

Abstract
Background
The mechanism by which heat sensitizes mammalian cells to ionizing radiation remains to be elucidated. We determined whether base excision repair (BER) is involved in heat-radiosensitization and report novel findings that provide insight regarding the role of BER in the radiation response of HeLa cells.
Materials and Methods
An siRNA approach was utilized to suppress expression of AP endonuclease (Ape1), a critical enzyme of BER. Clonogenic survival curves were obtained for HeLa cells expressing normal or reduced Ape1 content and which had been irradiated, and these were compared to survival curves from cells that were irradiated prior to hyperthermia treatment.
Results
The amount of heat-radiosensitization observed in Ape1-suppressed cells was similar to or slightly greater than that observed in cells expressing near-normal levels of Ape1. Interestingly, we also found that for unheated HeLa cells, suppressed expression of Ape1 resulted in enhanced resistance to X-rays.
Conclusions
The data suggest that Ape1, and therefore BER, is not involved in heat-radiosensitization. However, the observation that suppressed expression of Ape1 results in enhanced radioresistance supports the notion that BER may be detrimental to the survival of irradiated cells.
Keywords: Radiation, hyperthermia, DNA repair
Ionizing radiation (IR) induces a myriad of lesions in DNA. These lesions include various species of oxidative base damage and single- and double-strand breaks (SSB and DSB, respectively). While the DSB is believed to be the primary lesion responsible for radiation-induced cell killing, the majority of lesions are clustered damaged bases (1).
Removal of damaged bases resulting from insults such as IR is effected by the base excision repair (BER) pathway. In irradiated cells, removal of the base by DNA glycosylases results in the formation of an AP site. Subsequent to this, AP endonuclease 1 (Ape1) nicks the damaged DNA strand upstream of the AP site, creating a 3′-hydroxyl terminus and a 5′deoxyribose phosphate group flanking the gap. Thus, Ape1 is a 3-phosphodiesterase that can initiate repair of AP sites resulting from radical damage or glycosylases (2). Ape1 is associated with nearly all AP site excision activity [see (3) for a review of Ape1] and may also play a role in repairing SSBs containing 3′-8-oxoG as well as other oxidative lesions repaired via BER (4).
The Ape1 protein is essential, as mice nullizygous for Ape1 are embryonic lethal (5) and Ape1-deficient cell lines have not been isolated (6). However, nullizygous embryo fibroblasts have been created using a floxed Ape1 gene that resulted in an apoptotic response within 24 h after removal of the Ape1 gene (7). Thus, Ape1 appears to be the central protein involved in BER. Other functions unrelated to its role in DNA repair include redox activation of stress-inducible transcription factors and transcriptional repression (3).
When bacteria or mammalian cells are irradiated, some sites of clustered base damage may give rise to DSBs during BER if the sites are located within a few base pairs on opposite strands (8, and references therein). Therefore, BER may be deleterious to the survival of the cell after irradiation (8). However, evidence of a role for BER in the radiation response of mammalian cells is conflicting. While some studies have demonstrated the involvement of BER proteins in the repair of radiation damage, other studies have failed to establish a correlation with radiosensitivity (9). In regard to the relationship between Ape1 expression and sensitivity to IR, some studies have shown a positive correlation, but many have not [see (2) for a review]. While the role of BER in the radiation response of mammalian cells is somewhat incompletely understood, Ape1 is critical for BER and continued viability.
Hyperthermia sensitizes cells to IR (10). While heat-radiosensitization is often attributed to inhibition of DNA repair, the specific repair pathway(s) or proteins involved in heat-radiosensitization are not yet known with any certainty. Circumstantial evidence suggests the involvement of the BER pathway in heat-radiosensitization (11), but direct evidence is lacking. Thus, the role of BER in heat-radiosensitization remains unresolved. In this report, we utilized an siRNA approach to lower Ape1 expression prior to irradiation and heat treatments, and compared the extent of heat-radiosensitization in HeLa cells expressing normal amounts of Ape1 with cells deficient in Ape1.
Materials and Methods
Cell culture
HeLa CCL2 cells were grown in monolayer in McCoy's 5A media (Mediatech, Inc., Herndon, VA, USA) with 10% iron-supplemented calf serum (ISCS) (Hyclone, Inc., Logan, UT, USA) and maintained in a 5% CO2 atmosphere at 37°C. Twenty-four hours prior to transfection with siRNA or scRNA, 2 × 105 cells were plated into 25 cm2 tissue culture flasks containing 5 ml of medium supplemented with 10% ISCS.
Transfection and quantification of knockdown using Western blotting
Ape1 siRNA or scRNA, dissolved in water, was added to serum-free media and incubated for 5 min at room temperature (RT). APE1 siRNAs used to decrease AP endonuclease were obtained from Dharmacon Research, Inc. (Lafayette, CO, USA), deprotected and hybridized according to the manufacturer's directions. Sequences of the double stranded siRNAs are antisense (5′ GUCUGGUACGACUGGAGUACC 3′, 5′ UACUCCAGUCGUACCAGACCU 3′) and nonsense (5′ CCAUGAGGUCAGCAUGGUCUG 3′, 5′ GACCAUGCUGACCUCAUGGAA 3′). Scrambled (sc) APE1 siRNAs were used as negative controls. Final siRNA or scRNA concentration in medium was 40 nM. The sequences of scRNA were the reverse of the siRNA sequences. In a separate tube, 10 μl of Oligofectamine (Invitrogen, Carlsbad, CA USA) was incubated at RT for 5 min in serum-free media. After incubation, the aliquot of Oligofectamine was added to the siRNA or scRNA, and the medium was incubated for 20 min at RT before addition to monolayer cultures in 25 cm2 flasks. Prior to adding transfection reagents, monolayers were washed with serum-free media. After addition of transfection reagents, cells were incubated at 37°C for 4 h. Media was then replaced with McCoy's 5A with 10% ISCS and cells were incubated for 48 h. To determine levels of Ape1 using Western blotting, cells were trypsinized and washed once in phosphate- buffered saline (PBS) and Buffer A [10 mM HEPES, pH 7.9, 1.5 MgCl2, 10 mM KCl, 1 mM (PMSF), 2 μg/ml leupeptin], and resuspended in 1× Laemmli's Buffer. After heating (10 min, 100°C), the lysates were passed thrice through a 26-gauge syringe. Protein content was quantified and 15 μg of protein was loaded into lanes of a 10% SDS-polyacrylamide gel. Samples were electrophoresed at 125 V for 15 min, and then at 200 V for 45 min. Proteins were then transferred to nitrocellulose at 100 V for 1 h. Membranes were blocked at RT for 2 h with blotto [50 mM Tris (pH 7.5) 0.185 mM NaCl, 0.05% Tween-20, 3% nonfat dry milk]. Ape1 monoclonal antibody developed in our laboratory (Novus Biologicals, Littleton, CO, USA) was diluted in blotto and incubated with the membrane overnight at 4°C (1:1000). Membranes were washed thrice in blotto and incubated with secondary antibody (Amersham Biosciences, Piscataway, NJ, USA) (1:2000 anti-mouse-HRP in blocking solution) for 1 h at 4°C. Finally, membranes were washed thrice with blotto and twice with PBS before chemiluminescent detection of Ape1.
Heat and radiation treatments and analysis of clonogenic survival
After incubating for 48 h at 37°C, media were changed, flasks were placed on ice for 5 min and cells were irradiated on ice with 250 kVp x-rays (total time on ice was 15 min). After irradiation, media were replaced with fresh 37°C media. For some samples, cells were then heated for 2 h at 41.5°C. Following irradiation or irradiation and heating, cells were trypsinized, counted and plated for clonogenic survival. Cells were allowed to grow for 14-21 days at 37°C. Cells were then fixed with a 3:1 methanol/acetic acid mixture and stained with crystal violet to allow scoring of colonies. Only colonies with ≥50 cells were included in the survival analysis.
Results
Knockdown of Ape1 in HeLa CCL2 cells
To determine the role of Ape1 in the radiation response and heat-radiosensitization of HeLa cells, we first determined optimal parameters for lowering Ape1 expression. Ape1 protein expression was determined by Western blotting for untransfected, scRNA- or siRNA-transfected cells (Figure 1). Maximum suppression (with only minimal concomitant toxicity) of expression was observed after 48 h. By 48 h after transfection with Ape1 siRNA, Ape1 expression was routinely found to be within 25-30% of levels found in cells transfected with scRNA. Cells transfected with scRNA generally showed levels of Ape1 that were 90-95% of that found in cells that were not transfected with either siRNA or scRNA, but were only incubated with Oligofectamine.
Figure 1.
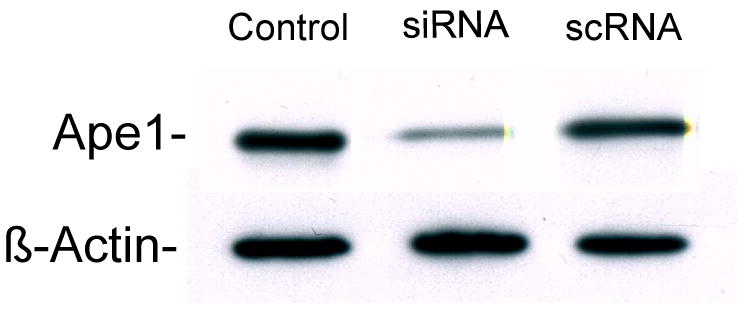
Representative Western blot showing the level of expression of Ape1 protein 48 h after transfection of HeLa CCL2 cells. Cells were transfected and processed for SDS-PAGE and Western blotting as described in Materials and Methods. Membranes were probed with anti-Ape1 antibody. β-actin was used as a loading control. The lane denoted as Control represents cells that were incubated with Oligofectamine, but not transfected. Cells transfected with scRNA showed Ape1 expression that was approximately within 95% of expression observed in untransfected control cells. Cells transfected with siRNA showed Ape1 expression that was approximately only 22% of that found in untransfected control cells, and expression in siRNA-transfected cells was reduced to 25-30% of that found in scRNA-transfected cells.
Radiation response of Ape1-suppressed cells
Prior to determining the role of Ape1 and BER in heat-radiosensitization, it was necessary to determine the consequences of reduced Ape1 expression on the survival of irradiated cells. Cells transfected with scRNA that expressed near-normal levels of Ape1, or cells transfected with Ape1 siRNA that expressed greatly reduced levels of Ape1 were exposed to various doses of x-rays. The resulting survival curves are shown in Figure 2. A computer program was used to calculate parameters of the cell survival curves using the “multi-target, single-hit model” to fit the data sets (12). Mean lethal dose (D0), extrapolation number (n), and quasi-threshold dose (Dq)[the latter two parameters being a measure of the shoulder region of the survival curve indicative of the ability of cells to accumulate or repair sublethal damage (13)], were calculated. Although the D0 doses for cells transfected with scRNA or siRNA were nearly identical (98.1 cGy vs. 97.5 cGy, respectively), cells with reduced Ape1 content were significantly more radioresistant to low doses of x-rays, as the survival curve had a well-defined shoulder compared to that for cells with near-normal levels of Ape1. Cells transfected with scRNA yielded a radiation survival curve with almost no shoulder. The Dq and n values for cells with reduced Ape1 content were 173 cGy and 4.7, respectively; Dq and n values for cells with near-normal Ape1 content were 74 cGy and 2.1, respectively. The survival curve for cells transfected with scRNA was similar to the curve obtained for non-transfected cells, and this was also reflected by the survival curve parameters (D0=100 cGy, Dq= 106 cGy, n= 2.9).
Figure 2.

Role of Ape1 in the radiation response of HeLa cells. Cells were transfected with Ape1 siRNA or scRNA and incubated for 48 h at 37°C. Cells were then irradiated with x-rays prior to plating for clonogenic survival. Ape1 expression in siRNA-transfected cells in each experiment was reduced to 25-30% of scRNA-transfected cells at the time of irradiation. A survival curve is also included for non-transfected cells (Control). The average plating efficiencies for each sample were: siRNA 0.11; scRNA 0.14; siRNA+heat 0.11; scRNA+heat 0.15; untransfected cells 0.36. Error bars represent the standard error of the mean (SEM) of at least three experiments. The survival curve for irradiated cells treated only with oligofectamine was nearly superimposable with that of untreated (untransfected) cells or cells transfected with scRNA (data not shown), indicating that Oligofectamine did not alter radiosensitivity.
We were concerned that cells rendered deficient in Ape1 might have become abnormally distributed throughout the cell cycle prior to irradiation. Synchronization, blockage, or lengthening of the transit time of cells in the G1 phase, if it occurred, could explain the observed radioresistance of cells which were knocked down for Ape1 just prior to irradiation, since cells in G1 are much more radioresistant compared to cells in G2 (14). However, flow cytometric analysis of cell cycle distributions of unirradiated cells which had been transfected with siRNA or scRNA for 48 h revealed only an 8% difference in the G1 phase content between the two populations of cells (siRNA=54% G1, scRNA=46%; average of two experiments, data not shown). This difference cannot account for the decrease in radiosensitivity (enhanced radioresistance) of cells with reduced Ape1 content.
Role of Ape1 in heat-radiosensitization
We next determined whether Ape1 content was a determinant in heat-radiosensitization. Cells transfected with siRNA which under-expressed Ape1 and cells transfected with scRNA which expressed Ape1 at near-normal levels were irradiated and surviving fractions were compared to those for siRNA- and scRNA-transfected cells that were irradiated and then heated for 2 h at 41.5°C. Survival curves for siRNA- and scRNA-transfected cells exposed to x-rays or x-rays and hyperthermia treatment are shown in Figure 3. Cells in which Ape1 was suppressed were more resistant to the combined treatment than cells expressing near-normal levels of Ape1. This trend was similar to that obtained for x-rays alone. Flow cytometric analysis of the G1-, S- and G2/M- phase DNA content of each population after heating indicated less than a 5% difference between each cell cycle phase of the respective populations (siRNA: G1=44%, S=32%, G2=24%; scRNA: G1=46%, S=27%, G2=27%, data not shown). Thus, again, the differences observed for Ape1-suppressed cells and cells with near-normal Ape1 content that were both irradiated and heated cannot be accounted for on the basis of cell cycle perturbations.
Figure 3.

Role of Ape1 in heat-radiosensitization. HeLa cells were transfected and irradiated, or irradiated and heated at 41.5°C for 2 h prior to plating for clonogenic survival. Ape1 expression in siRNA-transfected cells in each experiment was reduced to 25-30% of scRNA-transfected cells at the time of irradiation. Error bars represent the SEM of at least three experiments. Survival curves for irradiated and heated untransfected control cells or cells treated only with Oligofectamine were superimposable with the curve for scRNA-treated cells (data not shown).
When clonogenic survival curves obtained from irradiated vs. irradiated and heated Ape1-suppressed cells and those expressing near-normal levels of Ape1 are compared, the amount of heat-radiosensitization observed in suppressed cells is similar to or slightly greater than that observed in cells expressing near-normal levels of the protein (Figure 3). For example, at a surviving fraction of 0.01, the thermal enhancement ratio (TER) for siRNA-transfected cells is 1.34, while the TER for scRNA-transfected cells is 1.2. This result indicates that Ape1 is not involved in heat-radiosensitization. If Ape1 were a target for, or played a role in heat-radiosensitization, the survival curves of irradiated vs. irradiated and heated siRNA-transfected cells, when compared, would be superimposable, or nearly so; that is, a significant reduction of radiosensitization would have been observed in Ape1-suppressed cells compared to cells expressing near-normal amounts of Ape1.
Discussion
A significant proportion of the DNA damage induced by IR is repaired by BER. Several investigators have sought to determine the relationship between Ape1 content and sensitivity to IR in an attempt to examine the role of BER in the radiation response of mammalian cells. Unfortunately, the data are conflicting and no definitive conclusion has been reached. Several studies have shown little or no correlation between Ape1 expression and radiosensitivity, while some investigators have demonstrated a relationship (15-20). For example, tumors and cultured germ cells expressing high levels of Ape1 expression were reported to be less radiosensitive (15). Additionally, we previously showed that knocking down Ape1 in osteosarcoma cell lines increases sensitivity to IR (16). Chen and Olkowski (20) used an antisense approach to reduce Ape1 expression in human lung carcinoma cells to less than 50% of normal levels, and found a corresponding dramatic increase in radiosensitivity when clonogenic survival was measured. Interestingly, while the survival curve of control cells showed a significant shoulder, there was no shoulder for the Ape1-depleted cells; survival appeared to be an exponential function of radiation dose. Since lack of a shoulder usually corresponds to an inability of cells to accumulate or repair sublethal damage, the authors concluded that Ape1 plays a major role in the repair of IR-induced DNA damage.
The aforementioned studies are difficult to reconcile with our current data which indicate that cells with greatly reduced Ape1 levels are more resistant to IR than cells with near-normal levels of Ape1 (Figure 2). In contrast to data from Chen and Olkowski (20), we observed almost no shoulder in survival curves obtained from cells expressing near-normal levels of the protein. It is possible that the relationship between radiosensitivity and Ape1 expression may be dependent on cell type. We therefore attempted to demonstrate, using a second cell line routinely grown in our laboratory (U-1 human melanoma), that greatly reduced expression of Ape1 would result in decreased radiosensitivity. Unfortunately, we could not suppress the level of Ape1 in this second cell line, even after transfection with higher concentrations of Oligofectamine and a 10-fold greater concentration of siRNA compared to that which was used in our HeLa cell experiments (data not shown). However, it is also possible that differences in the efficiency, extent or duration of knockdown of Ape1 or the choice of assays used to measure radiosensititivity could account for the differences reported. For example, use of the MTT assay or measurement of apoptotic fraction as an indicator of cell killing (as used in other reports) could greatly underestimate the total cell death in a population of normal or Ape1-depleted cells.
While enhanced radioresistance associated with suppressed Ape1 expression may appear counterintuitive, our data are not surprising when considering the nature of IR-induced damage and how it is repaired. About 70% of DNA damage mediated by x-rays is attributed to free radicals produced by the radiolysis of water (21). The lesions produced, which predominantly consist of oxidized bases or apurinic/apyridimic (AP) sites, exist within damage clusters with radii up to 4 nm (1). Most of these lesions are repaired by BER. Weinfeld and co-workers used an in vitro system to show that DSBs may result at closely opposed base damage sites from the action of DNA glycosylases [22]. The BER pathway may also produce potentially lethal DSBs in vivo during the attempted repair of clustered free radical damage (8). Escherichia coli that were devoid of DNA glycosylases formed fewer strand breaks and were found to be more resistant to radiation-induced killing than wild-type cells, which continued to accumulate DSBs post-irradiation as a result of ongoing BER. Thus, at a clustered damage site containing damaged bases within a few base pairs on opposite strands, the formation of potentially lethal DSBs may occur should there be incision of opposite strands by enzymes recognizing the base damage. Ape1 nicks damaged DNA strands upstream of the AP site. If Ape1 normally makes excisions in cells with normal amounts of the enzyme, then DSBs could be generated if the base damage sites happen to be within a few base pairs of each other. These DSBs could be potentially lethal if unrepaired or misrepaired, and conceivably lead to formation of lethal exchange-type chromosome aberrations.
However, if BER were inhibited by the reduction of Ape1 levels resulting in a reduction of nicks produced on opposite strands within clustered damage sites induced by IR, some cells could be spared from killing, through reduction of DSBs produced indirectly through the BER process. Our finding that cells lacking Ape1 are more radioresistant supports this idea, as well as the notion that BER may be detrimental to the survival of mammalian cells exposed to IR. Conversely, there is the possibility that an inhibition of BER could also increase killing; BER may potentially generate intermediates (such as AP sites) which, if incompletely repaired, could be genotoxic in nature. This balance between sparing and killing of irradiated cells likely limits the extent of radioprotection that could be achieved by inhibition of BER.
Alternatively, our finding that cells with reduced Ape1 levels are more radioresistant (Figure 2) could be interpreted to mean that i) BER is not involved in the radioresponse and that several forms of base damage do not play a significant role in radiation-induced cell killing, at least in the HeLa CCL2 cell line, or ii) there is either an Ape1-independent (23) or a redundant pathway for repair of base damage (24) that may be utilized in the absence of BER. Either interpretation is important when considering the potential mechanisms of heat-radiosensitization.
We previously showed that the major non-homologous end joining (NHEJ) DSB repair pathway is not involved in heat-radiosensitization (25). In that study, we found that cells deficient in Ku80 or DNA ligase IV (2 proteins critical for NHEJ) were heat-radiosensitized by the same or a slightly greater amount as compared to wild-type cells. Limited data obtained using a chicken cell model suggest that homologous recombination may also not be involved, although this conclusion is somewhat controversial [see (11), and references therein]. While recent data suggest the existence of alternative pathways of NHEJ, and these pathways could potentially be involved in heat-radiosensitization (26), by deduction, some investigators, citing biochemical and correlative evidence, have proposed that inhibition of BER could be the critical step in thermal radiosensitization. Unfortunately, a lack of mutant cell lines had hampered efforts to definitively establish a role for or against involvement of BER. Using an siRNA approach, we have shown that cells deficient in Ape1 are radiosensitized to hyperthermia to an extent that is similar to cells expressing normal levels of the protein (Figure 3). A reduction in heat-radiosensitization of Ape1-deficient cells would have been suggestive of involvement of Ape1 in heat-radiosensitization. Our data suggest that Ape1 is not involved in heat-radiosensitization. Since Ape1 is critically involved in the BER pathway, by extension, then, our data also strongly suggest that Ape1-dependent BER is not involved in heat-radiosensitization (Figure 3).
Acknowledgments
This work was supported by NIH grants CA108582 (JRD) and CA094025, CA106298, CA114574, Ovar'coming Together and Riley Children's Foundation (MRK). We wish to thank Dr. Susan Wallace for providing helpful comments, Jennifer Lopez for technical support, and Dr. Marc Mendonca for assistance with survival analyses.
References
- 1.Brenner DJ, Ward JF. Constraints on energy deposition and target size of multiply damaged sites associated with DNA double-strand breaks. Int J Radiat Biol. 1992;61:737–748. doi: 10.1080/09553009214551591. [DOI] [PubMed] [Google Scholar]
- 2.Evans AR, Limp-Foster M, Kelley MR. Going Ape over Ref-1. Mutat Res. 2000;46:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 3.Fritz G, Grösch S, Tomicic M, Kaina B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology. 2003;193:67–68. doi: 10.1016/s0300-483x(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 4.Parsons JL, Dianova II, Dianov GL. APE1-dependent repir of DNA single-strand breaks containing 3′-end 8-oxoguanine. Nucleic Acids Res. 2005;33:2204–2209. doi: 10.1093/nar/gki518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA protein Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sc USA. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demple B, Sung J. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair. 2005;4:1442–1449. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Izumi T, Brown DB, Naidu CV, Bhakat KK, MacInnes MA, Saito H, Chen DJ, Mitra S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci USA. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaisdell JO, Wallace SS. Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli. Proc Natl Acad Sci USA. 2001;98:7426–7430. doi: 10.1073/pnas.131077798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vermeulen C, Verwijs-Janssen M, Cramers P, Begg AC, Vens C. Role for DNA polymerase beta in response to ionizing radiation. DNA Repair. 2007;6:202–212. doi: 10.1016/j.dnarep.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Hur E, Elkind MM, Bronk EV. Thermally enhance radioresponse of cultured Chinese hamster cells: inhibition of repair of sublethal damage and enhancement of lethal damage. Radiat Res. 1974;58:38–51. [PubMed] [Google Scholar]
- 11.Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (>43°C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia. 2004;20:131–139. doi: 10.1080/02656730310001627713. [DOI] [PubMed] [Google Scholar]
- 12.Albright N. Computer programs for the analysis of cellular survival data. Radiat Res. 1987;112:331–340. [PubMed] [Google Scholar]
- 13.Elkind MM, Whitmore GF. The Radiobiology of Cultured Mammalian Cells. New York: Gordon & Breach Science Publishers; 1967. [Google Scholar]
- 14.Terasima R, Tolmach LJ. X-ray sensitivity and DNA synthesis in synchronous populations of HeLa cells. Science. 1963;140:490–492. doi: 10.1126/science.140.3566.490. [DOI] [PubMed] [Google Scholar]
- 15.Robertson KA, Bullock HA, Xu Y, Tritt R, Zimmerman E, Ulbright TM, Foster RS, Einhorn LH, Kelley MR. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001;61:2220–2225. [PubMed] [Google Scholar]
- 16.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: Enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 17.Hughes-Davies L, Galanopoulos T, Harrison L, Maxwell M, Antoniades HN, Demple B. Expression of the human apurinic endonuclease gene in normal and malignant tissues. Int J Oncology. 1995;6:749–752. doi: 10.3892/ijo.6.4.749. [DOI] [PubMed] [Google Scholar]
- 18.Ono Y, Matsumoto K, Furuta T, Ohmoto T, Akiyama K, Seki S. Relationship between expression of a major apurinic/appyrimidnic endonuclease (APEX nuclease) and susceptibility to genotoxic agents in human glioma cell lines. J Neurooncology. 1995;2:183–192. doi: 10.1007/BF01053151. [DOI] [PubMed] [Google Scholar]
- 19.Herring CJ, West CM, Wilks DP, Davidson SE, Hunter RD, Berry P, Forster G, Mackinnon J, Rafferty JA, Elder RH, Hendry JH, Margison GP. Levels of the DNA repair enzyme human apurinc/apyrmidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. Br J Cancer. 1998;78:1128–1133. doi: 10.1038/bjc.1998.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen DS, Olkowski ZL. Biological responses of human apurinic endonuclease to radiation induced DNA damage. Ann NY Acad Sci. 1994;726:306–308. doi: 10.1111/j.1749-6632.1994.tb52834.x. [DOI] [PubMed] [Google Scholar]
- 21.Ward JF. Biochemistry of DNA lesions. Radiat Res. 1985;8:S103–111. [PubMed] [Google Scholar]
- 22.Weinfeld M, Rasouli-Nia A, Chaudhry MA, Britten RA. Response of base excision repair enzymes to complex DNA lesions. Radiat Res. 2001;156:584–589. doi: 10.1667/0033-7587(2001)156[0584:robere]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 23.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Wilson DM, III, Barsky D. The major human abasic endonuclaase: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 25.Dynlacht JR, Bittner ME, Bethel JA, Beck BD. The non-homologous-joining pathway is not involved in the radiosensitization of mammalian cells by heat shock. J Cell Physiol. 2003;196:557–564. doi: 10.1002/jcp.10334. [DOI] [PubMed] [Google Scholar]
- 26.Iliakis G, Wu W, Wang M. DNA double strand break repair inhibition as a cause of heat radiosensitization: re-evaluation considering backup pathways of NHEJ. Int J Hyperthermia. 2008;24:17–29. doi: 10.1080/02656730701784782. [DOI] [PubMed] [Google Scholar]