

Abstract
PURPOSE
To assess the presence of transforming growth factor-β (TGFβ) pathway markers in the epithelium of keratoconus patient corneas.
DESIGN
Retrospective, comparative case series of laboratory specimens.
METHODS
Immunohistochemistry results for TGFβ2, total TGFβ, mothers against decacentaplegic homolog (Smad) 2, and phosphorylated Smad2 was performed on formalin-fixed, paraffin-embedded sections of keratoconus patient corneas and normal corneas from human autopsy eyes. Keratoconus patient corneas were divided in two groups, depending on their severity based on keratometer readings and pachymetry. Autopsy controls were age-matched with the keratoconus cases. Immunohistochemistry signal quantification was performed using automated software. Real-time reverse-transcriptase polymerase chain reaction was performed on total ribonucleic acid of epithelium of keratoconus patient corneas and autopsy control corneas.
RESULTS
Immunohistochemistry quantification showed a significant increase in mean signal in the group of severe keratoconus cases compared with normal corneas for TGFβ2 and phosphorylated Smad2 (P < .05). Immunohistochemistry analysis using antibodies against total TGFβ and Smad2 did not show any significant increase in the keratoconus cases versus the autopsy controls. Reverse-transcriptase polymerase chain reaction exhibited elevated messenger ribonucleic acid levels of Smad2 and TGFβ2 in severe keratoconus corneal epithelium.
CONCLUSIONS
This work shows increased TGFβ pathway markers in severe keratoconus cases and provides the rationale for investigating TGFβ signaling further in the pathophysiology of keratoconus.
Keratoconus is a bilateral progressive corneal disease, leading to thinning, scarring, and protrusion of the central cornea.1 The origin and the pathogenesis of this disorder are not well understood. Although most often an isolated disease, it has been associated with several accompanying factors such as Down syndrome, contact lens wear, connective tissue disease, atopy, and eye rubbing, and it can occur in a familial setting. Keratoconus most likely is caused by multiple genes and may result from complex interactions between genes and environmental factors.2 Therapeutic measures focus first on the correction of refractive errors. Although preliminary results on riboflavin/ultraviolet-A-induced collagen-crosslinking suggest a favorable outcome, in the advanced stages, corneal transplantation is still the most effective treatment to date.3,4 Keratoconus in fact is the most common indication for keratoplasty.5 Gaining more insight into the mechanisms of keratoconus to find ways to prevent disease progression or to discover new treatment options therefore would be an important accomplishment.
Histologically, in the course of the disease, breaks in Bowman membrane and subepithelial scarring can be observed. Furthermore, the affected areas have marked alterations in the components of the extracellular matrix and show apoptotic cells, which, along with the thinning of the corneal stroma, suggest an increased activation of degrading enzymes and cell death resulting from oxidative stress.6 However, the exact mechanisms of the tissue breakdown remain unclear.
The signaling pathway of transforming growth factor-β (TGFβ) is a complex, multibranched signal transduction cascade that may modulate ECM alterations in keratoconus. TGFβ, with its 3 isoforms, TGFβ1, TGFβ2, and TGFβ3, is only one of numerous ligands of the TGFβ superfamily that bind to the TGFβ receptors that exist in 3 different isoforms. Binding of ligands to the TGFβ2 receptor, which has an intrinsic serin/threonine kinase activity, leads to recruitment and phosphorylation of the TGFβ1 receptor, which subsequently phosphorylates the mothers against decacentaplegic homolog (Smad) 2 and Smad3 proteins intracellularly. The Smad proteins are homologs of the Drosophila protein mothers against deca-pentaplegic and the C. elegans protein SMA. Phosphorylated Smad2 (pSmad2) forms a complex with the mediator Smad4 and is translocated into the nucleus, where it acts as a transcription factor for multiple TGFβ-dependent genes. Smad2 and Smad3 can be activated as well by non-TGFβ growth factors, which are capable of activating mitogen-activated protein kinases. These multiple growth factors include fibroblast growth factor, insulin-like growth factor-1, hepatocyte growth factor, and endothelial growth factor. Many of the cellular effects of the TGFβ pathway have in common their involvement in the restoration of normal tissue after injury by induction of both extracellular matrix and matrix-degrading enzymes.7–13 The involvement of the TGFβ pathway in the modulation and production of extracellular matrix suggests involvement in the pathogenesis of keratoconus, either in a causative role or a secondary repair response leading to structural changes in keratoconus. However, previous reports linking the TGFβ pathway with the pathogenesis of keratoconus have been inconclusive. Although Maier and associates found TGFβ2 levels to be elevated in the aqueous humor in keratoconus cases, immunofluorescence studies on TGFβ2 in patients with keratoconus did not show an increase in staining as compared with normal controls.14,15 This work attempts to elucidate the role of the TGFβ signaling pathway in keratoconus by focusing on the extracellular receptor ligands TGFβ and its isoforms, as well as the intracellular activation of Smad2, by immunohistochemistry and reverse-transcriptase polymerase chain reaction (RT-PCR) of keratoconus epithelium.
METHODS
PATIENTS AND CONTROLS
The clinical diagnosis of keratoconus was made by fellowship-trained corneal specialists. Diagnosis of keratoconus was based on corneal topography along with the presence of standard clinical signs. Cases for immunohistochemistry experiments were divided in two groups depending on disease severity. Severe cases were regarded as those with mean keratometry readings K ≥ 50 diopters (D) or a pachymetry reading of ≤ 400 μm, and mild cases were categorized as those with both K < 50 D and a pachymetry reading of > 400 μm. If both readings were not available, cases were classified on available data. Keratometry readings were obtained using the Pentacam (Oculus, Wetzlar, Germany) or Orbscan II (Bausch & Lomb, Rochester, New York, USA) devices, pachymetry was obtained using ultrasound (DGH Technologies, Exton, Pennsylvania, USA). Corneas from autopsy cases with no history of corneal disease served as controls. Patient and control characteristics are summarized in Table 1 (immunohistochemistry) and Table 2 (RT-PCR). No patients in this study were wearing contact lenses at the time of keratoplasty.
TABLE 1.
Autopsy Control and Keratoconus Patient Demographic Data for Immunohistochemistry Results
| Autopsy Patient Data | |||||
|---|---|---|---|---|---|
| Age (yrs) | Gender | Corneal Status | Cause of Death | Death to Preservation Time (hrs) | |
| 46 | Male | Normal | Multiple organ failure | 19 | |
| 40 | Male | Normal | Non-Hodgkin lymphoma | 21 | |
| 48 | female | Normal | Multiple organ failure | 6 | |
| 33 | female | Normal | Acquired immunodeficiency syndrome | 13 | |
| 48 | Male | Normal | Lung failure | 22 | |
| Mean | 43 | 16 | |||
| SD | 6 | 7 | |||
| Keratoconus Patient Data | ||||||
|---|---|---|---|---|---|---|
| Age (yrs) | Gender | Diagnosis | Keratometry (D) | Pachymetry (μm) | Severity | |
| 25 | Male | Keratoconus | 44 | 624 | Mild | |
| 35 | Female | Keratoconus | Unknown | 443 | Mild | |
| 27 | Female | Keratoconus | 43 | Unknown | Mild | |
| 39 | Female | Keratoconus | Unknown | 427 | Mild | |
| 42 | Female | Keratoconus | Unknown | 565 | Mild | |
| 16 | Male | Keratoconus | 42 | 538 | Mild | |
| Mean | 31 | 43 | 519 | |||
| SD | 10 | 1 | 83 | |||
| 54 | Female | Keratoconus | 63 | 321 | Severe | |
| 28 | Male | Keratoconus | 41 | 380 | Severe | |
| 68 | Female | Keratoconus | 63 | 420 | Severe | |
| 35 | Female | Keratoconus | 56 | 445 | Severe | |
| 17 | Male | Keratoconus | Unknown | 400 | Severe | |
| 26 | Female | Keratoconus | 51 | 275 | Severe | |
| 75 | Male | Keratoconus | 89 | 359 | Severe | |
| Mean | 43 | 61 | 371 | |||
| SD | 22 | 15 | 59 | |||
D = diopters; hrs = hours; SD = standard deviation; yrs = years.
TABLE 2.
Autopsy Control and Keratoconus Patient Demographic Data for Quantitative Reverse-Transcriptase Polymerase Chain Reaction
| Autopsy Patient Data | |||||
|---|---|---|---|---|---|
| Age (yrs) | Gender | Corneal Status | Cause of Death | Death to Preservation Time (hrs) | |
| 50 | Male | Normal | Hypertrophic cardiomyopathy | 26 | |
| 29 | Female | Normal | Metastatic gall bladder carcinoma | 15 | |
| 84 | Male | Normal | Coronary artery disease | 36 | |
| 58 | Female | Normal | Metastatic gastric carcinoma | 10 | |
| Mean | 55 | 22 | |||
| SD | 23 | 12 | |||
| Keratoconus Patient Data | ||||||
|---|---|---|---|---|---|---|
| Age (yrs) | Gender | Diagnosis | Keratometry (D) | Pachymetry (μm) | Severity | |
| 26 | Male | Keratoconus | 65 | 411 | Severe | |
| 46 | Female | Keratoconus | 70 | 277 | Severe | |
| 26 | Female | Keratoconus | 63 | 370 | Severe | |
| 58 | Female | Keratoconus | 65 | 323 | Severe | |
| 23 | Male | Keratoconus | 64 | 381 | Severe | |
| Mean | 36 | 65 | 352 | |||
| SD | 15 | 3 | 53 | |||
D = diopters; SD = standard deviation.
IMMUNOHISTOCHEMISTRY
All patient corneas had been diagnosed previously with keratoconus as described above, and these diagnoses were confirmed further by microscopic analysis. All control corneas had been assessed as unremarkable by an ophthalmic pathologist (C.G.E.) in the Wilmer Eye Pathology Laboratory. A total of 7 patients with severe keratoconus and 6 patients with mild keratoconus were evaluated (Table 1). Autopsy corneas were from individuals with an average age of 43 ± 6 years (mean ± standard deviation), and specimens from patients with keratoconus were from individuals with an average age of 37 ± 18 years (P > .05, compared with autopsy corneas). The mean age of the patients with severe keratoconus was 43 ± 22 years, and the mean age of the patients with mild keratoconus was 31 ± 10 years (P > .05, compared with severe cases). Mean pachymetry of patients with severe cases was 371 ± 59 μm, and that of patients with mild cases was 519 ± 83 μm (P < .05, compared with severe cases). Mean keratometry of severe keratoconus cases was 61 ± 15 D, and that of mild cases was 42 ± 1 D (P > .05, compared with severe cases).
Normal autopsy eyes and cornea specimens of patients with keratoconus were formalin fixed and paraffin embedded by the Johns Hopkins Pathology Laboratory. Unstained sections (5 μm) were deparaffinized and processed for immunohistochemistry using Elite ABC Peroxidase Kit (Vector Labs, Burlingame, California, USA) following the manufacturer’s instructions. Heat-induced epitope retrieval was performed in a buffer solution containing 10 mM Tris, 1 mM edetic acid, and 0.05% Tween 20 (Sigma-Aldrich Co, St Louis, Missouri, USA), pH 9.0. Sections were stained with diaminobenzidine substrate (Vector Labs) with addition of nickel solution and were counterstained with Mayer hematoxylin solution (Sigma-Aldrich Co.). Negative control slides using no primary antibody and nonspecific, species-matched, and isotype-matched antibodies were included. Antibodies (dilutions in brackets) included: mouse monoclonal anti-Smad2 (1/200; Santa Cruz Biotechnology, Santa Cruz, California, USA), rabbit-polyclonal anti-phos-pho-Smad2 (1/200; Millipore, Billerica, Massachusetts, USA), rabbit polyclonal anti-TGFβ (1/100; Abcam, Inc, Cambridge, Massachusetts, USA), mouse monoclonal anti-TGFβ2 (1/1000), normal rabbit immunoglobulin (Ig) G (1/50), normal mouse IgG (1/50; all from Santa Cruz Bio-technology), biotinylated anti-mouse IgG (1/200), and biotinylated anti-rabbit IgG (1/200; both from Vector Labs).
IMAGE ACQUISITION AND PROCESSING
Light microscopy images were obtained using an Olympus IMT-2 microscope (Olympus, Center Valley, Pennsylvania, USA) coupled to a ProgRes C5 digital camera system (Jenoptik, Jena, Germany). Images at × 200 magnification were used. For assessment of immunohistochemistry staining, a random field with intact epithelium from the central cornea was located at low magnification and was magnified to × 200. Subsequently, two directly adjacent fields with intact epithelium were imaged on either side of the original field. All three fields were used for evaluation of each patient or control.
IMMUNOHISTOCHEMISTRY QUANTIFICATION
Quantitative assessment of immunohistochemistry staining was performed using FRIDA Software (FRamework for Image Dataset Analysis, The Johns Hopkins University, http://bui2.win.ad.jhu.edu/frida/).16 This software provides a pixel color threshold mask as well as a freehand mask to mark regions of interest. A range of positive immunohistochemistry color signal is specified, and the software subsequently quantifies all pixels with the selected range of colors within an area of interest. Three adjacent fields of the corneal epithelium of a single immunohistochemistry-stained section were chosen as the region of interest for each individual patient or control. The same pixel color mask was applied to all samples being analyzed for a given antibody marker.
REVERSE-TRANSCRIPTASE POLYMERASE CHAIN REACTION
Total ribonucleic acid was prepared for real-time RT-PCR from epithelium of corneas from patients with keratoconus and autopsy control corneas (Table 2). A commercially available kit was used to isolate total ribonucleic acid (RNeasy Mini Kit; Qiagen, Valencia, California, USA). Real-time RT-PCR for Smad2, TGFβ 2, Vimentin, matrix metalloproteinase 9 (MMP9), and β-actin (housekeeping gene serving as positive control; all primers from IDT, Coralville, Iowa, USA) was performed on a thermal cycler (LightCycler; Roche, Mannheim, Germany). Experiments were performed twice, and in each experiment, all genes was tested in triplicate. Primers for all genes spanned introns to distinguish amplification of genomic deoxyribonucleic acid (DNA). Primer sequences were as follows: β-actin: sense primer, 5′-ccaaccgcgagaa-gatga-3′; antisense primer, 5′-ccagaggcgtacagggatag-3′. The expected product length was 97 base pairs. Primer sequences for TGFβ 2 were as follows: sense primer, 5′-ccaaagggtacaatgccaac-3′; antisense primer, 5′-cagat-gcttctggatttatggtatt-3′. The expected product length was 114 base pairs. Primer sequences for Smad2 were as follows: sense primer, 5′-aggcctttacagcttctctgaa-3′; antisense primer, 5′-gtggcaatccttttcgatg-3′. The expected product length was 77 base pairs. Primer sequences for Vimentin were as follows: sense primer, 5′-tacaggaagct-gctggaagg-3′; antisense primer, 5′-accagagggagtgaatc-cag-3′. The expected product length was 104 base pairs. Primer sequences for MMP9 were as follows: sense primer, 5′-gaaccaatctcaccgacagg-3′; antisense primer, 5′-gccacccgagtgtaaccata-3′. The expected product length was 67 base pairs. PCR was performed for 45 cycles using commercially available reagents and protocols (TaqMan Master Mix, hydrolysis probes; Roche). PCR steps were as follows: 1 cycle of 10 minutes at 95 C followed by 45 cycles of 10 seconds at 95 C, 40 seconds at 50 C, 1 second at 72 C, and 1 cycle of 40 seconds at 40 C. For quantification, the delta-delta method (2− Δ Δ CT) was used.17 Samples were electrophoresed in 1.5% agarose gels containing ethidium bromide and were photographed using standard methods.
STATISTICAL ANALYSIS
P values were calculated using the nonparametric Mann–Whitney U test (Instat; GraphPad software, San Diego, California, USA). P values less than .05 were considered statistically significant.
RESULTS
IMMUNOHISTOCHEMISTRY
Immunohistochemistry quantification for TGFβ2 in severe keratoconus corneal epithelium demonstrated a 1. 6-fold increase in signal compared with normal autopsy corneal epithelium (47 761 ± 47 378 [range, 5760 to 165 406] vs 29 376 ± 36 392 [range, 1004 to 111 889], mean ± standard deviation; P < .05; Figure 1A). Mild cases showed a statistically insignificant decrease compared with normal controls (20 650 ± 33 658 [range, 162 to 117 699] vs 29 376 ± 36 392; P > .05). Immunohistochemistry quantification for pSmad2 in severe keratoconus corneal epithelium demonstrated a 1.5-fold increase in signal compared with normal autopsy corneal epithelium (57 516 ± 51 910 [range, 5124 to 191 577] vs 37 163 ± 45 606 [range, 1132 to 142 866], mean ± standard deviation; P < .05; Figure 1B). Mild cases showed a statistically insignificant decrease compared with normal controls (16 126 ± 25 418 [range, 584 to 103 145] vs 37 163 ± 45 606; P > .05). Higher magnification (×400) showed specifically strong staining signal intranuclearly and perinuclearly, predominantly in the basal and suprabasal cell layers (Figure 2). The antibody against all isoforms of TGFβ (TGFβ1, TGFβ2, TGFβ3) demonstrated a statistically insignificant 2.3-fold increase in signal in severe keratoconus corneal epithelium compared with normal corneal epithelium (755 ± 1445 [range, 0 to 5911] vs 328 ± 220 [range, 25 to 804]; P > .05; Figure 1C).
FIGURE 1.

Immunohistochemistry for transforming growth factor-β (TGFβ) signaling pathway markers. TGFβ2, pSmad2, and TGFβ (total) were assessed in normal human corneas and corneas with keratoconus. Horseradish peroxidase (HRP)-positive signal was quantified as described in Methods. Error bars indicate standard deviation. *P < .05; **P < .01; ***P < .001; +P ≥ .05; Mann–Whitney U test. Original magnification ×200. (A and B): Immunohistochemistry for TGFβ signaling pathway markers (TGFβ2 and phosphorylated Smad2 [pSmad2]) in (Top left) normal human corneas and (Top right) in patients with mild and (Bottom left) severe keratoconus. (Bottom right) Signal quantification. (C): Immunohistochemistry results for TGFβ (total) in (Left) normal human corneas and (Middle) patients with severe keratoconus. (Right) Signal quantification.
FIGURE 2.
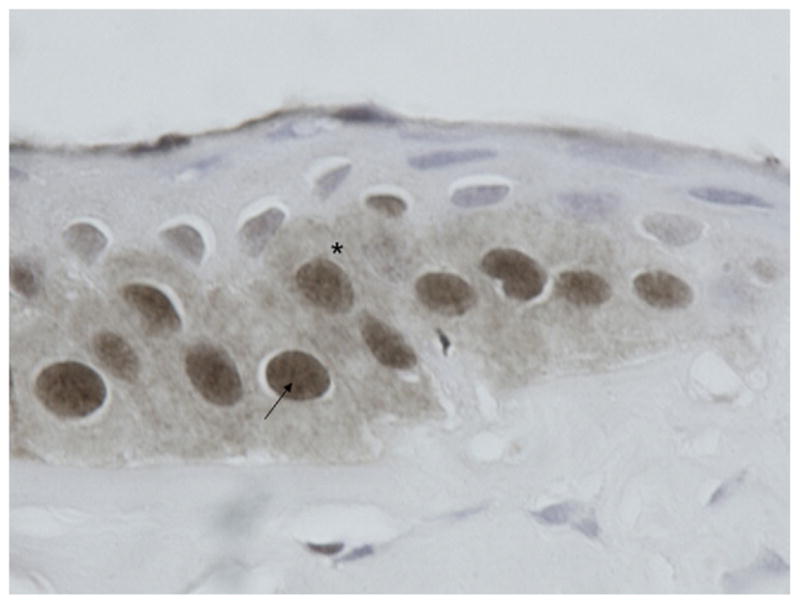
Photomicrograph showing immunohistochemistry results for phosphorylated Smad2 (pSmad2). Enlarged detail of keratoconus epithelium (×400 magnification) stained for pSmad2, showing a (*) perinuclear and (arrow) nuclear staining pattern.
Immunohistochemistry quantification for unphosphorylated Smad2 did not show an increase in the mild nor the severe keratoconus cases compared with the autopsy controls (results not shown). Negative control staining of severe keratoconus cases with no primary antibody or normal rabbit IgG as primary antibody showed 8.8-fold and 7.9-fold decreases in signal compared with anti-pSmad2 primary antibody (6532 ± 3825 [range, 1394 to 13,475] vs 57 516 ± 51 910 [range, 5124 to 191 577] and 7240 ± 9695 [range, 1650 to 36 917] vs 57 516 ± 51 910, respectively; P < .01 for both comparisons). Negative control staining with no primary antibody or normal mouse IgG as primary antibody showed 11.6-fold and 8.6-fold decreases in signal compared with TGFβ 2 (4122 ± 4251 [range, 372 to 17 306] vs 47 761 ± 47 378 [range, 5760 to 165 406], and 5539 ± 7798 [range, 0 to 32 653] vs 47 761 ± 47 378, respectively; P < 0.01 for both comparisons).
REAL-TIME POLYMERASE CHAIN REACTION
A total of 5 patients with severe keratoconus with an average age of 36 ± 15 years (mean ± standard deviation) and 4 autopsy corneas from individuals with an average age of 55 ± 23 (P > .05, compared with keratoconus cases) were evaluated. Real-time PCR showed an increase of expression of Smad2 (1.9-fold; range, 1.4 to 2.3) and TGFβ 2 (1.3-fold; range, 0.9 to 1.6) in keratoconus complimentary DNA (cDNA) compared with controls. Vimentin showed a decrease in gene expression (0.19-fold) in keratoconus cDNA. MMP9 gene expression was not detected in RT-PCR. Products of TGFβ 2 and Smad2 of RT-PCR were run on an agarose gel, which exhibited stronger bands for both genes in the severe keratoconus cDNA as compared with the control cDNA (results not shown).
DISCUSSION
The present study shows an increase of tgfb2 and pSmad2 signal in the epithelium of severe keratoconus cases. The increase in the immunohistochemistry experiments is confirmed by elevated expression of TGFβ 2 by RT-PCR. The increase in unphosphorylated Smad2 detected in RT-PCR could not be found in immunohistochemistry results, possibly because of the higher sensitivity of RT-PCR versus immunohistochemistry. The detected increase of both TGFβ 2 by immunohistochemistry and RT-PCR and intracellular un-phosphorylated (RT-PCR) and phosphorylated Smad2 (immunohistochemistry) suggests activation of the TGFβ pathway in the epithelium of keratoconus corneas.
Interestingly, the nonspecific antibody of TGFβ, which was used in immunohistochemistry experiments, did not show a significant increase in signal, suggesting a less important role of the isoforms TGFβ1 and TGFβ3 compared with that of TGFβ2. Although being structurally and functionally similar, the 3 TGFβ isoforms differ significantly in terms of potency, receptor affinity, and expression pattern.9,18,19 TGFβ1 is known to be as well involved in corneal fibrosis and scar formation,20 yet TGFβ2 may play a prominent role in the anterior eye because it is the predominant cytokine in the aqueous humor.9 It is as well known that mouse embryos that lack TGFβ2 develop multiple ocular defects—in particular a thin cornea with a loss of corneal endothelium—whereas ocular development is normal in the absence of TGFβ1 or TGFβ3.21 Moreover, the concentration of TGFβ2 increases in various other ophthalmic diseases such as in proliferative vitreoretinopathy, diabetic retinopathy, and open-angle glaucoma. In each of these diseases, the pathogenic role of TGFβ2 lies presumably in the modulation of extracellular matrix expression.9 It would be a worthwhile next step to correlate the positive staining results with studies on the concentration of TGFβ2 in the aqueous humor of keratoconus patients, as described by Maier and associates.14
It has been reported that Smad2 mediates expression of α-smooth muscle actin and MMP2.22 The presence of smooth muscle marker α-smooth muscle actin coincides with the fact that myofibroblasts are known to develop in scar tissue of corneas of patients with keratoconus.6 On the basis of the above, we performed RT-PCR for Vimentin, a marker of mesenchymal transformation (to detect myofibroblast formation), which, however, did not show an increase of expression.
Likewise, involvement of metalloproteinases in keratoconus is suggested by the disruption of Bowman and Descemet membranes.23–25 Zymography studies describe an upregulation of MMP2 levels and downregulation of tissue inhibitor of metalloproteinases-1 in clear keratoconic corneas.26 Whether this increase of proteinase activity is triggered by the TGFβ pathway or initiated by a different cytokine such as interleukin-1 or tumor necrosis factor-α requires further investigation.27–29 MMP9, which exerts along with MMP2 a key role in extracellular matrix remodeling, did not show an increase in the RT-PCR experiments. With regard to the literature, the previously described increase of MMP9 in keratoconus therefore may be limited exclusively to an increase in the tear fluid.24,29–31
To elucidate further the involvement of the TGFβ pathway, it may be rewarding to study the effects of its blockage (for instance, by use of TGFβ receptor kinase inhibitors or by angiotensin pathway blockade) on Bowman membrane integrity, scar formation, and other markers of keratoconus progression.32 The fact that angiotensin pathway antagonists are widely used in patients with hypertension could facilitate future research on TGFβ blockage, including possible prospective or retrospective studies on patients with keratoconus under angiotensin pathway antagonist treatment. In summary, current knowledge and the present study suggest that keratoconus pathogenesis could be related to activation of the TGFβ pathway; however, its causative involvement warrants further investigation.
Acknowledgments
PUBLICATION OF THIS ARTICLE WAS SUPPORTED BY GRANTS FROM THE OPENSHAW KERATOCONUS RESEARCH FUND, Baltimore, Maryland (A.S.J.); the Eye Bank Association of America, Washington, DC (C.E., C.K.); Grants EY015523 (A.S.J.) and EY11654 (S.C.) from the National Institutes of Health, Bethesda, Maryland; the Medical Illness Counseling Center, Chevy Chase, Maryland (A.S.J.); the National Marfan Foundation, Port Washington, New York (J.D.); Research to Prevent Blindness, Inc, New York, New York (A.S.J.); and the Wilmer Professors Research Fund, Baltimore, Maryland, and The Johns Hopkins Medical Institutions, Baltimore, Maryland (A.S.J.). The funding organizations had no role in the design or conduct of this research. The authors indicate no financial conflict of interest. Involved in Design of study (C.E., S.C., A.S.J.); Conduct of study (C.E., S.C., A.S.J.); Collection of data (C.E., S.C., J.D., H.M., A.S.J.); Management (S.C., C.G.E., H.M., C.K., W.S.S., A.S.J.), analysis (C.E., S.C., A.S.J.), and interpretation (C.E., C.S., A.S.J.) of data; and Preparation, review, or approval of the manuscript (C.E., S.C., J.D., C.G.E., H.M., C.K., W.S.S., A.S.J.). Johns Hopkins Institutional Review Board approval was obtained.
Biographies

Albert S. Jun, MD, PhD, is an Associate Professor of Ophthalmology at the Wilmer Eye Institute, Johns Hopkins School of Medicine, Baltimore, Maryland. His clinical and research interests include Fuchs dystrophy and endothelial keratoplasty. Dr. Jun completed residency at the Wilmer Institute and cornea fellowship at Moorfields Eye Hospital. His professional activities have been recognized with grants and awards from the National Institute of Health, the Heed Foundation, the Eye Bank Association of America, and the Association of University Professors of Ophthalmology.

Christoph Engler, MD, graduated from the University of Freiburg, Germany. He is currently working as a post-doctoral fellow in corneal research at the Wilmer Eye Institute, Johns Hopkins School of Medicine, in the laboratory of Dr. Albert Jun. Dr. Engler was awarded the Research Grant Award of the Eye Bank Association of America in 2008. His research interests include endothelial cell keratoplasty and pathophysiology of Fuchs corneal dystrophy.
References
- 1.Zhou L, Sawaguchi S, Twining SS, et al. Expression of degradative enzymes and protease inhibitors in corneas with keratoconus. Invest Ophthalmol Vis Sci. 1998;39(7):1117–1124. [PubMed] [Google Scholar]
- 2.Rabinowitz YS. The genetics of keratoconus. Ophthalmol Clin North Am. 2003;16(4):607, 20, vii. doi: 10.1016/s0896-1549(03)00099-3. [DOI] [PubMed] [Google Scholar]
- 3.Wittig-Silva C, Whiting M, Lamoureux E, et al. A randomized controlled trial of corneal collagen cross-linking in progressive keratoconus: preliminary results. J Refract Surg. 2008;24(7):S720–S725. doi: 10.3928/1081597X-20080901-15. [DOI] [PubMed] [Google Scholar]
- 4.Raiskup-Wolf F, Hoyer A, Spoerl E, et al. Collagen crosslinking with riboflavin and ultraviolet-A light in keratoconus: long-term results. J Cataract Refract Surg. 2008;34(5):796–801. doi: 10.1016/j.jcrs.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 5.Collier SA, Madigan MC, Penfold PL. Expression of membrane-type 1 matrix metalloproteinase (MT1-MMP) and MMP-2 in normal and keratoconus corneas. Curr Eye Res. 2000;21(2):662–668. [PubMed] [Google Scholar]
- 6.Maatta M, Vaisanen T, Vaisanen MR, et al. Altered expression of type XIII collagen in keratoconus and scarred human cornea: increased expression in scarred cornea is associated with myofibroblast transformation. Cornea. 2006;25(4):448–453. doi: 10.1097/01.ico.0000183537.45393.1f. [DOI] [PubMed] [Google Scholar]
- 7.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 8.Chakravarti S, Wu F, Vij N, et al. Microarray studies reveal macrophage-like function of stromal keratocytes in the cornea. Invest Ophthalmol Vis Sci. 2004;45(10):3475–3484. doi: 10.1167/iovs.04-0343. [DOI] [PubMed] [Google Scholar]
- 9.Saika S. TGFbeta pathobiology in the eye. Lab Invest. 2006;86(2):106–115. doi: 10.1038/labinvest.3700375. [DOI] [PubMed] [Google Scholar]
- 10.Kim IY, Kim MM, Kim SJ. Transforming growth factor-beta: biology and clinical relevance. J Biochem Mol Biol. 2005;38(1):1–8. doi: 10.5483/bmbrep.2005.38.1.001. [DOI] [PubMed] [Google Scholar]
- 11.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35(2):83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Klenkler B, Sheardown H. Growth factors in the anterior segment: role in tissue maintenance, wound healing and ocular pathology. Exp Eye Res. 2004;79(5):677–688. doi: 10.1016/j.exer.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Saika S, Yamanaka O, Sumioka T, et al. Fibrotic disorders in the eye: targets of gene therapy. Prog Retin Eye Res. 2008;27(2):177–196. doi: 10.1016/j.preteyeres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Maier P, Broszinski A, Heizmann U, et al. Active transforming growth factor-beta2 is increased in the aqueous humor of keratoconus patients. Mol Vis. 2007:131198–131202. [PubMed] [Google Scholar]
- 15.Saghizadeh M, Chwa M, Aoki A, et al. Altered expression of growth factors and cytokines in keratoconus, bullous keratopathy and diabetic human corneas. Exp Eye Res. 2001;73(2):179–189. doi: 10.1006/exer.2001.1028. [DOI] [PubMed] [Google Scholar]
- 16.Goodwin AC, Jadallah S, Toubaji A, et al. Increased spermine oxidase expression in human prostate cancer and prostatic intra-epithelial neoplasia tissues. Prostate. 2008;68(7):766–772. doi: 10.1002/pros.20735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Huh MI, Kim YH, Park JH, et al. Distribution of TGF-beta isoforms and signaling intermediates in corneal fibrotic wound repair. J Cell Biochem. 2009;108(2):476–488. doi: 10.1002/jcb.22277. [DOI] [PubMed] [Google Scholar]
- 19.Cheifetz S, Hernandez H, Laiho M, et al. Distinct transforming growth factor-beta (TGF-beta) receptor subsets as determinants of cellular responsiveness to three TGF-beta isoforms. J Biol Chem. 1990;265(33):20533–20538. [PubMed] [Google Scholar]
- 20.Karamichos D, Guo XQ, Hutcheon AE, et al. Human corneal fibrosis: an in vitro model. Invest Ophthalmol Vis Sci. 2010;51(3):1382–1388. doi: 10.1167/iovs.09-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124(13):2659 –2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans RA, Tian YC, Steadman R, et al. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of smad proteins. Exp Cell Res. 2003;282(2):90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 23.Kim WJ, Rabinowitz YS, Meisler DM, et al. Keratocyte apoptosis associated with keratoconus. Exp Eye Res. 1999;69(5):475–481. doi: 10.1006/exer.1999.0719. [DOI] [PubMed] [Google Scholar]
- 24.Collier SA. Is the corneal degradation in keratoconus caused by matrix-metalloproteinases? Clin Experiment Ophthalmol. 2001;29(6):340–344. doi: 10.1046/j.1442-9071.2001.d01-17.x. [DOI] [PubMed] [Google Scholar]
- 25.Teng CC. Electron microscope study of the pathology of keratoconus: I. Am J Ophthalmol. 1963:5518–5547. doi: 10.1016/0002-9394(63)91645-3. [DOI] [PubMed] [Google Scholar]
- 26.Smith VA, Matthews FJ, Majid MA, et al. Keratoconus: matrix metalloproteinase-2 activation and TIMP modulation. Biochim Biophys Acta. 2006;1762(4):431–439. doi: 10.1016/j.bbadis.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Olofsson EM, Marklund SL, Pedrosa-Domellof F, et al. Interleukin-1alpha downregulates extracellular-superoxide dismutase in human corneal keratoconus stromal cells. Mol Vis. 2007:131285–131290. [PubMed] [Google Scholar]
- 28.Xue ML, Wakefield D, Willcox MD, et al. Regulation of MMPs and TIMPs by IL-1beta during corneal ulceration and infection. Invest Ophthalmol Vis Sci. 2003;44(5):2020–2025. doi: 10.1167/iovs.02-0565. [DOI] [PubMed] [Google Scholar]
- 29.Lema I, Duran JA. Inflammatory molecules in the tears of patients with keratoconus. Ophthalmology. 2005;112(4):654–659. doi: 10.1016/j.ophtha.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 30.Lema I, Sobrino T, Duran JA, Brea D, Diez-Feijoo E. Subclinical keratoconus and inflammatory molecules from tears. Br J Ophthalmol. 2009;93(6):820–824. doi: 10.1136/bjo.2008.144253. [DOI] [PubMed] [Google Scholar]
- 31.Predovic J, Balog T, Marotti T, et al. The expression of human corneal MMP-2, MMP-9, proMMP-13 and TIMP-1 in bullous keratopathy and keratoconus. Coll Antropol. 2008;32(Suppl 2):15–19. [PubMed] [Google Scholar]
- 32.Campistol JM, Inigo P, Larios S, et al. Role of transforming growth factor-beta1 in the progression of chronic allograft nephropathy. Nephrol Dial Transplant. 2001;16(Suppl 1):114–116. doi: 10.1093/ndt/16.suppl_1.114. [DOI] [PubMed] [Google Scholar]