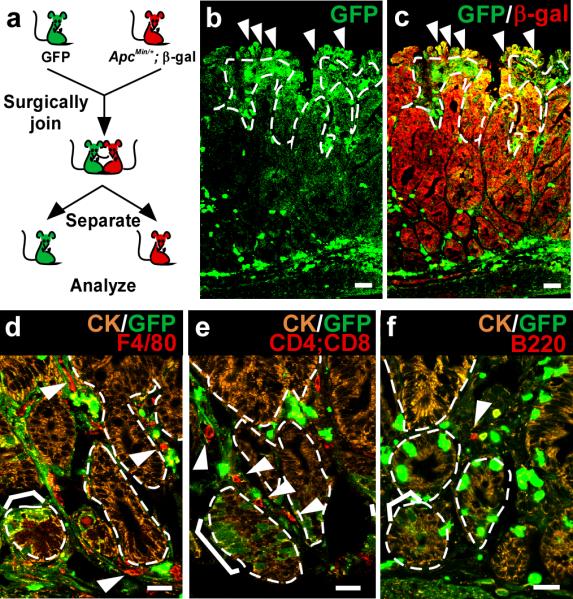
a, Parabiosis experimental design. GFP and ApcMin/+;ROSA26 mice were surgically joined. b-c, Cell fusion was observed in small intestinal polyps. Single plane confocal microscopy images of GFP (green) and β-galactosidase (red) detected by antibodies demonstrate fusion by co-localization in yellow (c). Arrowheads denote examples of fused cells. d-f, Lymphocytes and leukocytes are present within small intestinal polyps that have undergone fusion. Single plane confocal microscopy depicts fusion-derived (brackets) and unfused epithelia detected with antibodies to GFP (green) and cytokeratin (marking the epithelial compartment, orange). Arrowheads indicate F4/80+ macrophages (red) (d), CD4+ or CD8+ T cells (red) (e), or B220+ B cells (red) (f) in the tumor mesenchyme. Dashed white lines indicate epithelial/mesenchymal border. Bars=25μm.
Tumor cells are known to be highly fusogenic; this is especially evident in cell culture systems(1). Determining whether this is an active mechanism mediated by an equally fusogenic bone marrow-derived cell partner remains outstanding. Our previous work, examining cell fusion in the intestinal epithelium, revealed that the intestinal stem or progenitor cell is capable of fusion(4); however the BMDC fusion partner has yet to be identified. Therefore, to examine potential fusogenic candidate cells within the tumor microenvironment, we analyzed intestinal adenomas from parabiotic mice with antibodies to major lineages within the bone marrow: Macrophages, T and B cells (Fig.1d-f). All three blood-derived populations were present within the tumor microenvironment, suggesting that these populations may be poised for fusion with the tumor epithelium.
Identifying the BMDC fusion partner
It is possible that a number of mesenchymal cells are capable of cell fusion, as several BMDC lineages have previously been described to engage in fusion(10, 16, 17). Therefore, to determine which lineages contribute to intestinal cell fusion, we systematically surveyed the fusogenic capacity of GFP-expressing blood lineages isolated by Fluorescence-Activated Cell Sorting (FACS) in a mouse bone marrow transplantation system (Fig.2a). We have previously utilized gamma-irradiation and bone marrow transplantation to effectively promote cell fusion with the intestinal epithelium(4) and have documented that this approach results in high levels of inflammation and epithelial proliferation similar to a tumor microenvironment(3). Using this model system, we transplanted common myeloid and common lymphoid progenitors (CMP and CLP, respectively), as well as differentiated macrophages, B and T cells (Fig.2b) isolated by FACS using standard cell surface antigens(18-22) (Supplementary Fig.3, Supplementary Table 1). We then evaluated their ability to fuse with intestinal epithelium using a quantification scheme that accounts for fusion at the intestinal stem cell level (Supplementary Fig.1m). Interestingly, all five isolated populations displayed the ability to fuse with the intestinal epithelium to varying degrees. Fusion between CMPs (n=4) or CLPs (n=2) and the epithelium was detected, but was extremely rare (not shown). To further restrict possible candidates, mice were transplanted with Rag1-/-;GFP whole bone marrow (WBM) that is genetically devoid of mature B and T cells(7). Interestingly, robust epithelial cell fusion was observed (n=4; Supplementary Fig.4), suggesting that mature B and T cells were not required for cell fusion and that immature B or T cells and/or the myeloid lineage effectively contributed to cell fusion. When FACS-isolated macrophages, B or T cells were independently transplanted into our fusion model, all lineages were observed to participate in cell fusion (Fig.2d). However, very low levels of epithelial cell fusion were detected in B and T cell transplanted mice (n=4 each). Not surprisingly, the characteristically fusogenic macrophage(23) displayed the most robust cell fusion (n=14), resulting in levels that closely resembled WBM transplanted intestines (Fig.2c-d). Because the macrophage population is functionally diverse, we wondered if activated macrophages possessed different fusogenic capabilities compared to those isolated from WBM and peripheral blood or from those grown in culture. Interestingly, we found no difference in levels of cell fusion between these three macrophage populations (not shown). Although this might suggest that the type of macrophage is not important in cell fusion, it is likely that once transplanted into the recipient mouse, different macrophage populations are appropriately activated and thereby recruited for tumor cell fusion.
Figure 2. Macrophages are primarily responsible for fusion with injured intestinal epithelium.

a, Schematic representation of experimental design. Whole bone marrow or FACS-isolated blood populations were transplanted from GFP-expressing mice into lethally irradiated wild-type ROSA26 mice. b, Hematopoietic cell lineages. Red boxes indicate the isolated cell populations used for transplantation. c, Macrophage fusion with the intestinal epithelium in a ROSA26 mouse transplanted with 2×105 GFP-positive sorted macrophages. Single plane confocal microscopy image of GFP (green) and β-galactosidase (red) detected by antibodies demonstrates fusion by co-localization in yellow (bracket). Dashed white lines indicate epithelial/mesenchymal border. Bar=25μm. d, Fusion was observed with all transplanted lineages, however macrophage fusion was significantly more robust than in the B and T cell transplanted animals. (P<0.01, values reported with s.e.m., n is reported in text).
Macrophages are known to be actively recruited to the site of injury(24), and in this context may be stimulated to fuse with injured epithelium. Our previous work established that GFP-expressing BMDCs transit to the intestine after irradiation injury, just prior to detection of epithelial fusion(3). This suggests that specific pre-fusion mesenchymal actions facilitate events leading to fusion, including crossing of the basement membrane into the epithelial compartment. A detailed time course examining the arrival of GFP-expressing transplanted BMDCs into the intestine revealed a clustering of cells around the intestinal stem cell niche, forming a pre-fusion cluster, 4 days post-transplantation (Supplementary Fig.5). Confocal microscopy (Fig.3 a-g) and 3-dimensional reconstruction of individual intestinal crypts from 50μm thick tissue sections revealed that these BMDCs are juxtaposed to the basement membrane adjacent to the epithelial compartment (Supplementary Movie 1). Interestingly, all three lineages (macrophages, B and T cells) were present in these donor-marked cells clustered around the crypts (Fig.3 h-i; Supplementary Fig.6). Further, analysis of confocal serial planes through pre-fusion clusters surrounding intact crypts revealed rare instances where GFP-expressing macrophages were straddling or crossing the laminin-marked basement membrane, protruding into the epithelial space 4 days after transplantation (Supplementary Fig.7). In these confocal images, macrophages expressing GFP in the cytoplasm were identified by cell-surface F4/80 expression on both sides of the laminin-marked boundary delineating epithelium from mesenchyme in both the upper crypt and the lower stem cell region. While intraepithelial lymphocytes are located on the epithelial side of the basement membrane, macrophages are not known to reside within the epithelial compartment. It is possible that our data represents a snapshot of a macrophage in the process of antigen sampling, in which they have been described to extend lamellipodia across the basement membrane and between epithelial cells(25). However, active transit into the epithelial compartment is a likely prerequisite for fusion with the epithelial cell, therefore our data demonstrate the capacity for the macrophage to position itself in the proper location. While our focus on the macrophage does not exclude the possibility that B and T cells can also fuse, our limited-lineage transplantation analysis suggested that epithelial cell fusion involving these lymphocytes is significantly less frequent than with macrophages (Fig.2c). Regardless, capturing the act of pre-fusion macrophages invading the epithelial compartment supports our evidence that it is a primary partner for cell fusion with the intestinal epithelium.
Figure 3. Pre-fusion clusters of BMDCs contain macrophages.
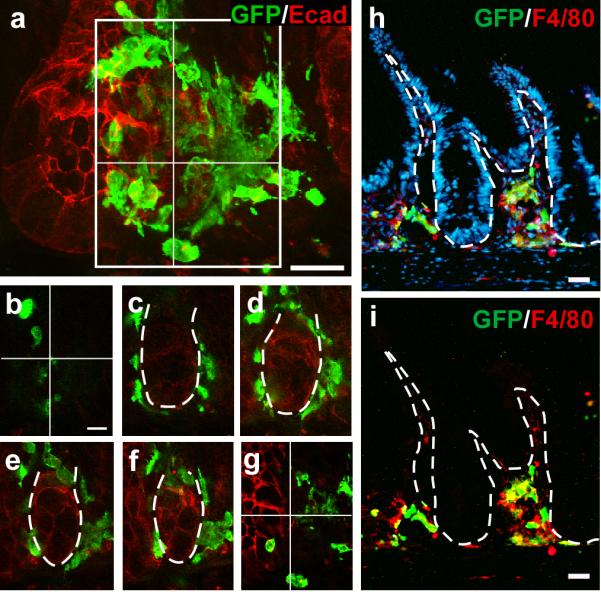
a, 50μm tissue section and 3-dimensional crypt reconstruction of a confocal Z-stack image illustrating GFP-positive BMDCs (green) surrounding the crypt/stem cell niche. Epithelial cells are marked with E-cadherin (red). b-g, Sequential sections through the Z-stack shown in (a). h-i, Pre-fusion cell clusters contain F4/80-positive macrophages (red) consisting of both donor-derived (yellow) and non-donor-derived cells. Nuclei are stained with Hoechst (h; blue). Dashed white lines indicate epithelial/mesenchymal border. Bars=25μm.
Epithelial cell fusion hybrids retain a macrophage identity
Identification of in vivo intestinal epithelial cell fusion partners provides important insight into the physiologic behavior of disease-generated cell fusion hybrids. Initial characterization of intestinal cell fusion hybrids revealed retention of an epithelial phenotype based upon their physiologic location, their epithelial protein expression, and the lack of the pan-lymphocytic donor marker CD45 expression(4). Additionally, in the experiments conducted here, cell fusion hybrids resided within the epithelial compartment and were indistinguishable from adjacent epithelial cells by H&E staining (Supplementary Figure 8). Evidence that the cell fusion hybrids divide and contribute to the intestinal epithelium is supported by GFP-expressing differentiated epithelial cells juxtaposed the stem cell compartment in our previously published data(3). Further, co-cultured MC-38H2BmRFP colon cancer cells and GFP-expressing macrophages displayed fusion readily detected by live-imaging (Figure 4a and Supplementary Movie 2). The fused cell harbors both nuclear and cytoplasmically localized GFP (green) and an RFP-expressing nucleus (yellow). Over the course of imaging (1.25h), the cell fusion hybrid functionally divides into two daughter cells, both retaining the dual markers, suggesting that cell fusion hybrids can contribute to the overall epithelial population. More importantly, these cell fusion hybrids identified by dual marker approaches do not represent macrophage phagocytosis of cancer cells where the RFP-expressing nucleus would be localized to phagosomes and rare division of the macrophage would not result in retention of the RFP-expressing nucleus.
Figure 4. Epithelial cell fusion hybrids express macrophage-specific markers.

a, Static images from a video of a cell fusion hybrid cell expressing both cytoplasmic GFP and nuclear H2BmRFP, undergoing mitosis. Each time point shows a maximum intensity projection of 5 Z-sections acquired in 2-μm intervals. b, The macrophage-specific protein F4/80 (red) is detectible in both the mesenchyme and epithelial compartment 7 days post-transplantation. c, F4/80-positive epithelium also expresses GFP (green), suggesting they are fusion products of donor-derived cells (yellow). Arrowheads indicate examples of fusion hybrid cells. Bars = 25 μm. d, qRT-PCR analysis of isolated GFP-positive epithelia (fusion hybrids) 4 weeks after transplantation revealed a significant expression of the macrophage marker F4/80, when compared to the adjacent GFP-negative epithelia P<0.01, values reported with s.e.m. for n=3; whereas the epithelial specific marker E-cadherin was expressed at a similar level .
Notably, these hybrid cells retain the transgenic donor marker, GFP, suggesting that some of the contributing donor cell transcriptome is preserved. To further explore this exciting possibility, newly generated cell fusion hybrids were assayed for expression of the macrophage-specific gene, F4/80. This membrane glycoprotein was co-expressed in GFP-expressing hybrid cells within the intestinal crypt shortly after fusion hybrids were first detectable (Fig.4b-c). Interestingly, although F4/80 is a cell surface protein in macrophages, it appeared to be localized to the cytoplasm in the epithelial-macrophage fusion cells. This reveals a potential inability of the epithelial-like cell fusion hybrid to traffic this protein to the proper macrophage location. At four weeks post-transplantation however, cell fusion hybrids lose the ability to express F4/80 protein (data not shown), but retained mRNA expression (Fig.4d), as detected by quantitative reverse transcriptase-PCR (qRT-PCR) in FACS-isolated GFP-positive cell fusion hybrids (Supplementary Fig.9). Although hybrid cells expressed a similar amount of the epithelial-specific transcript, E-cadherin, when compared to the adjacent GFP-negative wild-type epithelium, they expressed a statistically significant higher level of the F4/80 transcript (Fig.4d, n=3). It is possible that dynamic regulation of macrophage-specific genes occurs within the cell fusion hybrids, such that macrophage-specific gene expression is temporally modulated. Importantly, the hybrid cells retain the ability to express elevated F4/80 transcript levels even 4 weeks after fusion, suggesting that long-term reprogramming at the stem or progenitor level has occurred. Intriguingly, novel gene expression within this population of fused cells may have significant impact on cellular physiology and subsequent cellular behavior.
Acquired gene expression is an important step in tumor progression(26). Therefore, to determine if epithelial cells can acquire a broader macrophage-specific gene expression profile as a result of cell fusion, we compared the transcriptome profiles of three FACS-isolated populations: unfused intestinal epithelial cells (n=3 animals), unfused macrophages (n=3 animals) and epithelial-macrophage cell fusion hybrids (n=4 animals). To generate an unbiased and comprehensive analysis of the complete transcriptome, we used RNA-Seq(27), a genome analysis approach previously validated by microarray(28). Rigorous isolation procedures were followed to ensure purity of FACS-isolated populations (Supplementary Fig.10).
Comparative transcriptome analysis revealed that of ~20,000 transcripts analyzed, 20.8%, were differentially regulated between at least two populations at a statistically significant level (p<0.01). Of these differentially regulated transcripts, 32.8% were similarly regulated between cell fusion hybrids and wild-type intestinal epithelium (Fig.5a). Intriguingly, 4.0% of differentially-regulated transcripts were shared between cell fusion hybrids and blood-derived macrophages (Fig.5b). Most compelling was that 3.4% of the differentially regulated transcripts were uniquely expressed in the cell fusion hybrids, relative to either of the parental lineages (Fig.5c). To further validate the distinct transcriptome profiles, qRT-PCR expression analysis was employed (Fig.5d-f) on a subset of genes. Confirmation of the RNA-Seq results established that cell fusion hybrids retained transcriptome characteristics from both parental lineages while also developing an additional novel transcriptome profile, unique from either parental lineage. Cell fusion hybrids largely shared a similar gene expression pattern with the epithelial transcriptome; this is not surprising since these cells are morphologically epithelial in nature(4). However, retention of a macrophage-like transcriptome within hybrid cells provides the exciting possibility that these newly generated cells have acquired distinct physiologic potential to participate in disease progression. Interestingly, a number of genes know to be modulated in metastasis were similarly transcriptionally altered within the cell fusion hybrid population, lending support to the idea that cancer cells can gain metastatic capability through cell fusion (Supplementary Table 3). Significantly, a third subset of genes was uniquely expressed in cell fusion hybrids relative to expression within the epithelial and macrophage populations (Fig. 5c). This subset of genes suggests that some transcripts may be modulated in response to cell fusion. It is exciting to speculate that these genes may have important consequences for tumorigenesis and can facilitate specific identification of the tumorigenic cell fusion hybrid population as well as rational therapeutic targets.
Figure 5. Cell fusion hybrid epithelia express a unique transcriptome.

a-c, Heat maps illustrating subsets of cell fusion hybrid (CFH) Ref-Seq genes identified by RNA-Seq that harbor shared or unique transcriptional profiles with wild-type epithelium (EPI) and macrophages (MAC). d-e, Transcript expression from (a-c) confirmed by qRT-PCR. Three categories were confirmed: genes shared by CFH and EPI (d), genes shared by CFH and MAC (e) and genes unique to CFH (f). Data is represented as fold change of triplicate samples ± s.e.m. normalized to an internal reference gene and relative to expression in the CFH population.
Our data builds upon the demonstration that in vitro cell fusion can lead to transcriptional changes(29,30) by presenting a comprehensive, in vivo transcriptional analysis of cell fusion hybrids. The intriguing finding that products of cell fusion are genetically distinct from their parental populations provides mechanistic evidence for how tumor cells may acquire genetic heterogeneity. Additionally, our data illustrating cell fusion between tumor epithelium and macrophage populations provide an exciting explanation for how tumor cells gain the physical macrophage-attributed properties involved in tumor metastasis such as extravasation, migration and immune evasion. While the concept of fusion between tumor cells and blood cells as a mechanism for tumor progression was first proposed in 1911(1), the physical evidence for in vivo cell fusion driving tumorigenesis is only now emerging. Perhaps the best evidence to date for cell fusion in cancer comes from cell culture studies where donor gene expression can be detected in cancer cell fusion hybrids(30,31). In vivo examples of cell fusion in cancer have been observed in melanoma with acquisition of a myeloid-associated enzymatic activity and in renal cancer where detection of a donor Y-chromosome was detected in a female patient with renal carcinoma after bone marrow transplantation(32). However, presence of a donor-marker does not fully demonstrate the breadth of phenotypic alterations where cell fusion can lead. Importantly, our results demonstrating that in vivo-generated cell fusion hybrids can acquire macrophage transcriptional properties provide a critical piece of evidence supporting the impact of cell fusion on tumor progression. Our data provides a means by which tumor cells have the potential to acquire the ability to harness macrophage-specific properties including migration to a pre-metastatic niche(33) and evasion of the immune system. Although the mechanism for cell fusion requires further investigation, we show that it provides the basis for novel transcriptional expression patterns in tumor cells. Further, cell fusion, in a fashion similar to Darwin's theory of evolution, may allow for cells that have acquired distinct genetic changes to survive and adapt to the tumor microenvironment. This exciting possibility not only presents a potential paradigm shift in how we perceive metastatic spread, but opens new possibilities for inhibiting tumor-associated cell fusion as an additional preventative or therapeutic means.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Wong laboratory members for critical discussions of our work, Alexis Bailey for help with progenitor FACS-isolation, and Mandy Boyd and Pam Canaday for excellent assistance in FACS. This work was supported by MHW:R01DK068326, R01CA118235, AEP:T32HD409309, ECA:T32HL00781 and PSD:T32CA106195.
Footnotes
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
REFERENCES
- 1.Pawelek JM. Tumour-cell fusion as a source of myeloid traits in cancer. The lancet oncology. 2005;6(12):988–93. doi: 10.1016/S1470-2045(05)70466-6. [DOI] [PubMed] [Google Scholar]
- 2.Duelli D, Lazebnik Y. Cell fusion: a hidden enemy? Cancer cell. 2003;3(5):445–8. doi: 10.1016/s1535-6108(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 3.Davies PS, Powell AE, Swain JR, Wong MH. Inflammation and proliferation act together to mediate intestinal cell fusion. PloS one. 2009;4(8):e6530. doi: 10.1371/journal.pone.0006530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi AZ, Swain JR, Davies PS, et al. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells.. Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 6321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8(5):377–86. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 6.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature genetics. 1999;21(1):70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 7.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68(5):869–77. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 8.Moser AR, Pitot HC, Dove WF. Science. 4940. Vol. 247. New York, NY: 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. pp. 322–4. [DOI] [PubMed] [Google Scholar]
- 9.Nakanishi T, Kuroiwa A, Yamada S, et al. FISH analysis of 142 EGFP transgene integration sites into the mouse genome. Genomics. 2002;80(6):564–74. doi: 10.1006/geno.2002.7008. [DOI] [PubMed] [Google Scholar]
- 10.Willenbring H, Bailey AS, Foster M, et al. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nature medicine. 2004;10(7):744–8. doi: 10.1038/nm1062. [DOI] [PubMed] [Google Scholar]
- 11.Wong MH, Rubinfeld B, Gordon JI. Effects of forced expression of an NH2-terminal truncated beta-Catenin on mouse intestinal epithelial homeostasis. The Journal of cell biology. 1998;141(3):765–77. doi: 10.1083/jcb.141.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies PS, Dismuke AD, Powell AE, Carroll KH, Wong MH. Wnt-reporter expression pattern in the mouse intestine during homeostasis. BMC gastroenterology. 2008;8:57. doi: 10.1186/1471-230X-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storey J. A direct approach to false discovery rates. Journal of the Royal Statistical Society, Series B. 2002;64:479–798. [Google Scholar]
- 14.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson DA, Wu Y, Jiang S, et al. Donor marker infidelity in transgenic hematopoietic stem cells. Stem cells (Dayton, Ohio) 2005;23(5):638–43. doi: 10.1634/stemcells.2004-0325. [DOI] [PubMed] [Google Scholar]
- 16.Nygren JM, Liuba K, Breitbach M, et al. Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nature cell biology. 2008;10(5):584–92. doi: 10.1038/ncb1721. [DOI] [PubMed] [Google Scholar]
- 17.Johansson CB, Youssef S, Koleckar K, et al. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nature cell biology. 2008;10(5):575–83. doi: 10.1038/ncb1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91(5):661–72. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 19.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 20.Coffman RL. Surface antigen expression and immunoglobulin gene rearrangement during mouse pre-B cell development. Immunological reviews. 1982;69:5–23. doi: 10.1111/j.1600-065x.1983.tb00446.x. [DOI] [PubMed] [Google Scholar]
- 21.Zwadlo G, Brocker EB, von Bassewitz DB, Feige U, Sorg C. A monoclonal antibody to a differentiation antigen present on mature human macrophages and absent from monocytes. J Immunol. 1985;134(3):1487–92. [PubMed] [Google Scholar]
- 22.Godfrey DI, Kennedy J, Mombaerts P, Tonegawa S, Zlotnik A. Onset of TCR-beta gene rearrangement and role of TCR-beta expression during CD3-CD4-CD8-thymocyte differentiation. J Immunol. 1994;152(10):4783–92. [PubMed] [Google Scholar]
- 23.Chen EH, Grote E, Mohler W, Vignery A. Cell-cell fusion. FEBS Lett. 2007;581(11):2181–93. doi: 10.1016/j.febslet.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 24.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury.. Proceedings of the National Academy of Sciences of the United States of America; 2005. pp. 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31(3):368–76. doi: 10.1016/j.immuni.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. 2004;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–82. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 29.Palermo A, Doyonnas R, Bhutani N, Pomerantz J, Alkan O, Blau HM. Nuclear reprogramming in heterokaryons is rapid, extensive, and bidirectional. Faseb J. 2009;23(5):1431–40. doi: 10.1096/fj.08-122903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakraborty AK, Pawelek J, Ikeda Y, et al. Fusion hybrids with macrophage and melanoma cells up-regulate N-acetylglucosaminyltransferase V, beta1-6 branching, and metastasis. Cell Growth Differ. 2001;12(12):623–30. [PubMed] [Google Scholar]
- 31.Rupani R, Handerson T, Pawelek J. Co-localization of beta1,6-branched oligosaccharides and coarse melanin in macrophage-melanoma fusion hybrids and human melanoma cells in vitro. Pigment cell research / sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. 2004;17(3):281–8. doi: 10.1111/j.1600-0749.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 32.Yilmaz Y, Lazova R, Qumsiyeh M, Cooper D, Pawelek J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: visualization of putative BMT-tumor hybrids by FISH. Bone marrow transplantation. 2005;35(10):1021–4. doi: 10.1038/sj.bmt.1704939. [DOI] [PubMed] [Google Scholar]
- 33.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9(4):285–93. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.