

Abstract
Nitric oxide mediated activation of large conductance calcium-activated potassium (BK) channels is considered an important underlying mechanism of sepsis-induced hypotension. Indeed, the non-selective K channel inhibitor, tetraethylammonium chloride (TEA) has been proposed as a potential treatment to raise blood pressure in septic shock by virtue of its ability to inhibit BK channels. As experimental evidence has so far relied upon pharmacological inhibition, we examined the effects of channel deletion using BKα subunit knockout (α−/−, Slo−/−) mice in two mouse models of polymicrobial sepsis, namely intraperitoneal fecal slurry and cecal ligation and puncture (CLP). Comparison was made against TEA treatment in wildtype (WT) mice. Following slurry, BKα−/− and WT mice developed similar degrees of hypotension over 10 h with no difference in cardiac output as assessed by echocardiography between groups. TEA raised blood pressure significantly in septic WT mice, but had no effect upon survival. However, following CLP, a significantly reduced survival was seen in both BKα−/− mice and (high-dose) TEA-treated WT mice compared to untreated WT animals. In conclusion, the BK channel does not appear to be integral to sepsis-induced hypotension but does impact upon survival through other mechanisms. The pressor effect of TEA may be related to effects on other potassium channels.
Keywords: Large conductance calcium-activated potassium (BK) channels, BKα null mice, sepsis, tetraethylammonium chloride, hypotension, echocardiography, mortality
INTRODUCTION
Severe sepsis is a leading cause of mortality in intensive care (ICU) patients (1). In the UK approximately 27% of adult ICU patients meet severe sepsis criteria within the first 24 hrs of admission and approximately half of these will die (2). Given this high mortality rate, particularly in septic patients with refractory hypotension (3), dissecting the underlying pathophysiological mechanisms is of considerable importance.
Mechanisms implicated in sepsis-induced hypotension include overproduction of nitric oxide (NO), inappropriately low vasopressin levels and excessive potassium (K+) channel activation (4, 5). K+ channels regulate the resting membrane potential of vascular smooth muscle cells and can therefore influence vascular tone and blood pressure. Deletion of pore-forming or regulatory subunits of either the ATP-sensitive (KATP) or the large conductance calcium-activated (BK) channel causes hypertension in mice (6-8). Furthermore, abnormal activation of these channels has been implicated in meditating cardiovascular collapse in shock states (9, 10). The mechanism of channel activation is multi-factorial, but undoubtedly involves NO activation of the cyclic guanosine monophosphate (cGMP) pathway (9). Several animal models of sepsis have demonstrated that KATP channel inhibition reversed hypotension and vascular hyporeactivity to catecholamines (11, 12), although two human studies failed to show any pressor effect using the sulphonylurea inhibitor, glyburide (13, 14). BK channels are also activated following endotoxin (LPS) or peritonitis (15, 16); BK channel inhibition restored vascular responsiveness (17) and reduced LPS-induced mortality (18) when combined with the non-selective soluble guanylate cyclase and partial iNOS inhibitor, methylene blue. In addition, the non-selective K+ inhibitor tetraethylammonium (TEA) completely restored hyporeactivity to norepinephrine in healthy volunteers given endotoxin (19). The study concluded that this occurred via inhibition of the BK channel as the KATP inhibitor tolbutamide had no effect. BK channel inhibition therefore represents a potential therapeutic target for treatment of vascular dysfunction in septic shock.
The BK channel is widely expressed in tissues other than the vasculature (20) so channel inhibitors may have other actions that could prove beneficial (or harmful) in sepsis. Both TEA and paxilline (a more specific BK inhibitor) prevented early LPS-induced macrophage cytokine release (21, 22). However, macrophages from BK gene deleted and wild type (WT) mice demonstrated identical responses to LPS, suggesting that the anti-inflammatory properties of these agents is unrelated to effects on the BK channel (23). Mice have been generated in which the α (pore-forming) and β (regulatory) subunits of the BK have been deleted (7, 8). Knockout of the α subunit completely inhibits channel function, whereas β subunit knockout produces channels that are activated at much higher calcium concentrations (8). Neither has been studied in septic shock models, although the β subunit has been implicated in BK hypersensitivity in hemorrhagic shock (24). However, as NO phosphorylates the α subunit through cGMP-dependent protein kinase or tyrosine protein kinase, and cGMP dependent-relaxation is markedly attenuated in the α knockout, we chose this as the more appropriate knockout to study (7, 25).
We therefore undertook experiments to test (i) if BKα −/− mice would be resistant to sepsis-induced hypotension and (ii) whether this would confer a survival advantage over WT mice. In view of the controversy over whether the putative beneficial actions of BK inhibitors in sepsis actually relate to an action on the channel, we performed parallel studies using TEA. We chose TEA rather than a more specific BK channel inhibitor such as paxilline as the former has been proposed as an agent to reverse sepsis-induced hypotension in humans (19).
MATERIALS AND METHODS
Mice
Experiments were performed under UK Home Office approval according to the Animals (Scientific Procedures) Act 1986. Male mice were maintained under standard laboratory conditions with a 12 hour light/dark diurnal cycle and free access to food and water.
BK−/− mice were kindly provided by Dr Andrea Meredith, University of Maryland School of Medicine, Baltimore, MD (26). BKα−/− mice were confirmed by genotyping and we verified that the BKα−/− mice lacked BK functional activity by performing whole cell patch clamp recordings on freshly isolated aortic smooth muscle cells. As 40% of BKα−/− die of unknown cause at 10-12 weeks, mice living to 16-20 weeks were used for experimental purposes as these are considered to have normal organ development (26). Despite this claim, there was a significant difference (P < 0.001) in mean weight (±sem) between BKα−/− and their wild type (WT) littermates (21.4 ± 1g vs 36 ± 1.6g, respectively).
Genotyping
Primer sequences for polymerase chain reaction (PCR) are as follows: Wild type (WT) - Left Primer (5′-TTC ATC ATC TTG CTC TGG CGG AC-3′) and Right Primer (5′-CAA CAA CAA CAA CAA CAA CAA CA -3′). Knockout (KO) - Neo 5′ (5′-ATA GCC TGA AGA ACG AGA TCA GC-3′) and RA 14025 3′ (5′-CCT CAA GAA GGG GAC TCT AAA C-3′).
DNA was prepared using 100 μl of DirectPCR-Ear (PEQLAB Ltd, Fareham, UK) and 10 μl of Proteinase K per tube at 55°C for 2 h. Crude lysates were incubated at 85°C for 45 min, centrifuged for 10 s and supernatant removed. For WT 3μl of DNA was added to: Taq PCR Master Mix (Qiagen, Crawley, UK), primers, DMSO and water. PCR Protocol: 94°C, 2 min; 94°C, 30 s; 62-57°C, 30s × 5 cycles; 72°C, 2 min; 94°C, 30 s; 57°C, 30 s × 30 cycles; 72°C, 2 min; 72°C, 5 min: 10-15°C for 10-15 min (cooling). The WT amplified product was observed at 500bp. For the knockout mice, 1μl of DNA was added to pFu DNA Polymerase (Promega, Hampshire, UK) with buffer, primers, dNTP, DMSO and water. PCR Protocol: 94°C, 2 min; 94°C, 30 s; 55°C, 30 s × 5 cycles; 72°C, 2 min; 94°C, 30 s; 50°C, 30 s × 30 cycles; 72°C, 2 min; 72°C, 5 min; 10-15°C for 10-15 min (cooling). The KO allele amplified product was observed at 800bp.
Whole-cell recording
Mouse aortic cells were enzymatically isolated using papain as previously described (27) Membrane currents were recorded under voltage-clamp in whole-cell recording configuration of patch-clamp technique using Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Cells were clamped at 0 mV for 150 ms to inactivate delayed rectifier currents and the voltage stepped in 10 mV increments to +40 mV. Currents were filtered at 1 kHz and sampled at 2 kHz via a Digidata 1322A (Axon Instruments) interface. Data were acquired and analysed using pClamp8 computer software (Axon Instruments). Patch pipettes were made from thin walled (OD 1.5 mm) borosilicate glass capillaries (Harvard Apparatus, Edenbridge, Kent, UK). Whole-cell bath solutions (pH 7.4) contained (in mmol.L−1): 140 NaCl, 5 KCl, 10 HEPES, 1 MgCl2 and 1.8 CaCl2 glucose 10. Pipette solutions (pH 7.2) contained (mmol.L−1): 110 K-gluconate, 30 KCl, 5 HEPES, 1.2 MgCl2, 5 Na2ATP, 1 Na2GTP, I EGTA. All current measurements were made 10–15 min after ‘break-in’.
Model of fecal peritonitis with continuous blood pressure recording
Carotid arterial and jugular venous catheters were inserted aseptically under isoflurane anesthesia (1.5%) and tunnelled subcutaneously to the nape of the neck (procedure time approximately 30 min). This was connected to a lightweight swivel/tether system (Instech, Plymouth Meeting, PA) allowing continuous arterial monitoring and intravenous fluid administration. Catheters were kept patent with 0.1 ml.h−1 heparinized saline. Mean arterial pressure was measured (P23XL transducers, Viggo-Spectramed, Oxnard, CA) and recorded onto a precalibrated PowerLab system (ADInstruments, Sydney, Australia). The mice were allowed 24 h following instrumentation to acclimatize to tether and cage. For induction of fecal peritonitis (FP), fresh slurry from a healthy rat was administered intraperitoneally (ip) diluted in 0.5ml n-saline. Sham animals received 0.5 ml n-saline only. This dilution was determined from pilot characterization experiments. To standardize experiments as much as possible, 1-2 pairs of WT and BK−/− mice were tethered on each experimental day and received slurry from the same rat. Intravenous fluid resuscitation was commenced following ip injection of fecal slurry or n-saline. The infusion rate was 0.3 ml.h−1 using a 50:50 mix of 6% hydroxyethyl starch (Voluven®, Fresnius, Kabi Ltd, Runcorn, UK) and 5% glucose to prevent hypoglycemia.
Mice were removed from their cage just prior to injection of fecal slurry and 24 h after. Under isoflurane anesthesia, rectal temperature was measured and echocardiography performed. Two-dimensional images were recorded using a VIVID 7 Dimension machine (GE Vingmed, Horten, Norway) with an epicardial probe (model i13L, GE Vingmed). Ascending aortic flow velocity was determined with the probe positioned over the ascending aorta in Doppler pulsed wave mode. Beat-to-beat cycles of aortic blood flow velocity were recorded in three captured loops of five heart beat cycles. Peak aortic flow velocity and velocity time integral (VTI) were determined and stroke volume calculated as the product of VTI and aortic root cross-sectional area (data from SM Hollenberg, personal communication). The animals were allowed to recover from anesthesia after injection of slurry but were sacrificed at the 24 h timepoint with arterial blood sampled just prior. This blood was analyzed immediately for acid-base status using an iStat 300 (Abbott Diagnostics, Maidenhead, Berks, UK), or centrifuged (5000g for 10 min) with plasma stored at −80°C for cytokine analysis (Th1/Th2 10-plex FlowCytomix kit, Bender MedSystems, Vienna, Austria) and nitrite/nitrate levels (modified Greiss reaction).
As BKα−/− mice died at 12-14 h following induction of sepsis, the protocol was altered such that the observation period was shortened to 10 h with echocardiography performed at 6 h. As BKα−/− mice were ~40% smaller than WT mice, the volume of slurry was reduced similarly. In other experiments septic or sham WT mice were given an iv infusion of tetraethylammonium chloride (TEA, Sigma-Aldrich, Poole, UK) (10mg.kg.h−1 for 2 h) commencing at 6h.
Reverse Transcription-PCR analysis of BK channel α-subunit in mouse aorta
Reverse transcription PCR analysis was used to detect the presence of specific mRNA for mouse BKα−/− subunit in aorta from naïve (untethered) sham and septic mice. Aortae were removed at the end of the experiment, snap frozen in liquid nitrogen and stored at −80°C. RNA was isolated in standard fashion using trizol. β-actin was used as the housekeeping gene. First strand cDNA synthesis was created using Superscript™ II RT and real-time PCR was performed at 95°C for 10 min then 95°C for 15 seconds and then 60°C for 60 seconds × 40 cycles. Primer selection was based on previously published mouse sequences with left primer sequence – 5′-TCTGCCTGATGCAGTTTGAC-3′ and right – 5′-CCGGCTCATCTGTAAACAT-3′; both primers contained 50% GC content. The primer pair spans several exons of the BK channel gene to avoid amplification of genomic DNA.
Cecal ligation and puncture (CLP) model
In view of problems occurring in the septic tethered BKα−/− mice (see Results), an untethered cecal ligation and puncture model was used to assess survival compared to WT mice. Mice were anesthetized with isoflurane. The cecum was then exposed through a midline abdominal incision, ligated below the ileocecal valve without occluding the bowel, and perforated at two points with 21-gauge needle distal to ligation. A small amount of cecal content was squeezed out to ensure persistence of punctures. The bowel was repositioned and the abdomen closed. To induce high-grade sepsis, approximately 75% of cecum was ligated (28). Shams underwent the same procedure but without ligation or puncture. All animals received 1 ml of 5% glucose/hetastarch (50:50) subcutaneously at the time of surgery and every 8 h for 48 h. In these experiments, mice received either TEA at doses of 0, 12 or 50 mg.kg−1 ip (given in 0.5 ml n-saline) 4 h post CLP, in addition to fluid resuscitation as detailed above. There is very little abdominal inflammation at this time point and so it is likely that ip absorption is sufficient. Mice were observed for survival over a 72h period.
Data Analysis
Data are reported as mean ± standard error (s.e.m.) of n (number of animals) observations when normally distributed, or median with interquartile range (IQR) where data were not normally distributed. Statistical analysis was performed using GraphPad Prism 4 (GraphPad Software, La Jolla, CA). Survival curves was analysed by Kaplan-Meier curves with log rank testing for significance and mortality grouped at 6 h intervals. MAP was analysed by two way repeated measures ANOVA. To compare results between three groups, one-way ANOVA with repeated measures and post hoc Bonferroni correction was used. Student’s t test was used for two group comparisons. A P value < 0.05 was considered statistically significant.
RESULTS
Fecal slurry model of peritonitis
Characterization of model
Hemodynamic values for tethered WT sham mice (n=10) were unchanged throughout and similar to age-matched naive (untethered) mice (Figs. 1A-D).
Fig. 1. Hemodynamic measurements of (A) continuous blood pressure monitoring and intermittent echocardiography ((B) peak velocity, (C) stroke volume and (D) heart rate) in a fluid resuscitated conscious mouse model of fecal peritonitis (FP).
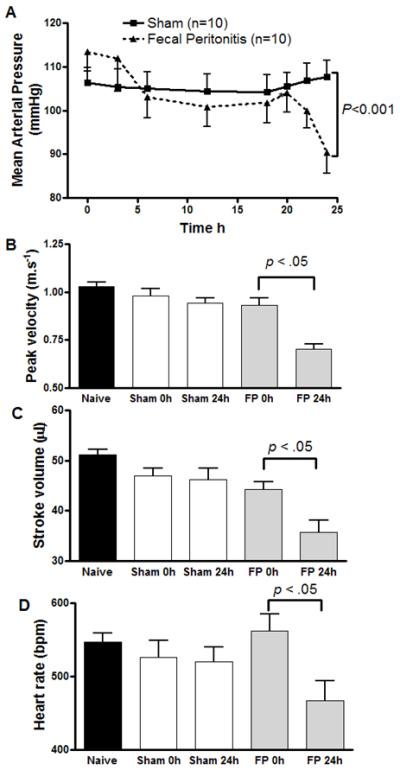
Echocardiography values displayed for naive (untethered) mice; pre FP/sham at time 0 h - (i.e. after 24 h on tether) and 24 h post FP/ sham. Fecal peritonitis (FP) induced a fall in mean arterial pressure (MAP) over 24h in contrast to sham mice (P < 0.001). In addition, at this timepoint FP induced a significant fall in aortic peak flow velocity, stroke volume and heart rate compared to shams (P < 0.05). Data are shown as mean ± sem with P values as shown (one way ANOVA or paired t-test for heart rate; n=10 for MAP and for echocardiography measurements n=13 fecal peritonitis, n=15 sham and n=15 naive mice).
Fecal peritonitis in the WT animals (n=10) induced an early fall in mean arterial pressure (MAP) between 0-6 h, followed by a further fall between 20-24 h despite fluid resuscitation (p<0.001 compared to sham controls, Fig 1A). At 24 h the septic animals showed significant falls in aortic peak flow velocity, stroke volume and heart rate (P < 0.05, Fig 1B-D). Despite keeping their cages on heated pads, temperature fell in the septic mice from 37.7±0.2 to 35±1.1 °C (P < 0.05, n=9), whereas this was unchanged in shams (37.5±0.3 °C) (Table 1). At 24 h plasma nitrite/nitrate levels, the pro-inflammatory cytokines interleukin-1β and -6 (IL-1β, IL-6), interferon-γ, and tumor necrosis factor alpha (TNF-α) (P < 0.05, n=6-9), and metabolic acidemia (P < 0.001, n=7, Table 1) were significantly elevated in septic compared to sham mice.
Table 1. Plasma pro-inflammatory cytokine values, nitrate/nitrite, base excess and core temperature in characterization studies with tethered sham and fecal peritonitis mice (FP) at 24h.
Data shown as mean (sem) for nitrite/nitrate, base excess and temperature as normally distributed and median (interquartile range-IQR) for cytokine data.
| Sham* | FP* | |
|---|---|---|
| IL-1β (pg.ml−) | 0 (0;0) | 19 (2;171) |
| IL-6 (pg.ml−) | 234 (76;646) |
2060 (467;9150) |
| TNF-α (pg.ml−) | 0 (0;0) | 46 (5;136) |
| IFN-γ (pg.ml−) | 0 (0;21) | 135 (39;197) |
| Nitrite / Nitrate (μM) | 73 (6.8) | 110 (12.5) |
| Arterial base excess | −2.2 (0.6) | −11 (0.9) |
| Core temperature ( □C) |
37.1 (0.2) | 36.45 (1.1) |
There was a significant difference between sham and FP mice for all values shown (P < 0.05).
Effect of fecal peritonitis on BK α–subunit gene expression in (non BK−/−) mice aorta
Quantification of the change in gene expression in sham and septic mice was evaluated relative to its expression in naïve mice (normalized to 1 using the 2−Δ ΔCT method (29)). This produced values of 1.33 for sham and 0.83 for septic mice, indicating that BK α–subunit gene expression was not substantially altered in aortae taken from septic mice compared to naïve or sham animals (samples pooled from 4 animals per group).
Effect of fecal peritonitis on hemodynamics in BKα−/− mice
Following the first seven successfully tethered BKα−/− mice, the next five exhibited distressing circling behavior so these experiments were abandoned. This behavior was not seen in the WT mice.
Baseline mean arterial pressure (MAP) was similar in BKα−/− (n=7) and WT (n=6-8) mice. Whereas MAP remained constant in both groups of sham mice, there was a significant albeit similar fall in both BKα−/− and WT mice following sepsis (approximately 20-25 mmHg at 10 h) (Figs. 2A, B and Table 2; representative traces shown in Fig. 3). Echocardiography-derived data measured at 6 h are shown in Table 2. Though absolute stroke volume was lower in the septic BKα−/− mice, when corrected for body weight the values were greater in the knockout animals (P < 0.01, n=5-6). Temperature fell to a similar extent in the septic BKα−/− and WT mice (Table 2).
Fig. 2. Mean arterial blood pressure readings following (A) fecal peritonitis (FP) or (B) sham insult in wild type (WT) or BKα−/− (KO) mice and (C) effect of TEA upon FP-induced hypotension.
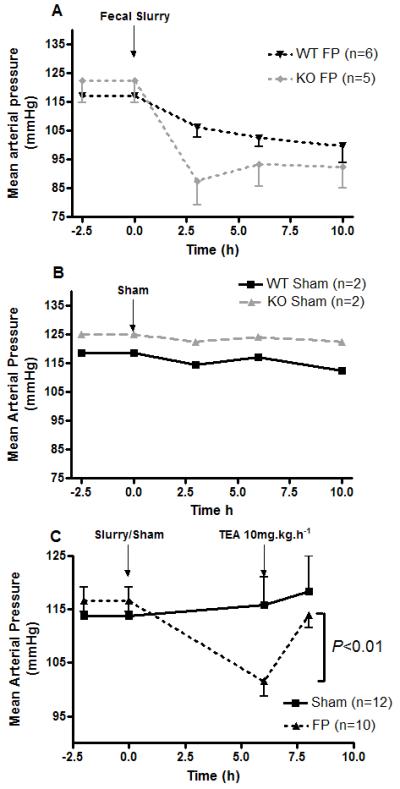
FP induced a fall in mean arterial blood pressure (MAP) in (A) BKα−/− mice and their WT litter mates compared to (B) sham animals. TEA (10mg.kg−1.h−1 iv for 2 h) reversed FP-induced hypotension at 6 h (C) with no effect on shams (P < 0.01). Data shown as mean ± sem, (except B - mean values only as n=2) with n values on graphs.
Table 2. Hemodynamic values and core temperature of tethered BKα−/− knockout mice and their wildtype (WT) litter mates following fecal peritonitis (FP).
Data shown as mean ± sem.
| Time 0 (after 24h on tether) |
6h post-fecal slurry | |||
|---|---|---|---|---|
| WT (n=6) | BKα−/− (n=5) | WT (n=6) | BKα−/− (n=5) | |
| Peak Velocity (m.s−1) | 0.84 ± 0.05 |
0.73 ± 0.03 | 0.84 ± 0.08 |
0.73 ± 0.07 |
| Stroke Volume (μl) | 41.3 ± 0.05 |
31.9 ± 0.05 | 43.8 ± 0.03 |
39.4 ± 0.03 |
| Stroke Volume.weight−1 (μl.mg−1) | 1.17 ± 0.13 |
1.41 ± 0.05 | 1.24 ± 0.08 |
1.73 ± 0.1 |
| Heart Rate (bpm) | 596 ± 19 | 600 ± 22 | 541 ± 18 | 518 ± 60 |
| Cardiac Output (ml.min−1) | 24.6 ± 3 | 19.1 ± 0.8 | 25.4 ± 1.8 | 20.4 ± 2.9 |
| Cardiac Output.weight−1 (ml.min−1.mg− 1) |
0.7 ± 0.09 | 0.85 ± 0.05 | 0.71 ± 0.05 |
0.9 ± 0.12 |
| Core temperature | 37.4 ± 0.5 | 37.4 ± 0.8 | 35.7 ± 0.6 | 35.7 ± 1 |
Fig. 3.
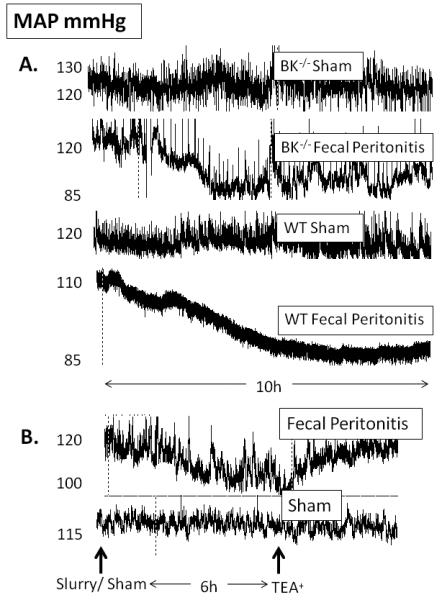
Representative traces of mean arterial pressure (MAP) readings from conscious mice. (A) BKα−/− and wild type mice following FP or sham and (B) effect of TEA 10 mg.kg.h−1 iv on MAP in FP and sham mice.
Effect of TEA upon blood pressure at 6 h post-fecal peritonitis in WT mice
TEA was infused for 2 h to non-BKα−/− mice beginning at 6 h after either injection of fecal slurry or sham insult (n=9 per group). Before TEA the MAP had fallen significantly in the septic mice (101.5±2.8 compared with 115.8±5.3 mmHg in sham mice, P < 0.001). Infusion of TEA resulted in a small but significantly greater (P < 0.01) increment in MAP in the septic mice (10.1±2 vs. 3.2±1.4 mmHg in shams, Fig. 2C).
Cecal ligation and puncture model (CLP)
WT and BKα−/− mice CLP survival studies
BKα−/− mice demonstrated significantly reduced survival CLP compared to WT mice with a median survival of 12 h (IQR 12-18) vs 30 h (IQR 18-33) (P<0.001, n=8-13, Fig. 4A).
Fig. 4.
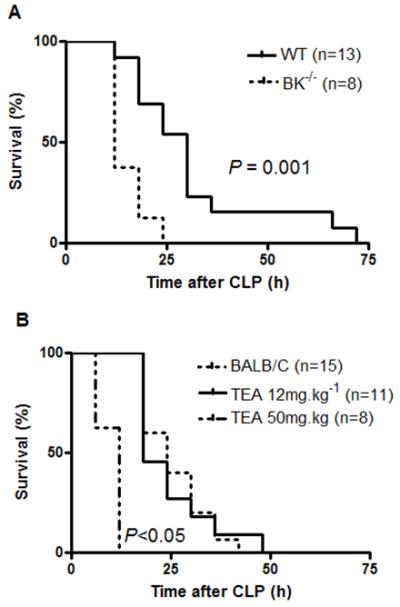
Effect of cecal ligation and puncture (CLP) upon survival in (A) BKα−/− mice and their wild type (WT) litter mates and in (B) mice ± TEA (data shown as mean ± sem. (A) BK−/− mice have significantly increased mortality compared to aged-matched WT litter mates in a CLP model (P < 0.001). (B) TEA 12 mg.kg−1 ip had no effect upon survival in a mouse model of CLP, whereas TEA 50mg.kg−1 ip significantly reduced survival following CLP (P < 0.05 compared to CLP, ANOVA, n values on graph). Data shown in A represent CLP experiments performed on 2 separate occasions while B represents 1 set of experiments.
Effect of TEA on survival and myocardial dysfunction following CLP in WT mice
Following sham surgery all mice survived to 72 h and showed similar hemodynamic values to naive mice (n=6, data not shown). Mice given TEA (12mg.kg−1 ip) 4h post-CLP showed no improvement in survival (median survival time 24h (IQR 18-30) versus 18h (IQR 18-30) in control animals, Fig. 4B). TEA did not attenuate the reduction in cardiac output observed at 8 h post-CLP (P < 0.001 compared to baseline pre-CLP values, n=8-15, Table 3). High-dose TEA (50mg.kg−1 ip) resulted in an increased mortality compared to TEA 0 or 12mg.kg−1, with a median survival time of 12 h (IQR 6-12; n=8-15, P < 0.05). There was a trend to a further reduction in cardiac output in mice given high-dose TEA but meaningful statistical comparison was precluded by only five such treated animals being alive at this point (Table 3).
Table 3.
Echocardiography values following cecal ligation and puncture (CLP) in BALB/C mice ± TEA. All echocardiograph values fell significantly at 8h following CLP ± TEA (12 or 50 mg.kg−1). Data shown as mean ± sem.
| Pre-CLP* (Time 0) (n=34) |
8 h post CLP* | |||
|---|---|---|---|---|
| No treatment (n=14) |
+ TEA 12 mg.kg−1 (n=10) |
+ TEA 50 mg.kg−1 (n=5) |
||
| Peak Velocity (m.s−1) | 0.85 ± 0.01 | 0.65 ± 0.02 | 0.69 ± 0.07 | 0.56 ± 0.06 |
| Stroke Volume (μl) | 45 ± 0.9 | 28 ± 0.9 | 32 ± 2 | 23.5 ± 1.8 |
| Heart Rate (bpm) | 544 ± 13 | 499 ± 13 | 475 ± 17 | 452 ± 12 |
| Cardiac Output (ml.min−1) | 24.6 ± 0.7 | 14.1 ± 0.8 | 15.9 ± 1.7 | 10.7 ± 1 |
| Cardiac Output.weight−1 (ml.min−1.mg−1) |
1.29 ± 0.05 | 0.7 ± 0.04 | 0.76 ± 0.08 | 0.57 ± 0.05 |
P < 0.001
DISCUSSION
To test the hypothesis that BKα−/− mice would be resistant to sepsis-induced hypotension, we developed an invasive model of fecal peritonitis in conscious mice with continuous mean arterial pressure (MAP) monitoring and intravenous fluid resuscitation. Both wildtype and BKα−/− mice developed similar degrees of hypotension and myocardial dysfunction, with similar increases in inflammatory cytokine levels, nitric oxide production and acidemia. A small but statistically significant increase in blood pressure was seen with the non-specific BK potassium channel blocker, tetraethylammonium chloride (TEA) in the wildtype mice. Survival time was however reduced in septic BKα−/− mice and, in a separate cecal ligation and puncture model, in wild type mice given high-dose TEA.
The difference in resting blood pressure between wildtype and BKα−/− animals was small (~5 mmHg) and similar to that reported by Sausbier et al using radiotelemetry arterial blood pressure readings (7). Contrary to expectation, we found septic BKα−/− mice developed hypotension (secondary to reduced total peripheral resistance) to a similar degree to their WT littermates. In addition, gene transcript expression of the BK α-subunit was not increased in aortae taken from septic compared to sham or naïve animals. Therefore, we conclude that activation of BK channels is not a crucial determinant of hypotension in septic mice. Our findings contradict several studies showing activation of the channel in various experimental models of inflammation, with inhibition partially or fully reversing hyperpolarisation or vascular hyporeactivity (15, 17-18). However, these studies have largely used a single intravenous bolus of endotoxin (or TNF-α) as a model of systemic inflammation. Such insults produce a severe acute inflammatory hit that is not representative of true sepsis where bacteria are present and proliferating, and disease progression is more gradual. Equally the study in a rodent model of peritonitis used aortic blood vessels for their in vitro experiments which, as a conduit vessel, has little influence upon blood pressure (16). Alternatively, despite these plausible explanations for differences, the loss of the BK channel may be compensated for by up-regulation of other potassium channels or other vasodilatory mechanisms.
The non-selective K+ channel inhibitor, TEA raised blood pressure in the septic mice, albeit to a small degree only (10.1 ± 1.9 mmHg). Although the concentration of TEA used would adequately inhibit BK channels (20), its effect is in stark contrast to the sepsis-induced hypotension seen in the BKα−/− mice which was identical to WTs. This is an important finding as reversal by TEA of endotoxin-induced vascular hyporeactivity to norepinephrine in healthy volunteers was claimed to be evidence for nitric oxide-mediated BK channel activation and thus a potential therapeutic modality (19). While TEA may indeed be effective in raising blood pressure, it would appear to do so via other mechanisms. TEA inhibited ATP-sensitive (KATP) potassium channels in vitro with an EC50 of 7 mM, and could also block the in vivo effects of KATP channel activators (30). Therefore, inhibition of other K+ channels, or other pathways, cannot be excluded. Furthermore, endotoxin-mediated TNFα production in macrophages was prevented by BK inhibitors (21, 22), though this was effective even in BKα−/− mice (23), perhaps TEA has another as yet unknown effect.
Our second prediction that BKα−/− mice would exhibit improved survival following sepsis was also not supported by our experiments. As the size difference between the BKα−/− and WT mice is a potential confounding influence, we used mice of similar weight to the mutants for the CLP±TEA experiments. Clayton et al found that TEA not only failed to reverse endotoxin (LPS)-induced hypotension in rats but also reduced survival by 50% (31) However, their study period was short (45 min) and, as LPS administration causes profound myocardial depression, a vasoconstrictor agent in these circumstances might be predicted to have adverse effects. In our model, moderate dose (12 mg.kg−1 ip) TEA had no effect on survival yet high-dose TEA was injurious. The high dose of TEA (50 mg.kg−1) we administered had previously been used in combination with methylene blue to improve endotoxin and TNFα-induced mortality (18). However, at this concentration, TEA actually worsened survival in our CLP model. We hypothesized that decreased survival related to TEA was due to excessive arterial vasoconstriction leading to increased coronary afterload and/or coronary artery vasospasm in view of the fall in cardiac output observed (from 15.9 ± 0.1 to 10.9 ± 1.7 ml.min−1). (Only five mice were alive at this point and so statistical analysis was not performed). To confirm this, we would have liked to measure blood pressure in these animals but the insertion of arterial and venous lines in combination with CLP surgery would have been too physiologically stressful for the mice. Equally, given the ability of TEA to inhibit early macrophage cytokine production (21, 22), we cannot discount the possibility that impaired survival may have been related to excessive dampening of macrophage cytokine production, thereby inhibiting the animal’s ability to mount an efficient inflammatory response.
Given the increased stroke volume and maintained cardiac output observed at 6 h, systemic vasodilatation, even in the absence of functional BK channels, is likely responsible for the sepsis-induced hypotension seen in the BKα−/− mice. However, the BK channel is widely expressed in tissues other than the vasculature (e.g. brain, bladder and bowel) (20); although we controlled for size differences, it is possible that phenotypic characteristics other than absence of vascular BK channels may have contributed to the increased mortality following CLP. Moreover, the BKα−/− mice did not appear unwell – their resting temperature and behaviour was the same as WT mice and their stroke volume corrected for size was actually greater than WT mice (Table 2). Given the ubiquitous nature of this channel, a smooth muscle specific mutant may be a better model to study, however this was not available to us.
Although both sepsis models used represent polymicrobial insults, the time course differs between the two. While CLP is considered the gold standard for sepsis studies (32) and survival is the most important outcome in sepsis studies, we felt that this was an appropriate model to use. In tethered mice we considered the additional surgery required for CLP would make the model too severe, and thus used a slurry injection that took seconds to perform. The insertion of arterial and venous lines in these small animals may have had a continuing physiological effect even though animals were given 24 h to recover. Although more invasive, mice required a week to recover following insertion of an implantable telemetric device (33). Echocardiography values at 24 and 48 h in shams were identical to those seen in naives while blood pressure was unchanged. Likewise there were no adverse cardiovascular effects from the tether at 6 h in BKα−/− and WT mice. We thus feel that though this model is invasive, it is robust.
Unfortunately, we could complete experiments in only seven tethered BKα−/− mice due to animal discomfort. Spontaneous circling has been described in 6% of mutants and is likely a functional deficit (26). As BKα−/− mice have ataxia and deficits in motor performance, it seems likely that the additional balance required with the tether in situ proved overwhelming. We may have thus missed significant effects due to this study being underpowered, however the hypotensive effect of the FP insult in the five BKα−/− mice was quite homogenous and more severe than in the six WT mice. We thus regard this as strong evidence that BKα−/− mice were not resistant to hypotension following FP. Crucially, we were unable to administer TEA to FP BKα−/− mice as this would have clarified whether any reversal of sepsis-induced hypotension was related to BK channel inhibition or not.
We conclude that polymicrobial sepsis-induced hypotension in mice is not mediated by activation of large conductance calcium-activated potassium channels. However, the non-selective K+ channel inhibitor TEA reversed sepsis-induced hypotension in non-BKα−/− mice so this effect is probably unrelated to BK channel inhibition. While the mechanism remains to be elucidated, it could relate to inhibition of other potassium channels regulating blood pressure. Regardless, the administration of TEA did not improve survival in our CLP model, suggesting it may well be another vasoactive agent that will not prove beneficial in the treatment of sepsis.
Supplementary Material
Acknowledgments
This work was funded by a grant from the University College London Hospitals Special Trustees. Dr Orie is funded by the British Heart Foundation (PG/07/078/2355).
Funding: This work was funded by a grant from the University College London Hospitals Special Trustees. The work was undertaken at UCLH/UCL which receives a proportion of its funding from the Department of Health’s NIHR Comprehensive Biomedical Research Centre funding scheme. LHC and MS also receive funding support from the Wellcome Trust, British Heart Foundation and (UK) Medical Research Council.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Padkin A, Goldfrad C, Brady AR, Young D, Black N, Rowan K. Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med. 2003;31(9):2332–8. doi: 10.1097/01.CCM.0000085141.75513.2B. [DOI] [PubMed] [Google Scholar]
- 3.Parker MM, Shelhamer JH, Natanson C, Alling DW, Parrillo JE. Serial cardiovascular variables in survivors and nonsurvivors of human septic shock: heart rate as an early predictor of prognosis. Crit Care Med. 1987;15(10):923–9. doi: 10.1097/00003246-198710000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Russel JA. The current management of septic shock. Minerva Med. 2008;99(5):431–58. [PubMed] [Google Scholar]
- 5.Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365(9453):63–78. doi: 10.1016/S0140-6736(04)17667-8. [DOI] [PubMed] [Google Scholar]
- 6.Kane GC, Lam CF, O’Cochlain F, Hodgson DM, Reyes S, Liu XK, Miki T, Seino S, Katusic ZS, Terzic A. Gene knockout of the KCNJ8-encoded Kir6.1 K(ATP) channel imparts fatal susceptibility to endotoxemia. FASEB J. 2006;20(13):2271–80. doi: 10.1096/fj.06-6349com. [DOI] [PubMed] [Google Scholar]
- 7.Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, Feil S, Kamm S, Essin K, Sailer CA, Abdullah U, Krippeit-Drews P, Feil R, Hofmann F, Knaus HG, Kenyon C, Shipston MJ, Storm JF, Neuhuber W, Korth M, Schubert R, Gollasch M, Ruth P. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation. 2005;112(1):60–8. doi: 10.1161/01.CIR.0000156448.74296.FE. [DOI] [PubMed] [Google Scholar]
- 8.Plüger S, Faulhaber J, Fürstenau M, Löhn M, Waldschütz R, Gollasch M, Haller H, Luft FC, Ehmke H, Pongs O. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87(11):E53–60. doi: 10.1161/01.res.87.11.e53. [DOI] [PubMed] [Google Scholar]
- 9.Buckley JF, Singer M, Clapp LH. Role of KATP channels in sepsis. Cardiovasc Res. 2006;72(2):220–30. doi: 10.1016/j.cardiores.2006.07.011. 1. [DOI] [PubMed] [Google Scholar]
- 10.Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345(8):588–95. doi: 10.1056/NEJMra002709. [DOI] [PubMed] [Google Scholar]
- 11.Vanelli G, Hussain SN, Aguggini A. Glyburide, a blocker of ATP-sensitive potassium channels, reverses endotoxin-induced hypotension in pig. Exp Physiol. 1995;80(1):167–170. doi: 10.1113/expphysiol.1995.sp003832. [DOI] [PubMed] [Google Scholar]
- 12.O’Brien AJ, Thakur G, Buckley JF, Singer M, Clapp LH. The pore-forming subunit of the KATP channel is an important molecular target for LPS-induced vascular hyporeactivity in vitro. Br J Pharmacol. 2005;144(3):367–375. doi: 10.1038/sj.bjp.0706065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morelli A, Lange M, Ertmer C, Broeking K, Van Aken H, Orecchioni A, Rocco M, Bachetoni A, Traber DL, Landoni G, Pietropaoli P, Westphal M. Glibenclamide dose response in patients with septic shock: effects on norepinephrine requirements, cardiopulmonary performance, and global oxygen transport. Shock. 2007;28(5):530–535. doi: 10.1097/shk.0b013e3180556a3c. [DOI] [PubMed] [Google Scholar]
- 14.Warrillow S, Egi M, Bellomo R. Randomized, double-blind, placebo-controlled crossover pilot study of a potassium channel blocker in patients with septic shock. Crit Care Med. 2006;34(4):980–5. doi: 10.1097/01.CCM.0000206114.19707.7C. [DOI] [PubMed] [Google Scholar]
- 15.Yakubovich N, Eldstrom JR, Mathers DA. Lipopolysaccharide can activate BK channels of arterial smooth muscle in the absence of iNOS expression. Biochim Biophys Acta. 2001;1514(2):239–52. doi: 10.1016/s0005-2736(01)00378-9. [DOI] [PubMed] [Google Scholar]
- 16.Kuo JH, Chen SJ, Shih CC, Lue WM, Wu CC. Abnormal activation of potassium channels in aortic smooth muscle of rats with peritonitis-induced septic shock. Shock. 2009;32(1):74–9. doi: 10.1097/SHK.0b013e31818bc033. [DOI] [PubMed] [Google Scholar]
- 17.da Silva-Santos JE, Terluk MR, Assreuy J. Differential involvement of guanylate cyclase and potassium channels in nitric oxide-induced hyporesponsiveness to phenylephrine in endotoxemic rats. Shock. 2002;17(1):70–6. doi: 10.1097/00024382-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 18.Cauwels A, Brouckaert P. Critical role for small and large conductance calcium-dependent potassium channels in endotoxemia and TNF toxicity. Shock. 2008;29(5):577–82. doi: 10.1097/shk.0b013e31815071e9. [DOI] [PubMed] [Google Scholar]
- 19.Pickkers P, Dorresteijn MJ, Bouw MP, van der Hoeven JG, Smits P. In vivo evidence for nitric oxide-mediated calcium-activated potassium-channel activation during human endotoxemia. Circulation. 2006;114(5):414–21. doi: 10.1161/CIRCULATIONAHA.105.590232. [DOI] [PubMed] [Google Scholar]
- 20.Ghatta S, Nimmagadda D, Xu X, O’Rourke ST. Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol Ther. 2006;110(1):103–16. doi: 10.1016/j.pharmthera.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Papavlassopoulos M, Stamme C, Thon L, Adam D, Hillemann D, Seydel U, Schromm AB. MaxiK blockade selectively inhibits the lipopolysaccharide-induced I kappa B-alpha /NF-kappa B signalling pathway in macrophages. J Immunol. 2006;177(6):4086–93. doi: 10.4049/jimmunol.177.6.4086. [DOI] [PubMed] [Google Scholar]
- 22.Scheel O, Papavlassopoulos M, Blunck R, Gebert A, Hartung T, Zähringer U, Seydel U, Schromm AB. Cell activation by ligands of the toll-like receptor and interleukin-1 receptor family depends on the function of the large-conductance potassium channel MaxiK in human macrophages. Infect Immun. 2006;74(7):4354–6. doi: 10.1128/IAI.01783-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Essin K, Gollasch M, Rolle S, Weissgerber P, Sausbier M, Bohn E, Autenrieth IB, Ruth P, Luft FC, Nauseef WM, Kettritz R. BK channels in innate immune functions of neutrophils and macrophages. Blood. 2009;113(6):1326–31. doi: 10.1182/blood-2008-07-166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao G, Zhao Y, Pan B, Liu J, Huang X, Zhang X, Cao C, Hou N, Wu C, Zhao KS, Cheng H. Hypersensitivity of BKCa to Ca2+ sparks underlies hyporeactivity of arterial smooth muscle in shock. Circ Res. 2007;101(5):493–502. doi: 10.1161/CIRCRESAHA.107.157271. [DOI] [PubMed] [Google Scholar]
- 25.Zhou R, Liu LM, Hu DY. Effect of nitric oxide tyrosine phosphorylation of calcium activated potassium channel alpha subunit on vascular hyporesponsiveness in rats. Chin J Traumatol. 2005;8(4):209–15. [PubMed] [Google Scholar]
- 26.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279(35):36746–52. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 27.Wilson AJ, Jabr RI, Clapp LH. Calcium modulation of vascular smooth muscle ATP-sensitive K(+) channels: role of protein phosphatase-2B. Circ Res. 2000;87(11):1019–25. doi: 10.1161/01.res.87.11.1019. [DOI] [PubMed] [Google Scholar]
- 28.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Quayle JM, Bonnev AD, Brayden JE, Nelson MT. Pharmacology of ATP-sensitive K+ currents in smooth muscle cells from rabbit mesenteric artery. Am J Physiol. 1995;269(5 Pt 1):C1112–8. doi: 10.1152/ajpcell.1995.269.5.C1112. [DOI] [PubMed] [Google Scholar]
- 31.Clayton NP, LeDuc BW, Kelly LJ. Effect of potassium channel and cytochrome P450 inhibition on transient hypotension and survival during lipopolysaccharide-induced endotoxic shock in the rat. Pharmacology. 2005;73(3):113–20. doi: 10.1159/000081631. [DOI] [PubMed] [Google Scholar]
- 32.Otero-Antón E, González-Quintela A, López-Soto A, López-Ben S, Llovo J, Pérez LF. Cecal ligation and puncture as a model of sepsis in the rat: influence of the puncture size on mortality, bacteremia, endotoxemia and tumor necrosis factor alpha levels. Eur Surg Res. 2001;33(2):77–9. doi: 10.1159/000049698. [DOI] [PubMed] [Google Scholar]
- 33.Zanotti-Cavazzoni SL, Guglielmi M, Parrillo JE, Walker T, Dellinger RP, Hollenberg SM. Fluid resuscitation influences cardiovascular performance and mortality in a murine model of sepsis. Intensive Care Med. 2009;35(4):748–54. doi: 10.1007/s00134-008-1360-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.