

Abstract
Hypoxia-ischemia (HI) in the neonate leads to white matter injury and subsequently cerebral palsy. We find that expression of bone morphogenetic protein 4 (BMP4) increases in the neonatal mouse brain after unilateral common carotid artery ligation followed by hypoxia. Since signaling by the BMP family of factors is a potent inhibitor of oligodendroglial differentiation, we tested the hypothesis that antagonism of BMP signaling would prevent loss of oligodendroglia (OL) and white matter in a mouse model of perinatal HI. Perinatal HI was induced in transgenic mice in which the BMP antagonist noggin is overexpressed during oligodendrogenesis (pNSE-Noggin). Following perinatal HI, pNSE-Noggin mice had more oligodendroglial progenitor cells (OPCs) and more mature OL compared to wild type (WT) animals. The increase in OPC numbers did not result from proliferation but rather from increased differentiation from precursor cells. Immunofluorescence studies showed preservation of white matter in lesioned pNSE-Noggin mice compared to lesioned WT animals. Further, following perinatal HI, the pNSE-Noggin mice were protected from gait deficits. Together these findings indicate that the BMP-inhibitor noggin protects from HI-induced loss of oligodendroglial lineage cells and white matter as well as loss of motor function.
Keywords: hypoxia-ischemia, bone morphogenetic protein, noggin, oligodendrocyte progenitor cells, oligodendrocytes
Introduction
Perinatal hypoxic-ischemic brain injury (HI) is an important mechanism in the development of white matter injury (WMI) in preterm newborns (Volpe, 2001). The clinical correlate of WMI is cerebral palsy, a non-progressive spastic disorder of motor control (Johnston and Hoon, 2006). Cerebral palsy affects more than half of preterm babies weighing <1500g at birth (Volpe, 2001), and larger late preterm neonates are also at increased risk for cerebral palsy (Winter et al., 2002). There are currently no medical therapies to prevent or treat WMI.
The persistence of neural stem/progenitor cells (NSPC) within postnatal brain, including postnatal human brain, makes them attractive as potential replacement cells for regenerating damaged white matter. During development NSPCs migrate from the subventricular zone (SVZ) to cortex and subcortical areas (Menn et al., 2006) where they may either differentiate into oligodendroglia or reside as uncommitted progenitors within the cortex (Dawson et al., 2003). Migration of oligodendrocyte progenitor cells (OPCs) from the SVZ to injured areas is not significantly upregulated following postnatal HI in the wild-type mouse (Dizon et al. 2010), although migration of OPCs from the SVZ is upregulated following BMP antagonism in the context of a demyelination injury (Jablonska et al., 2010). Thus, the most relevant NSPCs for potentially contributing to an endogenous white matter reparative response following HI are likely those that reside locally near the site of injury. We therefore sought to test whether these NSPCs could be induced to commit to an oligodendroglial fate thereby protecting white matter from HI injury.
Bone morphogenetic proteins (BMPs), a subclass of the transforming growth factor beta (TGFβ) superfamily, are present in the brain throughout development. BMP4 is one of two major family members expressed within the cortex, and transcripts for BMP4 are preferentially localized to bipotent oligodendroglial-astroglial progenitors. During mouse development, BMP4 expression peaks in all brain regions at postnatal day 4 (Mehler et al., 1997). BMP signaling negatively regulates an oligodendroglial fate choice by both multipotential and bipotential neural progenitor cells (Mabie et al., 1997). Conversely, downregulation of BMP signaling by the BMP antagonist noggin, which binds BMP4 with high affinity (Mehler et al., 1997; Zimmerman et al., 1996), increases oligodendroglial lineage commitment by cultured neural progenitor cells (Gross et al., 1996; Mabie et al., 1999; Mabie et al., 1997). Thus the BMP signaling pathway is a tractable potential target for enhancing the production of replacement oligodendrocytes in vivo after HI. In adult rodents, components of BMP signaling pathways, including BMP7 and the type II BMP receptor (BMPRII), increase in response to stroke, and BMPRII is also upregulated in response to transient global HI (Charytoniuk et al., 2000). Recently, our lab showed that transgenic expression of noggin decreases infarct size and improves motor function in an adult stroke model (Samanta et al. 2010). We therefore analyzed the effects of neonatal global HI on transgenic mice that overexpress noggin. Our data show that downregulation of BMP signaling increases survival of both OPCs and more mature oligodendroglia with preservation of white matter and of motor function.
Materials and Methods
Animals
The generation of neuron specific enolase promoter driven (pNSE)-Noggin transgenic mice has been previously described(Gomes et al., 2003; Guha et al., 2004; Kan et al., 2004). pNSE-Noggin mice were generated on an FEV1 background and backcrossed to C57Bl/6 background for at least 6 generations. NSE-Noggin males were crossed with CD-1 females (Charles River) to render offspring more vulnerable to HI (NOG). C57BL6 males (Charles River) were crossed with CD-1 females for wild-type controls (WT). The F1 generation of either cross was used for experiments. CD-1 mice were used for quantitative RT-PCR and Western blot experiments. All mice were housed in a facility with a 12hr light/dark cycle and allowed access to food and water ad libitum. Experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee and Northwestern Center for Comparative Medicine.
Neonatal Hypoxic-Ischemic Lesions
Postnatal day 7 (P7) mice were anesthetized with isoflurane, then the right common carotid artery was ligated and transected. After 2hr recovery, pups were placed in humidified hypoxia chambers (8% oxygen/92% balanced air) kept at 37°C for 60min, were allowed to recover for 1hr, then returned to the dam as previously described (Ditelberg et al., 1996; Plane et al., 2004). Non-lesioned controls underwent sham surgeries. Mice were sacrificed 3–28 days post lesion (dpl).
BrdU administration
BrdU (Sigma, B5002) 50mg/kg was injected intraperitoneally b.i.d. beginning on the day of lesion, continuing for 48hr and mice sacrificed 3dpl or b.i.d. beginning 5dpl, continued until mice sacrificed 7dpl.
Quantification of infarcted brain
Mice were anesthetized with pentobarbital IP, brains removed and chilled in ice-cold PBS for 5min, cut into 1mm sections using a stainless steel mouse brain slicer (Harvard Apparatus NP72-5032), incubated in 2% Triphenyltetrazolium chloride (TTC) (Sigma T8877) in PBS for 20min at 37°C, then stored in 4% PFA. Images were taken using a Nikon Camera and Spot software (Nikon) and analyzed using Axiovision software (Carl Zeiss Vision). Cross-sectional areas of infarction were measured indirectly and infarct volumes calculated from these; that is, the infarct volume of right hemisphere was calculated by subtracting the measured non-infarcted cortex volume of the right hemisphere from the measured total cortical volume of the left hemisphere (Lin et al., 1993; Sola et al., 2005).
Quantitative RT-PCR
Mice were anesthetized with pentobarbital IP, then were decapitated, brains were removed and chilled in ice-cold PBS for 5min, cut into 1mm sections using a stainless steel mouse brain slicer, then an approximately 15mg piece of intact peri-infarct cortex/subcortical white matter was dissected using tungsten needles at 24, 48, and 72hr and 7days after lesioning. Tissue was collected into PBS. Total RNA was obtained using the RNAqueous-4PCR kit (Ambion). A total of 1µg of RNA was used for generating cDNA using the Thermoscript reverse transcriptase and oligo-dT primers (Invitrogen). cDNA was stored at − 80°C until used. Quantitative real-time PCR was performed with the SybrGreen Master Mix (Applied Biosystems) and Realplex2 Mastercycler (Eppendorf) using the following cycling parameters: 95°C ×10min, then 95°×15sec, 60° ×1smin for 40 cycles, 95°C ×15sec, 60°C ×15sec, 20min melting curve, followed by 95°C ×15sec. A total of 4µl of cDNA was used per PCR. The following primers were used: BMP4 qRT 7+ (forward) CAG CCG AGC CAA CAC TGT GAG, BMP4 qRT 10- (reverse) GTC CCT GGG ATG TTC TCC AGA TGT T.
Protein Extraction and Western Blot Analysis
Tissue was collected as for quantitative RT-PCR except that tissue was collected into Tissue Protein Extraction Reagent (Thermo Scientific) with 1× HALT protease inhibitor (Thermo Scientific) and frozen at −80°C until used. Protein was quantified using the BCA Protein Assay Kit (Thermo Scientific). Protein extracts (15µg) were separated by gel electrophoresis on 4–20% Tris-HCl SDS-polyacrylamide gels (Bio-Rad). Separated protein was electrophorectically transferred to PVDF membranes. Membranes were first blocked with TBS/0.1% Tween containing 5% non-fat dry milk, then immunoblotted with anti-BMP4 antibody (Chemicon MAB1049) and anti-β actin antibody (Santa Cruz sc-1616). Membranes were then probed with secondary antibodies raised against the appropriate species. After washing with TBS/0.1% Tween, membranes were developed using enhanced chemilumninescence (ECL) (Pierce). Blots were imaged using photographic film and results were quantified using ImageJ software (NIH).
Immunofluorescence
Mice were anesthetized with pentobarbital IP, transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde (PFA) in PBS, brains were removed, post-fixed for 2hr in ice cold 4% PFA, cryoprotected in 30% sucrose in PBS, embedded and frozen in O.C.T. Compound (Sakura Finetek 4583), cut into 10µm sections using a cryostat, and immunostained as described previously using antigen retrieval (Samanta et al., 2007). Antibodies used were as follows: BrdU (Accurate OBT-0030), BrdU (Chemicon MAB3510), CNPase (Sternberger SMI91), MBP (Sternberger SMI99), Olig1 (Chemicon MAB5540), Olig2 (Chemicon AB9610), PDGFRA (Fitzgerald CD140a), AlexaFluor 488 Goat anti-Rabbit, AlexaFluor 594 Goat anti-Mouse IgG2a, AlexaFluor 594 Goat anti-Mouse IgG2b and AlexaFluor 647 Goat anti-Rat (Invitrogen).
Digital Gait Analysis
Digital video images of the underside of mice walking at a speed of 8cm/sec were collected and analyzed using the DigiGait Imaging System and software (Mouse Specifics, Boston, MA) as previously described (Hampton et al., 2004). Mouse paw area over time was used to calculate swing duration (sec), stride duration (sec), stride length (cm) and stride frequency (steps per sec). Results of lesioned animals were normalized to the results of non-lesioned animals.
Statistics and Analysis
Three to eight mice were used per treatment group. Mid-striatal coronal sections were collected (between +0.98mm anterior to Bregma and +0.38 posterior to Bregma). For each animal, four 40x fields were captured from corpus callosum and the supracallosal radiations, using Axiovision 4.6 software (Carl Zeiss Vision). Image files were encoded to mask the study group, areas were measured and cells were counted using Axiovision 4.6 and Image J software (NIH) by investigators blinded to the treatment group. After counts were completed, the data was decoded, cell counts per area were calculated for each field and these were averaged. Relative fluorescence was measured using Metamorph software (Molecular Devices, Sunnyvale, CA) Counts and relative immunofluoresence were analyzed by ANOVA, using Fisher’s PLSD for post-hoc comparisons with statistical significance set at p<0.05. For two group comparisons, the Student’s t-test was used and the significance level was set at p<0.05.
Results
Postnatal HI does not change levels of BMP4 transcript but increases BMP4 protein
The effects of postnatal HI on expression of BMP4 were examined using quantitative reverse transcription PCR (qRT-PCR) and Western blot analysis. Although there was a bimodal trend towards increased levels of BMP4 transcript in the lesioned hemisphere 24–48hr and 7 days following postnatal HI (Fig. 1A) the increase was not significant. By contrast levels of BMP4 protein were significantly increased at 7 days following postnatal HI (Fig.1B’) compared to 24hr non-lesioned controls, 24hr lesioned mice, 48hr non-lesioned controls, 72hr non-lesioned controls, 72hr lesioned mice and 7day non-lesioned controls. BMPs are produced initially as a preproprotein, then cleaved to a proprotein, then a propeptide (Hogan, 1996). The Western blots displayed a single band at a weight of approximately 25kD, the size of the propeptide (Fig 1B).
Figure 1. Postnatal HI increases BMP4 protein expression.

A) Quantitated RT-PCR output for P8, 24hr post sham surgery control (CONT), 24hr post lesion (LES), 48hr CONT, 48hr LES, 72hr CONT, 72hr LES, 7d CONT and 7d LES. Only a non-significant trend of increased BMP4 transcript at 24–48hr and 7 days post lesion compared to 24hr post sham surgery was observed, n=6 mice per each group. B) A representative Western blot is shown. B’) Quantitated Western blot: CONT=sham surgery, LES=lesion. In contrast to BMP4 transcript, significant increases in BMP4 protein was seen at 7 days after lesion compared to 24hr CONT (*p=0.02), 24hr LES (*p=0.005), 48hr CONT (*p=0.005), 72hr CONT (*p=0.006), 72hr LES (*p=0.005) and 7d CONT (*p=0.01), n=3 mice per each group.
Noggin overexpression reduces infarct size following postnatal HI
Postnatal day 7 (P7) pNSE-Noggin (NOG) and wildtype (WT) mice were subjected to postnatal HI, and triphenyltetrazolium chloride (TTC) staining was performed on brain slices 48hr after injury. Infarction was demonstrated by lack of TTC staining and presence of white color. The infarct volume was significantly smaller in NOG mice (8.2 ±1.4 mm2) versus WT (17.1 ±2.3 mm2), p=0.005, n=9 NOG mice and 10 WT mice. Consistent with this finding, NOG mice were more likely to survive the insult (90.0% versus 58.8%). Thus, noggin overexpression decreases infarct size and confers a survival advantage.
Noggin overexpression results in increased Olig1+ cells following postnatal HI
We performed immunofluorescence for Olig1 which marks all oligodendroglial lineage cells and evaluated 2 distinct regions (Fig. 2A), corpus callosum (a) and subcortical white matter (b), that were possibly affected differently due to their distance from the infarction and to their different cell phenotypic compositions. There were no differences in the numbers of Olig1+ cells in non-lesioned WT and non-lesioned NOG mice. However at 7 days post lesion (dpl), the number of Olig1+ cells within the corpus callosum of lesioned NOG mice was significantly increased 1.5-fold compared to both lesioned WT mice and non-lesioned WT mice as well as non-lesioned NOG mice (Fig. 2B–B’’’’). Similarly within the subcortical white matter, numbers of Olig1+ cells were significantly increased 1.4-fold in lesioned NOG compared to lesioned WT mice (Fig.2C–C’’’’). These findings suggest that in the presence of noggin, HI leads to an increase in the sum of committed oligodendroglial progenitors, immature oligodendrocytes and mature oligodendrocytes whereas HI leads to no change in cell number in the absence of noggin.
Figure 2. Noggin overexpression increases oligodendroglial lineage cells following postnatal HI.
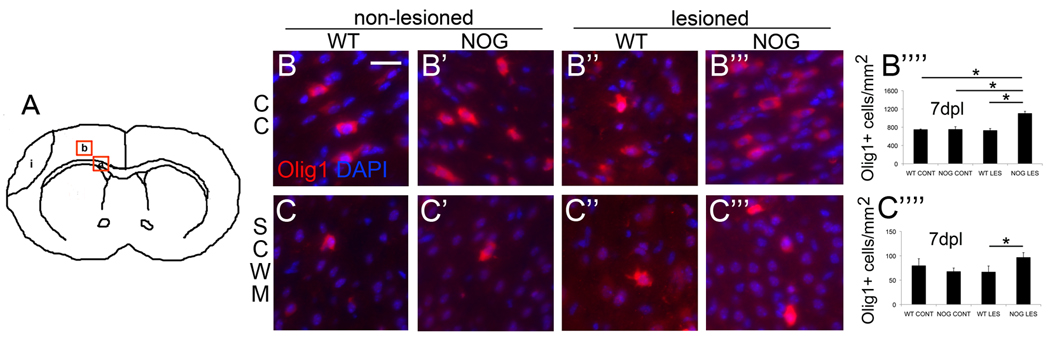
(A) Four fields from 2 distinct regions were imaged for each animal. These regions are delineated by red boxes and include a) corpus callosum (CC), and b) subcortical white matter (SCWM). Representative micrographs of Olig1+ cells within CC 7dpl show more Olig1+ cells in lesioned NSE-Noggin pups compared to all other groups: B) wild type control (WT CONT), (magnification 40x, scale bar = 25 µm), B’) NSE-Noggin control (NOG CONT), B’’) WT lesion (WT LES), B’’’) NSE-Noggin lesion (NOG LES). (B’’’’) Quantitated Olig1+ cells within CC : error bars represent s.e.m., NOG LES versus WT CONT, *p=0.0005, n=4 NOG LES and 3 WT CONT; NOG LES versus NOG CONT, *p=0.0001, n=4 NOG LES and 6 NOG CONT; NOG LES versus WT LES, *p=0.0002, n=4 NOG LES and 4 WT LES. Representative micrographs of Olig1+ cells within subcortical white matter (SCWM) show more Olig1+ cells in lesioned NSE-Noggin pups compared to WT lesioned pups: C) WT CONT, C’) NOG CONT, C’’) WT LES, C’’’) NOG LES. (C’’’’) Quantitated Olig1+ cells within SCWM, NOG LES versus WT LES, *p=0.05, n=4 NOG LES and 4 WT LES.
Noggin overexpression results in increased Olig2+ cells following postnatal HI
Previously, we had reported an increase in Olig2+ cells within cortex and striatum but not corpus callosum of lesioned wild-type mice as compared to non-lesioned controls following postnatal HI (Dizon et al. 2010). Here, we again used immunofluorescence to probe for Olig2+ cells in order to characterize changes in oligodendroglial progenitor cells (OPCs) in response to postnatal HI in NOG versus WT mice. At 7dpl we found significant increases in Olig2+ cells within corpus callosum of NOG mice compared to WT, both non-lesioned and lesioned: there was a 1.3-fold increase in non-lesioned NOG compared to non-lesioned WT mice, a 1.9-fold increase in lesioned NOG mice compared to non-lesioned WT mice, a 1.4-fold increase in lesioned NOG compared to non-lesioned NOG, and a 1.7-fold increase in lesioned NOG mice compared to lesioned WT mice (Fig.3A’’’’). We also found significant increases in Olig2+ cells within subcortical white matter of NOG mice compared to WT mice, both non-lesioned and lesioned: there was a 1.4-fold increase in non-lesioned NOG mice versus non-lesioned WT mice, a 1.5-fold increase in non-lesioned NOG mice versus lesioned WT mice, a 2.6 increase in lesioned NOG mice versus non-lesioned WT mice, a 1.8-fold increase in lesioned NOG mice versus non-lesioned NOG mice, and a 2.6-fold increase in lesioned NOG mice versus lesioned WT mice (Fig.3B’’’’).
Figure 3. Noggin over-expression increases Olig2+ cells following postnatal HI.

Representative micrographs of Olig2+ cells within corpus callosum (CC) 7dpl: A) wild type control (WT CONT), (magnification 40x, scale bar = 25µm), A’) NSE-Noggin control (NOG CONT), A’’) WT lesion (WT LES), A’’’) NSE-Noggin lesion (NOG LES). (A’’’’) Quantitated Olig2+ cells in CC, error bars represent s.e.m.: NOG CONT versus WT CONT, *p=0.004; NOG LES versus WT CONT, *p<0.0001; NOG LES versus NOG CONT, *p=0.0001; NOG LES versus WT LES, *p<0.0001, n= 3 WT CONT, 4 WT LES, 6 NOG CONT, 4 NOG LES. Representative micrographs of Olig2+ cells within subcortical white matter (SCWM) 7dpl: B) WT CONT, B’) NOG CONT, B’’) WT LES, B’’’) NOG LES. (B’’’’) Quantitated Olig2+ cells in SCWM: NOG CONT versus WT CONT, *p=0.0002; NOG CONT versus WT LES, *p=0.0001; NOG LES versus WT CONT, *p<0.0001; NOG LES versus NOG CONT, *p<0.0001; NOG LES versus WT LES, *p<0.0001, n=4 WT CONT, 4 WT LES, 6 NOG CONT and 4 NOG LES. (C) Quantitated PDGFRA+/Olig2+ cells in CC: WT LES versus WT CONT, *p=0.0006; WT LES mice versus NOG CONT, *p=0.02; NOG LES versus NOG LES, *p=0.002; n=5 WT CONT, 5 WT LES, 6 NOG CONT and 6 NOG LES.
In both regions, non-lesioned NOG mice possessed more Olig2+ cells compared to non-lesioned WT mice. However, persistence of Olig2+ cells in NOG mice compared to WT following injury cannot simply be attributed to a greater starting number of Olig2+ cells because, in fact, lesioned NOG mice possess significantly greater numbers of Olig2+ cells compared to non-lesioned NOG mice. Despite their differences in proximity to the lesion and native population of oligodendroglial cells, in both regions an interaction between noggin overexpression and hypoxia-ischemia induced the production of more Olig2+ cells.
To evaluate whether the change in Olig2+ cells represented a change in committed oligodendroglial progenitors rather than uncommitted neural progenitors, we counted PDGFRA+/Olig2+ cells at corpus callosum 7dpl. We found no difference in PDGFRA+/Olig2+ cell numbers between unlesioned NOG and WT mice. There was a significant loss in PDGFRA+/Olig2+ cells in lesioned compared to non-lesioned WT mice (35 ±12 cells/mm2 v. 162 ±24 cells/mm2). Likewise, there was a significant loss in PDGFRA+/Olig2+ cells in lesioned WT mice compared to non-lesioned NOG mice (35 ±12 cells/mm2 v. 143 ±10 cells/mm2). Importantly, there was a significant increase in PDGFRA+/Olig2+ cells in lesioned NOG mice compared to lesioned WT mice (141 ±28 cells/mm2 v. 35 ±12 cells/mm2).
Increased Olig2+ cells in noggin overexpressing mice do not result from increased proliferation
We marked proliferative cells with BrdU using 2 different paradigms: 1) injections twice daily for 48hrs starting 24hr after lesion with sacrifice 3dpl (Fig.4A), or 2) injections twice daily on days 5, 6 and 7 post-lesion with sacrifice 7dpl (Fig.4A’). Animals were sacrificed 3dpl in order to avoid missing cells that may have proliferated early but then failed to survive to 7dpl. The earlier BrdU paradigm captured a significant decrease in proliferation of Olig2+ cells within corpus callosum in lesioned WT mice compared to non-lesioned WT mice. There were also significantly fewer proliferative Olig2+ cells within corpus callosum in lesioned WT mice compared to lesioned NOG mice (Fig.4B). At 3dpl within subcortical white matter, there was a significant increase in proliferative Olig2+ cells in lesioned WT mice compared to non-lesioned WT mice, in lesioned WT mice compared to non-lesioned NOG mice, and in lesioned WT mice compared to lesioned NOG mice (Fig. 4C).
Figure 4. Increased Olig2+ cells following postnatal HI do not result from proliferation.

Proliferative cells were marked with BrdU using 2 different paradigms: A) bid injections for 48hrs starting 24hr after lesion with sacrifice at 3dpl, or A’) bid injections on days 5, 6 and 7 post-lesion with sacrifice at 7dpl.
(B) Quantitated of Olig2+ BrdU+ cells in corpus callosum, error bars represent s.e.m.: WT LES versus WT CONT at 3dpl, *p=0.007; NOG LES versus WT LES at 3dpl, *p=0.03, n=5 WT CONT, 5WT LES, 5 NOG CONT and 6 NOG LES. NOG LES versus WT LES at 7dpl, *p=0.02, n=4 WT CONT, 8WT LES, 3 NOG CONT and 5 NOG LES.
(C) Quantitated of Olig2+ BrdU+ cells in subcortical white matter: WT LES versus WT CONT at 3dpl, *p=0.01; WT LES versus NOG CONT at 3dpl, *p=0.02; WT LES versus NOG LES at 3dpl, *p=0.006, n=4 WT CONT, 8WT LES, 3 NOG CONT and 5 NOG LES.
NOG LES versus NOG CONT at 7dpl, *p=0.02, n=4 WT CONT, 8WT LES, 3 NOG CONT and 5 NOG LES.
With the later BrdU paradigm, significant differences were also seen. At 7dpl within the corpus callosum, a decrease in proliferative Olig2+ cells was seen in lesioned NOG mice as compared to lesioned WT mice (Fig.4B). At 7dpl within subcortical white matter, a decrease in proliferative Olig2+ cells was seen in lesioned NOG mice versus non-lesioned NOG mice (Fig.4C).
These data show an early loss of proliferative Olig2+ cells in large white matter tracts but perhaps an early increase in proliferative Olig2+ cells in subcortical white matter in response to HI that is not sustained. Overall, Olig2+BrdU+ cell counts suggest that noggin overexpression, if anything, coincides with decreased proliferation at both 3dpl and 7dpl.
Noggin overexpression results in preservation of white matter following postnatal HI
P7 mice were lesioned and sacrificed 14dpl for evaluation of white matter. We performed immunofluorescence studies for CNPase (Fig.5A–A’’’) and myelin basic protein (MBP) (Fig.5BB’’’). Quantification of relative fluorescence by MetaMorph (Molecular Devices) showed a significant decrease in CNPase staining (0.42-fold) in lesioned WT animals compared to non-lesioned WT animals. There was no difference in CNPase staining between non-lesioned NOG mice and non-lesioned WT mice. There was a decrease (0.53-fold) between non-lesioned NOG mice and lesioned WT mice, but a 1.3-fold increase when comparing lesioned NOG mice to non-lesioned NOG mice. There was a 1.7-fold increase when comparing lesioned NOG mice to non-lesioned WT mice. Finally and most importantly, there was a 1.8-fold increase in CNPase staining when comparing lesioned NOG mice to lesioned WT mice (Fig.5A’’’’). MetaMorph quantification of MBP staining showed similar although less pronounced differences. Significant changes included a 1.5-fold increase in MBP staining in lesioned NOG mice compared to non-lesioned WT mice. Most importantly, there was a 1.4-fold increase in lesioned NOG mice compared to lesioned WT mice (Fig.5B’’’’).
Figure 5. Noggin overexpression results in preservation of white matter following postnatal HI.

Representative micrographs of corpus callosum are shown for CNPase: A) WT CONT, A’) NOG CONT, A’’) WT LES, A’’’) NOG LES; and for MBP: B) WT CONT, B’) NOG CONT, B’’) WT LES, B’’’) NOG LES. (A’’’’) Quantitated relative fluorescence for CNPase, error bars represent mean ±s.e.m.: WT LES versus WT CONT, *p=0.03; NOG CONT versus WT LES, *p=0.001; NOG LES versus NOG CONT, *p=0.02; NOG LES versus WT CONT, *p=0.0007; NOG LES versus WT LES, *p<0.0001, n=4 WT CONT, 4 WT LES, 4 NOG CONT and 4 NOG LES. (B’’’’) Quantitated relative fluorescence for MBP: NOG LES versus WT CONT, *p=0.01; NOG LES versus WT LES, p=0.02, n=4 WT CONT, 4 WT LES, 4 NOG CONT and 4 NOG LES.
Noggin overexpression ameliorates the negative impact of postnatal HI on ambulation
P7 mice were lesioned and allowed to recover for 7 days. Their ability to ambulate was then assessed using the DigiGait system. At 7dpl, NOG mice were able to walk easily upon a treadmill at a speed of 8cm/sec, however none of the WT mice were able to complete this task. At 14 days, WT mice had recovered sufficiently to be able to complete this task, nonetheless their gait was not normal with abnormalities evident at the contralateral forelimb. Lesioned mice spent 65.6% ±16% more time in swinging their left forelimb (Fig.6C) and 42.7% ±12% more time in left forelimb stride (Fig.6D). As a result, with a fixed treadmill speed, their stride length was 40.4% ±12% longer (Fig.6E) and stride frequency was 30.5% ±5.5% slower (steps per sec) (Fig. 6F). By contrast, lesioned NOG mice did not differ significantly from non-lesioned NOG mice either at 7dpl or 14dpl (Fig.6C–F).
Figure 6. Noggin overexpression ameliorates the negative impact of postnatal HI on ambulation.

Representative raw (un-processed) gait tracings for WT CONT (A) and WT LES mice (B) at 14dpl are shown. Differences in gait were confined to the contralateral forelimb. Lesioned wild-type mice (WT LES) spent more time in swinging their forelimb compared to non-lesioned wild-type mice (WT CONT), *p=0.02 (C), and more time in stride, *p=0.03 (D); stride length was longer, *p=0.03 (E), and stride frequency was slower, *p=0.02 (F), n=4 WT LES and 4 WT CONT. Measurements of left forelimb swing time, stride time, stride length and stride frequency did not differ for NOG LES compared to NOG CONT at 14dpl, n=6 NOG LES and 4 NOG CONT (C–F).
Discussion
Previously, others have shown that cells of the oligodendroglial lineage mount a limited endogenous regenerative response to postnatal HI. Two weeks after moderate but not severe injury, MBP immunostaining is restored (Liu et al., 2002). Four weeks after injury, newly born OL are found in the striatum, corpus callosum and infarcted cortex (Ong et al., 2005; Zaidi et al., 2004). One proposed explanation for these finding is that early OPC accelerate their maturation following HI (Back et al., 2002). However, we have shown an increase in OPCs one week following injury 2000 (Dizon et al. 2010). This observation suggests an alternative explanation: increased commitment by uncommitted neural progenitors to the oligodendroglial fate. Indeed, neurospheres generated from the subventricular zone of lesioned mice contain OL more often than neurospheres from non-lesioned mice (Felling et al., 2006) (Yang and Levison, 2006). Furthermore, cells dissociated from these neurospheres, upon differentiation yield more newly born OL (Yang and Levison, 2006).
It is likely that NG2+ cells are the progenitor cells giving rise to OL in these models and to increased OPCs in our in vivo injury model. NG2+ cells are found throughout the postnatal CNS and give rise predominantly to OL. In the NG2CreBAC:Z/EG mouse, EGFP+ cells residing in white matter give rise to OL, whereas those residing in gray matter give rise to OL and some astrocytes (Zhu et al., 2008). NG2+ cells in adult mice co-express PDGFRA and vice versa. In the PDGFRACreERT2:Rosa26-YFP mouse, YFP+ cells give rise to OL in the corpus callosum and to projection neurons in the piriform cortex but never astrocytes (Rivers et al., 2008). Infusion of the BMP-antagonist chordin in the setting of LPC-induced demyelination increases SVZ-derived (GAD65-GFP+) NG2+ and Olig2+ progenitors as well as CC1+ and CNPase+ OL in the corpus callosum (Jablonska et al. 2010)
We reasoned that if we could increase the commitment of uncommitted neural progenitors to the oligodendroglial fate, then we might be able to protect from hypoxia-ischemia-induced white matter loss. Oligodendrogenesis has been shown to be increased in vivo through manipulation of BMP signaling, namely by conditional deletion of BMPRIa in developing mice (Samanta et al., 2007) and by conditional deletion of Smad 4 in adult mice (Colak et al., 2008). Furthermore, our lab has shown an increase in PDGFRα+ cells in the ischemic boundary zone following stroke in noggin overexpressing adult mice (Samanta et al. 2010). Since BMP4 is the family member most predominantly represented postnatally and since it is avidly bound by noggin, we chose the strategy of noggin overexpression to block BMP signaling.
Ordinarily, noggin is prominently expressed by neurons of the olfactory bulb, piriform cortex, lateral septal nucleus, the supraoptic and paraventricular nuclei of the hypothalamus and the cerebellum (Valenzuela et al., 1995). In the transgenic mouse line we used, noggin expression is under the control of the neuron specific enolase promoter that is specifically active in all terminally differentiated neurons. Thus in our transgenic line, noggin is ectopically expressed. It is not known whether endogenous noggin expression is increased in response to HI.
Surprisingly, the results were not exactly what we anticipated. Rather, we observed an interaction between HI and noggin overexpression that resulted in more white matter following injury compared to noggin overexpression alone without HI. HI in combination with noggin overexpression provoked an increase in neural progenitors (Olig2+ cells) at 7dpl. Moreover, HI in combination with noggin overexpression protected from loss of committed OPCs (PDGFRA+/Olig2+ cells). Our interpretation is that noggin removes BMP-mediated inhibition of oligodendroglial fate choice by uncommitted neural progenitors while, simultaneously, HI provides a stimulus for accelerated maturation of progenitors. Independent of HI, downregulation of BMP signaling by noggin may allow for accelerated maturation of OPCs to myelinating OLs as well. Although it is likely that noggin effects are mediated via BMP4, it is possible that noggin effects are also mediated via other BMPs or even other molecules.
Noggin may also expand the local NSPC population as has been shown in slowly dividing cells of the hippocampus (Bonaguidi et al., 2008). However, our studies showed that proliferation was not the mechanism for increased OPCs, and instead the increase must have been derived by differentiation of precursors to these cells. Additionally, noggin may increase emigration of NSPC from the SVZ (Colak et al., 2008), resulting in more Olig2+ cells in corpus callosum and subcortical white matter. From our data it not clear whether local neural progenitors or progenitors recently emigrated from the SVZ are the source of increased Olig2+ cells.
In adult rodents, components of BMP signaling including BMP7 and BMPRII increase in brain after experimental stroke (Chang et al, 2003;Charytoniuk et al, 2000), and we found an increase in levels of BMP4 after perinatal HI. Moreover, administration of BMP6 and BMP7 prior to adult stroke decreases infarct volumes and neurologic deficits (Chang et al., 2003; Chang et al., 2002; Wang et al., 2001). However the BMP antagonist noggin decreases infarct size and improves motor function in an adult stroke model (Samanta et al. 2010). There are several possible explanations for these apparently divergent findings. First, BMP4 preferentially binds to BMPRIA and/or BMPRIB whereas BMP6 and BMP7 most readily bind to ACVR1 as well as BMPRIB (Aoki et al., 2001; Ebisawa et al., 1999; ten Dijke et al., 1994; Yamashita et al., 1995). Noggin binds with highest affinity to BMP4 and BMP2, with lesser affinity for BMP7, and even less for BMP6. Thus different complexes of receptors were likely altered in the different studies. Second, the BMP7 and BMP6 were given systemically, and it is possible that effects on the nervous system indirectly reflected actions of the BMPs in the periphery. In fact the best results with BMP7 administration required administration prior to the ischemic insult. Alternatively, the peripherally administered factors only appeared to enter the ischemic region of brain whereas noggin was present throughout the brain. Thus some of the effects of noggin may have occurred outside of the immediate area of ischemia.
Notably noggin overexpression not only reduced the lesion volume after perinatal HI but also increased survival. The increased lesion volume in WT mice is unlikely to secondarily reflect changes in survival since death of the most severely affected WT mice would have been expected to bias towards a smaller lesion volume rather than a larger one. The mechanisms underlying the survival effect of noggin are unclear.
Conclusions
Using a genetic model, we have shown that noggin overexpression protects white matter from perinatal hypoxic-ischemic injury by increasing the number of oligodendrocyte progenitors and total oligodendroglial lineage cells, in turn resulting not only in the preservation of, but also the production of, more white matter. These findings explain the protection from motor deficits observed in lesioned pNSE-Noggin mice compared to non-lesioned wildtype mice.
Acknowledgements
This work was supported by NIH KO8 NS053529 (Dizon) and by NIH NINDS RO1 NS20013 and NS20778 (Kessler).
Abbreviations Footnote
- BMP
bone morphogenetic protein
- HI
hypoxia-ischemia
- WMI
white matter injury
- NSPC
neural stem/progenitor cells
- TGFβ
transforming growth factor beta
- BMPR
BMP receptor
- dpl
days post lesion
- SCWM
subcortical white matter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki H, et al. Synergistic effects of different bone morphogenetic protein type I receptors on alkaline phosphatase induction. J Cell Sci. 2001;114:1483–1489. doi: 10.1242/jcs.114.8.1483. [DOI] [PubMed] [Google Scholar]
- Back SA, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, et al. Noggin expands neural stem cells in the adult hippocampus. J Neurosci. 2008;28:9194–9204. doi: 10.1523/JNEUROSCI.3314-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CF, et al. Intravenous administration of bone morphogenetic protein-7 after ischemia improves motor function in stroke rats. Stroke. 2003;34:558–564. doi: 10.1161/01.str.0000051507.64423.00. [DOI] [PubMed] [Google Scholar]
- Chang CF, et al. Bone morphogenetic proteins are involved in fetal kidney tissue transplantation-induced neuroprotection in stroke rats. Neuropharmacology. 2002;43:418–426. doi: 10.1016/s0028-3908(02)00092-8. [DOI] [PubMed] [Google Scholar]
- Charytoniuk DA, et al. Distribution of bone morphogenetic protein and bone morphogenetic protein receptor transcripts in the rodent nervous system and up-regulation of bone morphogenetic protein receptor type II in hippocampal dentate gyrus in a rat model of global cerebral ischemia. Neuroscience. 2000;100:33–43. doi: 10.1016/s0306-4522(00)00246-3. [DOI] [PubMed] [Google Scholar]
- Colak D, et al. Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J Neurosci. 2008;28:434–446. doi: 10.1523/JNEUROSCI.4374-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MR, et al. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, et al. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Dizon M, et al. Hypoxia-ischemia induces an endogenous reparative response by local neural progenitors in the postnatal mouse telencephalon. Dev Neurosci. 2010;32:173–183. doi: 10.1159/000313468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisawa T, et al. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J Cell Sci. 1999;112(Pt 20):3519–3527. doi: 10.1242/jcs.112.20.3519. [DOI] [PubMed] [Google Scholar]
- Felling RJ, et al. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, et al. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Gross RE, et al. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 1996;17:595–606. doi: 10.1016/s0896-6273(00)80193-2. [DOI] [PubMed] [Google Scholar]
- Guha U, et al. Bone morphogenetic protein signaling regulates postnatal hair follicle differentiation and cycling. Am J Pathol. 2004;165:729–740. doi: 10.1016/S0002-9440(10)63336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton TG, et al. Gait dynamics in trisomic mice: quantitative neurological traits of Down syndrome. Physiol Behav. 2004;82:381–389. doi: 10.1016/j.physbeh.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hogan BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- Jablonska B, et al. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Hoon AH., Jr Cerebral palsy. Neuromolecular Med. 2006;8:435–450. doi: 10.1385/NMM:8:4:435. [DOI] [PubMed] [Google Scholar]
- Kan L, et al. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol. 2004;165:1107–1115. doi: 10.1016/S0002-9440(10)63372-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, et al. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res. 2002;51:25–33. doi: 10.1203/00006450-200201000-00007. [DOI] [PubMed] [Google Scholar]
- Mabie PC, et al. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, et al. Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial-astroglial progenitor cells. J Neurosci. 1997;17:4112–4120. doi: 10.1523/JNEUROSCI.17-11-04112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehler MF, et al. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997;20:309–317. doi: 10.1016/s0166-2236(96)01046-6. [DOI] [PubMed] [Google Scholar]
- Menn B, et al. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong J, et al. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res. 2005;58:600–606. doi: 10.1203/01.PDR.0000179381.86809.02. [DOI] [PubMed] [Google Scholar]
- Plane JM, et al. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–595. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Rivers LE, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, et al. Noggin protects against ischemic brain injury in rodents. Stroke. 2010;41:357–362. doi: 10.1161/STROKEAHA.109.565523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, et al. BMPR1a signaling determines numbers of oligodendrocytes and calbindin-expressing interneurons in the cortex. J Neurosci. 2007;27:7397–7407. doi: 10.1523/JNEUROSCI.1434-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola A, et al. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57:481–487. doi: 10.1203/01.PDR.0000155760.88664.06. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, et al. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem. 1994;269:16985–16988. [PubMed] [Google Scholar]
- Valenzuela DM, et al. Identification of mammalian noggin and its expression in the adult nervous system. J Neurosci. 1995;15:6077–6084. doi: 10.1523/JNEUROSCI.15-09-06077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Bone morphogenetic protein-6 reduces ischemia-induced brain damage in rats. Stroke. 2001;32:2170–2178. doi: 10.1161/hs0901.095650. [DOI] [PubMed] [Google Scholar]
- Winter S, et al. Trends in the prevalence of cerebral palsy in a population-based study. Pediatrics. 2002;110:1220–1225. doi: 10.1542/peds.110.6.1220. [DOI] [PubMed] [Google Scholar]
- Yamashita H, et al. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J Cell Biol. 1995;130:217–226. doi: 10.1083/jcb.130.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Levison SW. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience. 2006;139:555–564. doi: 10.1016/j.neuroscience.2005.12.059. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, et al. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, et al. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]