

Abstract
CD36, a class B scavenger receptor present in microglia, endothelium and leukocytes, plays key role in ischemic brain injury by promoting the expression of inflammatory genes and production of reactive oxygen species (ROS). However, it is not known whether ischemic brain damage is mediated by CD36 activation in resident brain cells, i.e., microglia, or by blood-borne cells that infiltrate the brain. To address this question, we studied oxygen-glucose deprivation (OGD) in hippocampal slice cultures, a model of ischemic injury that does not involve cells extrinsic to the brain. We found that CD36 gene knockout does not afford protection of hippocampal slices to OGD-induced cytotoxicity. In contrast, immunoactivated bone marrow-derived monocytes-macrophages (BMM) from wild type (WT) mice trigger hippocampal damage when incubated with brain slices via a mechanism that is prevented in CD36−/− BMM. The neurotoxic activity of CD36+/+ BMM was attributed to reactive oxygen species (ROS), since it was concomitant with increased ROS production and could be prevented by treatment with a selective ROS scavenger, MnTBAP, or a peroxynitrite decomposition catalyst, FeTPPS. Importantly, ROS production and accumulation 3-nitrotyrosine in hippocampal proteins (a hallmark of peroxynitrite production) was significantly dampened in immunoactivated CD36−/− BMM, whereas production of NO-derived metabolites (nitrite and nitrate) was unaltered. We conclude that CD36 signaling may not contribute to injury induced by OGD in the brain itself, but is involved in the neurotoxicity mediated by activated BMM. These findings are consistent with the hypothesis that CD36 in infiltrating inflammatory cells drives peroxynitrite-mediated ischemic brain damage. Accordingly, targeting CD36 in the vascular compartment may protect against neurotoxicity in the ischemic brain.
Introduction
Ischemic brain injury results from the concerted action of multiple pathogenic factors acting in a well-defined temporal sequence (Moskowitz et al. 2010) Whereas energy failure, glutamate and Ca2+ dysregulation mediate the early phase of the injury, activation of inflammatory pathways contributes to the expansion of the damage in the late stages of the ischemic cascade (Moskowitz et al. 2010). After cerebral ischemia, cytokines, adhesion molecules and chemokines lead to influx of circulating leukocytes into the brain and activation of resident inflammatory cells, which contribute to the damage (Iadecola 2004). In experimental animals, inhibition of post-ischemic inflammation ameliorates ischemic injury with an extended time widow {Wang, 2007 #5; Iadecola et al. 2004), but initial attempts to treat stroke patients using similar approaches have not been successful (Sughrue et al. 2004). This failure reflects, in part, our incomplete understanding of the biological processes underlying post-ischemic inflammatory signaling (Furuya et al. 2001; Sughrue et al. 2004; Lakhan et al. 2009). In particular, the relative roles that infiltrating blood-borne cells and resident brain cells play in the mechanisms of the injury remain to be elucidated.
The scavenger receptor CD36 has emerged as a key factor in post-ischemic inflammatory signaling. CD36, expressed in monocytes-macrophages, endothelial cells and microglia, recognizes a wide variety of ligands, like modified lipids, β-amyloid, apoptotic cells, and anti-angiogenic factor thrombospondin-1 (Silverstein and Febbraio 2009). CD36, acting in concert with other scavenger receptors and toll-like receptors, may recognize molecular patterns that are generated by cellular damage (Akashi-Takamura and Miyake 2008) and trigger adaptive cellular responses, including production of reactive oxygen species (ROS) and expression of proinflammatory genes (Silverstein and Febbraio 2009). With excessive activation, however, these cellular responses may turn maladaptive and mediate cytotoxicity (Nathan and Ding 2010). Indeed, CD36 has been found to play a critical role in the inflammatory reaction to cerebral ischemia. After occlusion of the middle cerebral artery (MCA), inflammatory gene expression, infiltration of blood-borne leukocytes and microglial activation is dampened in CD36−/− mice, effects related to suppression of the activity of the pro-inflammatory transcription factor NF-κB (Kunz et al. 2008). Consequently, ROS production, infarct volume and motor deficits produced by MCA occlusion are reduced in CD36−/− mice (Cho et al. 2005). However, it remains to be determined whether these effects are mediated by CD36 activation in intrinsic brain cells, mainly microglia, or in blood-borne cells such as monocytes-macrophages that infiltrate the post-ischemic brain.
In the present study we used oxygen-glucose deprivation (OGD) in hippocampal slice cultures to investigate whether CD36 in resident brain cells contributes to hypoxic-ischemic damage. We found that hippocampal slices lacking CD36 are not protected from the OGD-induced injury. However, neurotoxicity produced by application of immunoactivated bone marrow-derived monocytes-macrophages (BMM) to the slices was markedly attenuated with CD36−/− BMM and this cytoprotection was associated with diminished ROS production and accumulation of proteinaceous 3-nitrotyrosine (3-NT). These findings highlight the potential importance of CD36 signaling in infiltrating hematogenous cells in the component of the injury mediated by oxidative and nitrative stress.
Material and Methods
Materials and reagents
Hank’s balanced salt solution (HBSS), Eagle’s Basal Medium, Propidium iodide (PI) and other tissue culture supplies were from Invitrogen (Carlsbad, CA). Millicell CM membrane insert and polyvinylidene difluoride (PVDF) membrane for western blot were from Millipore (Bedford, MA). Horse serum and other chemicals were from Sigma (St. Louis, MO). Granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from R & D (Minneapolis, MN). MnTBAP (Mn(III)tetrakis(4-benzoic acid)porphyrin Chloride) and FeTPPS (5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrinato Iron (III), Chloride) are from Calbiochem (San Diego, CA). Pam3CSK4 is from Invivogen (San Diego, CA).
Methods
Organotypic mouse hippocampal slice cultures
All procedures involving newborn mice for hippocampal slices cultures were approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee. We used CD36−/− or iNOS−/− mice congenic with the C57BL6 strain. C57BL6 mice were used as wild type (WT) controls. Hippocampal slice cultures were prepared according to the procedure of Muller et al (Muller et al. 2001) as previously described (Kawano et al. 2006; Kim et al. 2007; Yin et al. 2010). Briefly, hippocampi from 5–6 day old mouse pups were dissected out aseptically and coronal slices of 350 μm in thickness were obtained using a tissue chopper (Mc Ilwain tissue chopper, Vibratome Company, St. Louis, MO). Slices were transferred to the Millicell CM membrane insert (30 mm, 0.4 μm) placed in a 6-well plates filled with 1 ml medium (25% horse serum, 50% Eagle’s Basal Medium, 25% HBSS, 5 mg/ml glucose). Six slices were seeded in each well. The slice cultures were maintained in a humidified chamber at 37°C with 5% CO2 and culture medium was changed twice a week. The slices were cultured for 14 days before being used for experiments. CD36 mRNA was found to be significantly expressed in hippocampi from 6 day-old mouse pups (supplemental figure S1).
Oxygen-glucose deprivation and cell death assessment in hippocampal slices
Slices were rinsed twice with warm OGD buffer (125 mM NaCl, 5 mM KCl, 1.2 mM Na2PO4, 26 mM NaHCO3, 1.8 mM CaCl2, 0.9 mM MgCl2, 10 mM Hepes, pH 7.4) plus 10 mM glucose and incubated for 30 min with the same rinsing buffer. The slices were then rinsed with OGD buffer twice and transferred to the OGD chamber (Billups-Rothenberg, San Diego, CA) and flushed with anoxic gas (95% N2 and 5% CO2) at 21 Liter/min for 5 min. The chamber was then sealed and placed into 37°C incubator for 1 hr. Sham controls were rinsed with OGD buffer but incubated under normoxia. After OGD, the slices were transferred to regular culture medium and incubated for a period of time (24, 48 and 72 hrs) before cell death assessment. Before fluorescence images were taken, propidium iodide (PI) was added to the culture medium (5 μg/ml final concentration) for 1 hr and then washed away. Longer incubation times were avoided as they result in false positives (unpublished observations). Cell death in slices was assessed based on PI staining, as previously described (Kawano et al. 2006). Briefly, slices were imaged using a Nikon inverted fluorescence microscope equipped with a digital camera operated by a Mac computer using IPLab software. The slices were then treated with 100 μM NMDA for an additional 24 hrs and, after re-incubation with PI for 1 hr, images were recorded to serve as maximum fluorescence intensity in the slices. The same camera settings (exposure time, filter setting, gain) were used throughout all experiments. To calculate the percentage of cell death after OGD in the hippocampal CA1 region, digital images of PI staining at three different time points (before and after OGD, and after NMDA treatment) were outlined to define the same CA1 region and mean fluorescence intensity at corresponding time points: Fbasal, FOGD and Fmax. The percent cell death was calculated using the formula (FOGD - Fbasal)/(Fmax - Fbasal) X100% (% of max cell death) (Kawano et al. 2006); (Kim et al. 2007; Yin et al. 2010). The extent of the injury assessed by this method corresponds well to that assessed by immunostaing with NeuN and the nuclear marker To-Pro3 (data not shown). Pam3CSK4 (5μg/ml) was added to CD36+/+ and −/− slices 2 hrs before OGD and cell death was assessed 24 hrs after OGD.
Microglial expansion in hippocampal slices
Microglial cells in hippocampal slices were expanded by incubation with GM-CSF (10 ng/ml) for three days as previously described (Duport and Garthwaite 2005; Yin et al. 2010). The microglial cell population was activated by treatment with a combination of LPS (1 μg/ml) and IFN-γ (100 units/ml) for 3 days. Cell death was assessed as described above.
Bone marrow derived monocyte-macrophages
BMM were isolated and cultured according to an established protocol (Zhang et al. 2008). Briefly, femur bones from the CD36+/+, CD36−/− or iNOS−/− mice were dissected out and both ends cut open. The bone marrow was flushed out with a syringe needle filled with phosphate-buffered saline (PBS). The bone marrow was washed, centrifuged, and resuspended in BMM culture medium (DMEM/F12 medium supplemented with 10% FBS and 30% L-929 cell conditioned medium as a source of M-CSF) (Austin et al. 1971). After counting cell numbers, bone marrow cells were seeded in 15 cm non-tissue culture dishes at a density of 4×106 cells/dish in BMM medium. BMM were cultured for 7 days in a tissue culture incubator and harvested for use in experiments by trituration with PBS. Histological analysis following Giemsa staining revealed that cells in culture were essentially pure monocytes-macrophages (data not shown). For studies testing the effect of BMM on hippocampal cell viability, BMM were added to the slices at a density of 105 cells/slice in the absence or presence of LPS and IFN-γ (100 units/ml). Tissue damage was assessed 5 days later using PI as described for OGD. Because the cell death caused by activated BMM is not restricted to CA1, in these experiments maximum cell death was obtained by treatment with 0.1% Triton x-100 before PI incubation.
Flow cytometry
Cells were harvested and resuspended in flow cytometry buffer (PBS without Ca2+ and Mg2+, supplemented with 2% FBS and 0.05% sodium azide). 5 × 105 cells were incubated with anti-mouse specific antibodies or adequate Ig isotype controls for 20 minutes at 4°C as follows: 0.05 μg anti-Ly6C-FITC, clone HK1.4, Biolegend; 0.1 μg anti-CD11c-FITC, clone N418, eBioscience; 0.1 μg anti-CD11b-PE, clone M1/70, BD Bioscience; 0.05 μg anti-CD115-PE, clone AFS98, Biolegend; 0.1 μg anti-CD45-APC, clone 30F-11, eBioscience; 0.1 μg anti-CD54-AlexaFluor 647, clone YN1/1.7.4, Biolegend. After two washes cells were analyzed on an Accuri C6 flow cytometer (Accuri Cytometers Inc, Ann Arbor, MI). Data were analyzed and graphed with FlowJo Vers. 7.6.1 software package (Fl-A = fluorescent peak area).
Measurement of NO metabolites and superoxide from activated BMM
An equal number of BMM were transferred into tissue culture plates and activated with LPS and IFN-γ (see above). Nitrate and nitrite, the stable end products of NO metabolism, were measured in the culture medium 1, 2, 3, 4, 5 days later, based on the method of Griess (Grisham et al. 1996). Briefly, culture medium from BMM was collected, centrifuged and aliquoted into triplicate wells in a 96-well plate. Nitrate (NO3−) in the medium was reduced by nitrate reductase to nitrite (NO2−). The concentration of total nitrite was determined by adding Griess reagent and quantifying A540 in a microplate reader, vs. a standard curve constructed using linear dilutions of nitrate and nitrite stock solutions.
ROS production by immunoactivated BMM was measured based on reaction with a fluorescence dye, dihydroethidium (DHE, Invitrogen), according to the following protocol: BMM were counted and seeded in equal number in tissue culture plates and immunoactivated as described above. After 1, 2, 3, 4, and 5 days in culture, the cells were incubated with DHE (2 μM) for 60 min before being harvested for analysis. Cells were washed, scraped off the plates, collected in PBS, and centrifuged at 4°C for 5 min @ 1000 rpm. Cell supernatants were removed and the cell pellets were resuspended and fixed in 5 ml of 4% paraformaldehyde for 10 min. The cells were collected by centrifugation, resuspended in PBS, and counted. Aliquots of cells (5×105) were placed into 96-well plate in duplicate and volumes were adjusted with PBS to 125 μl/well. Fluorescence was measured in a plate reader using EX 340 nm and EM 595 nm filters. Data were averaged and presented as relative fluorescence intensity.
Quantification of 3-NT by HPLC with electrochemical detection (ECD)
The protocol for 3-NT quantification by HPLC with electrochemical detection was similar to that previously described (Nuriel et al. 2008) with the following modifications. Protein samples (a minimum of 200 μg/sample) were digested with proteinase K (150 U/mg; 8 hr at 55°C). Cooled digests were precipitated with 3 volumes of ice-cold buffer (0.1 M phosphoric acid and 0.23 M TCA), vortexed, incubated on ice for 5 min, and centrifuged at 12,000 x g for 5 min at 4°C. The resulting supernatants were extracted with 2 volumes of chloroform each. The aqueous fractions containing 3-NT were dried at room temperature (SpeedVac) followed by reconstitution with vacuum-filtered (0.2-μm nylon membrane) and degassed HPLC mobile-phase buffer containing 90 mM sodium acetate, 35 mM citric acid, 130 μM EDTA, and 460 μM sodium octane sulfonate (pH 4.35) prepared in 18-MΩ resistance water. An isocratic HPLC system with a multichannel electrochemical (EC) CoulArray detector and EC cell (ESA, Inc.) was used to resolve 3-NT (+700 mV, RT = 15 min) from other species using a 100-mm C18 column (Microsorb-MV, Varian) and flow rate at 0.75 ml/min.
Statistical analysis
Data are presented as mean ± SEM. Two-group comparisons were evaluated by the unpaired t-test. Multiple pairwise comparisons were evaluated by one way analysis of variance followed by Newman-Keuls test.
Results
1. The damage produced by OGD is not attenuated in CD36−/− hippocampal slices
Organotypic hippocampal slice cultures from WT and CD36−/− mice were subjected to ODG. In WT slices, OGD produced damage predominantly in the CA1 region of the hippocampus, as previously described (Kawano et al. 2006; Kim et al. 2007; Yin et al. 2010). In CD36−/− slices, the magnitude and spatial distribution of the damage did not differ from that of WT slices both at 24 and 72 hrs after OGD (Fig. 1A, B). To rule out the possibility that the lack of effect of CD36 was due the fact that its ligands were not generated, we incubated the slices with Pam3CSK4, a synthetic triacylated lipopeptide that specifically activates the macromolecular complex formed by CD36 with TLR2/1 (Hoebe et al. 2005; Abe et al. 2010). Pam3CSK4 did not affect cell viability in sham-treated slices (data not shown) and did not enhance the damage induced by OGD in CD36+/+ or CD36−/− slices (Fig. 1C). Therefore, absence of CD36 failed to protect hippocampal slices from OGD-induced cytotoxicity.
Figure 1.
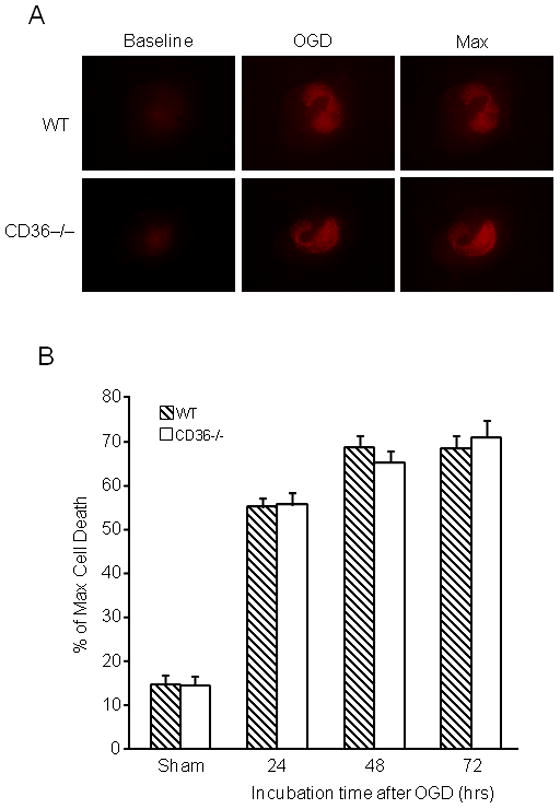
Lack of CD36 in hippocampal slices does not confer neuroprotection against OGD. Hippocampal slice cultures were cultured for 14 days and subjected to OGD for 45 min, followed by 23 hrs of normoxic incubation in culture medium (reperfusion). Cell death was assessed based on propidium iodide (PI) uptake. No difference in cell death was observed between CD36 KO and WT slices. (A): representative photomicrographs of hippocampal slices from CD36 KO and WT mice under the indicated treatment conditions. Baseline: PI uptake before OGD. OGD: Slices treated with OGD for 45 min. Max: maximum degree of cell death (PI uptake), observed after overnight incubation with 1mM of NMDA. (B). Quantitative assessment of cell death at 24, 48 and 72 after OGD expressed as % of max PI uptake (see above). *p<0.05 from the corresponding sham group; analysis of variance and Newman-Keuls test; n: 35–50 slices/group, from 7 separate experiments. (C) Effect of Pam3CSK4 on cell death after OGD. Pam3CSK4 did not increase the injury in CD36+/+ or CD36−/− slices (p>0.05; n=18–24, from 3 experiments).
2. Role of CD36 in the hippocampal damage produced by microglial activation
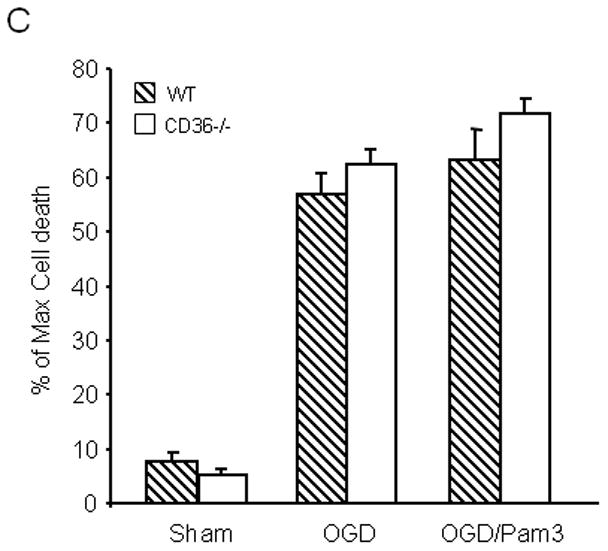
Microglial cells, which express CD36, have been implicated in tissue damage produced by cerebral ischemia and other brain injury modalities (Mabuchi et al. 2000; Block et al. 2007; del Zoppo et al. 2007). To determine whether CD36 is required for the cytotoxicity of immunoactivated microglia in hippocampi, we investigated the effect of microglial activation with LPS/INF-γ in WT and CD36−/− slices (Duport and Garthwaite 2005). As illustrated in Figure 2A, microglial activation induced cell death, but the magnitude of this effect did not differ in WT and CD36−/− slices. To increase the extent of tissue damage to a level comparable to that produced by OGD (Fig. 1), we used GM-CSF to expand the population of resident microglia prior to activation with LPS/INF-γ, as previously described (Duport and Garthwaite 2005; Yin et al. 2010). Microglial expansion increased the damage in WT slices, an effect that was slightly attenuated in CD36−/− slices (Fig. 2B). In contrast, the hippocampal damage induced by expanded and activated microglia was completely blocked in iNOS−/− slices (Fig. 2B), consistent with a major role of iNOS-derived NO in this injury model.
Figure 2.
Effect of activation of resident microglia on cell death in hippocampal slices. (A) Hippocampal slices, after culture for 14 days, were treated with LPS (10μg/ml) and IFN-γ (100Units/ml) for 5 days to activate resident microglial cells. (B) The microglia population in 12 day old hippocampal slice cultures was expanded by treatment with GM-CSF (10 ng/ml) for three days, followed by activation with LPS/IFN-γ treatment (LPS: 10 μg/ml; INF-γ: 100 Units/ml) for 5 days. At the end of the indicated treatment period, cell viability in hippocampal slices was determined based on propidium iodide (PI) uptake. *p<0.05 from WT; #p<0.05 from WT (vehicle), $p<0.05 from CD36−/− (vehicle) and WT (LPS/IFN-γ+GM-CSF). n= 30–36 slices in each group from 5 sets of separate experiments.
3. CD36-deficient BMM produce less hippocampal damage relative to CD36+/+ BMM
Activated mononuclear cells enter brain after cerebral ischemia-reperfusion and contribute to ischemic brain injury (Gelderblom et al. 2009; Hyman et al. 2009; Felger et al. 2010). In these experiments, we sought to determine whether activated BMM induce tissue damage in hippocampal slices, and if so, whether the cytotoxic effect is CD36 dependent. First, we used flow cytometry to characterize WT and CD36−/− BMM. No differences in major surface antigens that define the BMM cell population were found between CD36+/+ and CD36−/− BMM (Fig. 3). Next, BMM from CD36+/+ and CD36−/− mice were applied to WT slices and tissues damage was assessed 5 days later. Without immunoactivation, neither CD36+/+ nor CD36−/− BMM caused hippocampal cell damage when incubated with the slices (Fig. 4A, C). However, incubation of immunoactivated CD36+/+ BMM with CD36+/+ slices resulted in extensive hippocampal cell damage (Fig. 4B, C). The spatial distribution of the injury involved all hippocampal sectors and was not predominantly limited to the CA1 region, as observed for OGD (Fig. 1). Notably, hippocampal damage was markedly attenuated when CD36−/− BMM were applied to the CD36+/+ slices (Fig 4B). In contrast, the cytotoxicity produced by activated CD+/+ BMM in CD36−/− slices (80±3% of max. cell death; n=12) was indistinguishable from that observed in CD36+/+ slices (79±4%; n=10; p>0.05). These results demonstrate that CD36 in BMM, but not the brain, contributes to the immunoactivation-induced cytotoxicity in hippocampal slices. The hippocampal damage induced by BMM lacking iNOS was also markedly attenuated compared to iNOS+/+ BMM (Fig. 4B, C). Therefore, CD36 and iNOS are both dominant contributors to the cytotoxicity of activated BMM in hippocampal slices.
Figure 3.
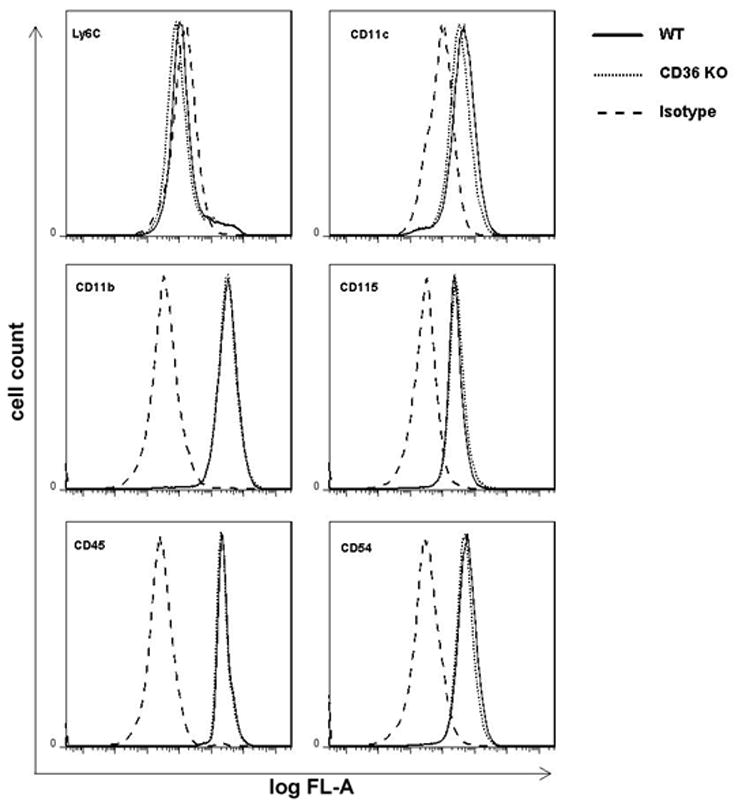
Flow cytometry of BMM derived from WT and CD36 −/− mice. BMM (5 × 105 cells/assay) were cultured for 7 days and labeled with fluorescently-conjugated primary antibodies to monocytes/macrophages surface markers as described in Methods. Isotype specific immunoglobulins were used in each sample to demonstrate the specificity of the profile. Results show that CD+/+ and CD36−/− BMM were indistinguishable, considering their expression of specific cell surface markers.
Figure 4.
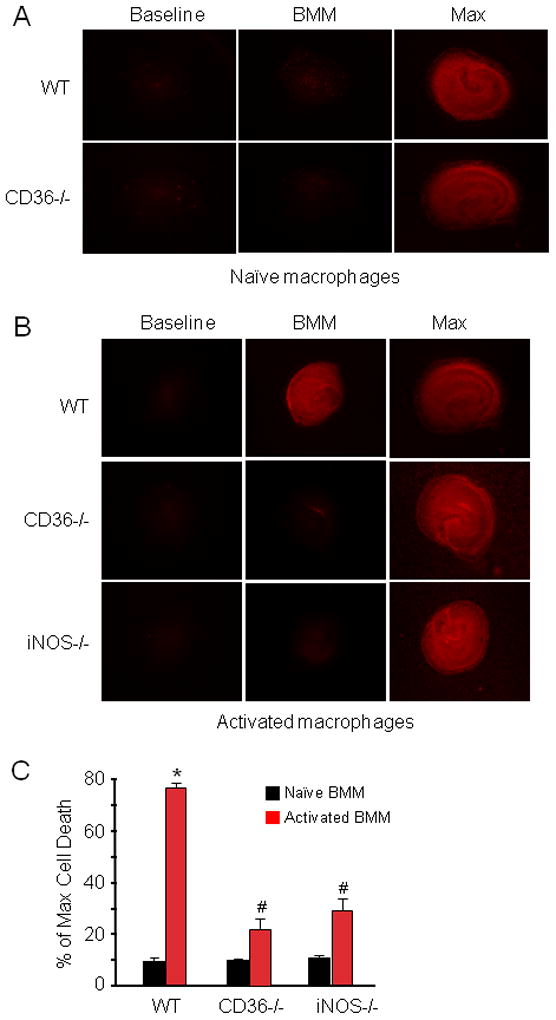
BMM activation induces marked cell death in hippocampal slices in a CD36-dependent manner. (A): WT or CD36−/− naïve (not-activated) BBM were applied to hippocampal slices (14 days in culture, 1×105 cells/slice) and cultured for 5 days. (B): WT, CD36−/− or iNOS−/− BMM were applied to the slices and activated with LPS (0.5μg/ml) and IFN-γ (10Units/ml). Cell viability was assessed five days later. WT, but not CD36−/− or iNOS−/− BMM, induce cell death. (C) Quantitative analysis of cell death expressed as percentage of maximum induced by 0.1% Triton-x100 incubation. *p<0.05 from WT naïve BMM and from WT activated BMM; # p<0.05 from naïve BMM; n=24 slice/group from 4 separate experiments.
4. ROS production, but not NO production, is attenuated in CD36−/− BMM
NO and ROS are major effectors of monocytic cell mediated cytotoxicity (Pacher et al. 2007). Therefore, we sought to determine whether the attenuated cytotoxicity of immunoactivated CD36−/− vs CD+/+ BMM was associated with a deficit in NO and/or ROS production. Toward this end, CD36+/+ and CD36−/− BMM were immunoactivated with LPS/IFN-γ and culture medium was collected at different time points and assayed for the NO metabolites nitrite and nitrate (Grisham et al. 1996), while ROS production in BMM was assessed using the superoxide-sensitive dye DHE (Benov et al. 1998). In these experiments BMM were not applied to brain slices. As illustrated in figure 5A, activation of WT BMM increased the accumulation of NO metabolites, which was first observed on culture day 1 and reached a plateau at day 2. The increase in NO metabolites did not differ between CD36+/+ and CD36−/− BMM. In control experiments, as expected, NO production was markedly suppressed in iNOS−/− (CD36+/+) BMM (Fig. 5A), attesting to the fact that iNOS is the major NO source in this model. Immunoactivated CD36+/+ BMM increased ROS production, which started at day 1 and remained elevated throughout the experiment (Fig. 5B). This increase in ROS was not observed in CD36−/− BMM. In contrast, activated iNOS−/− BMM exhibited an increase in ROS that was comparable to that of CD36+/+ BMM (Fig. 5B). These observations suggest that CD36 is required for the ROS production in this model where as iNOS is the major source of NO.
Figure 5.
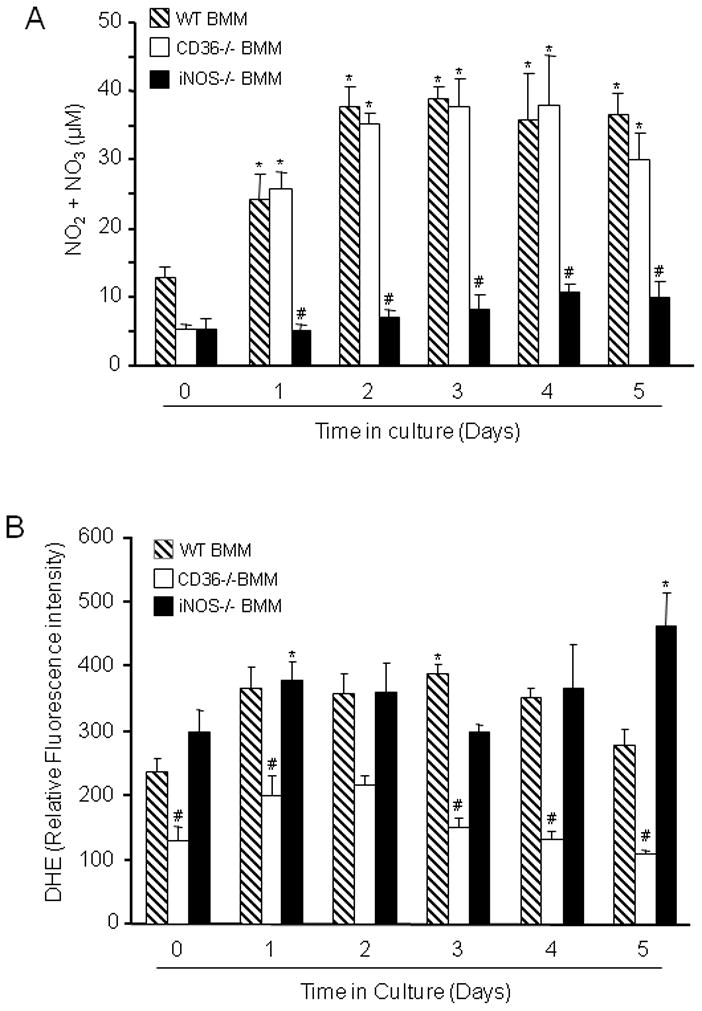
Measurement of NO metabolites and ROS in LPS/IFN-γ activated WT, CD36−/− and iNOS−/− BMM. (A): Nitrite and nitrate concentration was measured in the culture medium by the Griess reaction. *p<0.05 from WT time 0; #p<0.001 from WT and CD36−/−; analysis of variance and Newman-Keuls test; n=8 from 4 sets of separate experiments. (B): ROS were measured in BMM suspensions using DHE as an indicator. ROS production is suppressed in CD36−/− BMM. *p<0.05 from WT time 0; #p<0.05 from WT and iNOS−/− BMM; analysis of variance and Newman-Keuls test; n=8 slices/group from 4 separate experiments.
5. The hippocampal damage produced by BMM is attenuated by the ROS scavenger MnTBAP and the peroxynitrite decomposition catalyst FeTPPS
The lack of increase in ROS production and cytotoxicity with CD36−/− BMM raises the possibility that ROS contribute to the cytotoxicity of activated CD36+/+ BMM. Therefore, we examined the effect of the ROS scavenger MnTBAP on the extent of hippocampal damage produced by CD36+/+ BMM. As illustrated in figure 6A, MnTBAP markedly attenuated the cytotoxicity, implicating ROS in the cell death mechanisms. Since superoxide reacts at a near diffusion-limited rate with NO to form peroxynitrite, a highly toxic chemical species (Pacher et al. 2007), we hypothesized the peroxynitrite is a key contributor to CD36+/+-dependent neurotoxicity. To this end, we investigated whether the peroxynitrite decomposition catalyst FeTPPS affords protection against hippocampal cell death produced by CD36+/+ BMM. As shown in Fig. 6B, FeTPPS attenuated the hippocampal cell death caused by CD36+/+ BMM to a level comparable to that obtained with MnTBAP.
Figure 6.
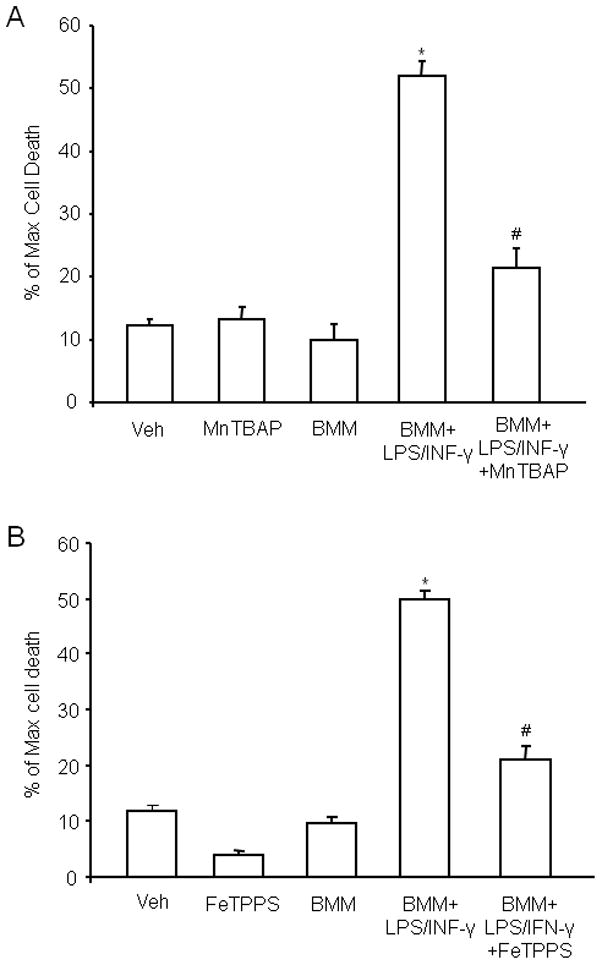
The ROS scavenger MnTBAP and the peroxynitrite decomposition catalyst FeTPPS counteract the hippocampal damage produced by activated WT BMM. MnTBAP or FeTPPS (100 μM on 1st day and 50 μM subsequent days until day 4 for both reagents) was added to the culture medium. Cell viability in slices was measured at day 5 by PI uptake. (A): MnTBAP attenuates BMM-induced cell death. *p<0.05 from vehicle (veh), MnTBAP, and non-activated BMM (BMM); #p<0.05 from BMM+LPS/INFγ. (B) FeTPPS attenuates the cell death produced by activated BMM. *p<0.05 from veh, FeTPPS and BMM; #p<0.05 from BMM+LPS/INFγ; for both A and B: analysis of variance and Newman-Keuls test; n=28–35 slices/group from 4–5 separate experiments.
6. Immunoactivated BMM increase the levels of protein-incorporated peroxynitrite marker 3-NT in a CD36-dependent manner
The observations presented above implicate peroxynitrite in the hippocampal cell death produced by activated CD36+/+ BMM. Considering that CD36−/− BMM fail to produce ROS when activated, it is conceivable that their attenuated cytotoxic activity is a consequence of a diminished capacity for triggering peroxynitrite formation. To test this possibility, we used HPLC with ECD to quantify the content of 3-NT, a hallmark reaction product of peroxynitrite, in protein from hippocampal slices after incubation with CD36−/− vs. CD36+/+ BMM. 3-NT was assessed at 5 days after application of activated macrophages because tissue damage was assessed at this time. Whereas CD36+/+ BMM markedly increased proteinaceous 3-NT in the slices (Fig. 7), this increase in 3-NT was virtually abolished in slices treated with immunoactivated CD36−/− BMM.
Figure 7.
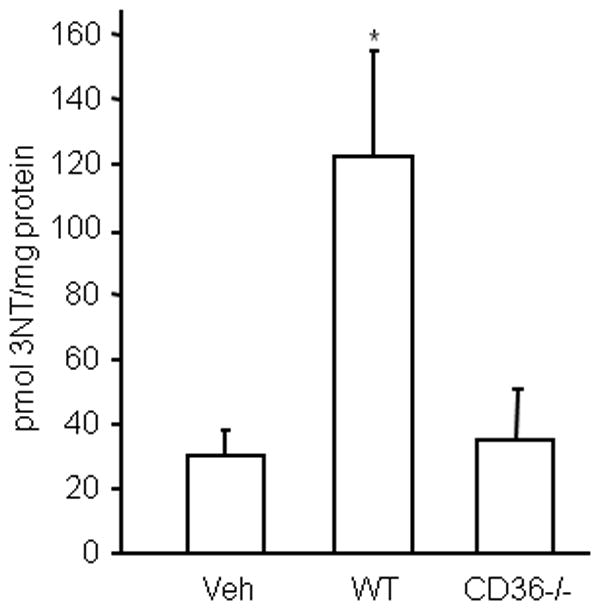
BMM increase 3NT levels in hippocampal slices in a CD36-dependent manner. Hippocampal slices were overlaid with BMM from WT and CD36 KO mice and activated with LPS/INFγ. Five days later, slices were collected for 3-NT assay by HPLC with electrochemical detection. *p<0.05, from vehicle (veh); n=5/group from 3 sets of separate experiments.
Discussion
We used an in vitro model of hypoxia-ischemia to examine whether or not the contribution of CD36 to ischemic brain injury is attributable to CD36 activation in cells intrinsic to the brain. We found that the OGD-induced hippocampal damage was not attenuated in CD36−/− slices, even in the presence of a CD36-TLR2/1 ligand, indicating that CD36 is unlikely to contribute to the tissue damage induced in this model of hypoxia-ischemia. Because mononuclear cells invade the post-ischemic brain and are known to participate in the injury (Gelderblom et al. 2009; Hyman et al. 2009; Felger et al. 2010), we further examined whether the cytotoxicity of BMM in hippocampal slices is CD36-dependent. Incubation of CD36−/− BMM with CD36+/+ hippocampal slices produced substantially less neurotoxicity than CD36+/+ BMM. Notably, the cytotoxicity of iNOS−/− BMM was also attenuated, demonstrating a prominent role of iNOS-derived NO in this brain injury model. Whereas immunoactivation of CD36−/− BMM generated levels of NO metabolites comparable to those of CD36+/+ BMM, we observed that ROS production was markedly suppressed, implicating ROS as a potential mediator of the injury. Consistent with this hypothesis, the damage produced by BMM was attenuated by the ROS scavenger MnTBAP. Peroxynitrite, the product of the reaction of NO with superoxide, is known to be involved in the cytotoxicity of monocytes-macrophages (Pacher et al. 2007). Therefore, we further examined whether the dampened cytotoxicity of CD36−/− BMM is related to a decrease in peroxynitrite production. First, we established that the peroxynitrite decomposition catalyst FeTPPS attenuated the hippocampal injury produced by CD36+/+ BMM, in accord with the role of peroxynitrite in the tissue damage. Next, we found that levels of 3-NT, in proteins from hippocampal slices were diminished after exposed to CD36−/− vs. CD36+/+ BMM. Collectively, these observations are consistent with the hypothesis that resident brain cells expressing CD36, particularly microglia, may not contribute to tissue damage induced by OGD. Rather, our findings support the hypothesis that CD36 in infiltrating leukocytes, possibly monocytes-macrophages, induce cytotoxicity in the post-ischemic brain through the accelerated production of superoxide- and NO-derived peroxynitrite.
Microglia are resident brain cells involved in immune surveillance and regulation (Perry et al. 2010). Activated microglial cells can contribute to the cytotoxicity of acute inflammation in the setting of ischemic brain injury and other neurological pathologies (Brown and Neher 2010). However, microglia can also counteract the deleterious effects of inflammation by producing IL-10 and play a role in tissue repair by producing TGF-β and other growth factors (Brown and Neher 2010). In microglia, CD36 is involved in phagocytic activity and in inflammatory signaling induced by β-amyloid (Moore et al. 2002; Zhao et al. 2007), but its role in neurotoxicity remains unclear. Our finding that CD36−/− hippocampal slices are not protected from OGD suggests that CD36 expression in resident inflammatory cells, predominantly microglia, is unlikely to participate in this injury modality. Such lack of involvement in OGD was not due to the fact that CD36−/− microglia is not cytotoxic in brain slices, because activation of resident microglia was able to produce tissue damage both in WT and CD36−/− slices, but not in iNOS−/− slices. Although we cannot rule out the possibility that CD36 ligand(s) were not generated during OGD, the fact that the CD36-TLR2/1 activator Pam3CSK4 does not enhance the damage would suggest that CD36 plays a relatively minor role compared to iNOS in the cytotoxicity of resident microglia and are in agreement with findings in other models (Hooper et al. 2009). On the other hand, increasing evidence supports the involvement of BMM in ischemic injury. Experiments in mice transplanted with GFP positive bone marrow have clearly shown the presence of BMM in the peri-infarct regions in rodent models of focal cerebral ischemia (Schilling et al. 2003; Tanaka et al. 2003). Recent data indicate that BMM may contribute to tissue damage by releasing IL-23 to promote cytotoxicity by γ–δT-lymphocytes (Shichita et al. 2009).
Activated BMM exhibited an increase in ROS and iNOS-derived NO, assessed by the accumulating metabolites nitrite and nitrate. Both ROS and NO were required for the cytotoxicity because the hippocampal damage produced by BMM was attenuated by treatment with the ROS scavenger MnTBAP as well as in iNOS−/− slices, in which NO production was suppressed. Furthermore, the hippocampal damage produced by activated CD36−/− BMM was markedly attenuated, an effect associated with a reduction in ROS. Such lack of cytotoxicity was not due to a reduced level of activation because activated CD36−/− BMM produced levels of NO metabolites comparable to those of WT BMM. Therefore, CD36 is required for the ROS production, which is essential for the cytotoxicity of activated BMM. This finding is consistent with previous data showing that the both NO and ROS are needed to induce neurotoxicity in vitro (Mander and Brown 2005). Therefore, iNOS-derived NO is not sufficient to induce toxicity in the absence of CD36-dependent ROS production. The reaction of superoxide with NO to form peroxynitrite is critical for the cytotoxicity of monocytes and macrophages (Pacher et al. 2007). Using 3-NT as a peroxynitrite marker, we found that hippocampal slices treated with CD36+/+ BMM generate peroxynitrite, an effect not observed with CD36−/− BMM. Attesting to the pathogenic role of peroxynitrite in this model, the peroxynitrite decomposition catalyst FeTPPS markedly attenuates the cytotoxicity of CD36+/+ BMM.
We have previously demonstrated that CD36 plays a key role in the mechanisms of ischemic brain injury (Cho et al. 2005; Kunz et al. 2008; Abe et al. 2010). The role of CD36 is related to the mechanisms of post-ischemic inflammation, and, specifically, to the activation of NF-κB and to a reduction in post-ischemic ROS production (Cho et al. 2005; Kunz et al. 2008). Our present results suggest that CD36 expression in resident brain cells is not required for the full expression of hypoxic-ischemic brain injury. Rather, CD36 is essential for the ROS production and cytotoxicity exerted by mononuclear cells coming in contact with the brain. Considering that the damage of post-ischemic inflammation is driven in part by leukocytes invading the post-ischemic brain (Iadecola et al. 2004; Wang et al. 2007; Lakhan et al. 2009), our results now suggest that the deleterious role of CD36 in ischemic injury is related to the cytotoxicity of infiltrating mononuclear cells. With that in mind, a few caveats should be pointed out. First, as stated above, it is unclear whether CD36 ligands are generated in hippocampal slices during OGD. In the absence of ligands, CD36 would remain quiescent and not play a role in the mechanisms of the damage. Second, the number of BMM added to the slice is likely to be larger than the number of hematogenous cells infiltrating a comparable volume of ischemic brain, resulting in a more intense cytotoxic stimulus in our model system. Third, the blood-borne cells invading the post-ischemic brain over time are likely to be more diverse and under different activation states than the BMM used in the present model. Despite these limitations, the observation that lack of CD36 in BMM prevents their ability to kill brain cells attests to the critical role of this scavenger receptor in the cytotoxicity of activated BMM. This finding, in concert with the absence of protection in CD36−/− slices exposed to OGD is consistent with the hypothesis that CD36 in invading hematogenous cells is responsible for the deleterious effect of this scavenger receptor on the post-ischemic brain.
In conclusion, we have demonstrated that CD36 may not play an essential role in the cell death induced by OGD in hippocampal slices. However, this scavenger receptor is a key factor in the hippocampal damage produced by BMM, an effect mediated through peroxynitrite generated from ROS and iNOS-derived-NO. The contribution of CD36 is related to the fact that the ROS production by BMM is dependent upon the presence of this scavenger receptor. These findings, in concert with those of previous studies (Cho et al. 2005; Kunz et al. 2008) point to a fundamental role of CD36 in the mechanisms responsible for the infiltration of inflammatory cells into the post-ischemic brain. Inasmuch as these in vitro observations relate to the pathobiology of ischemic stroke, our findings suggest that targeting CD36 in peripheral inflammatory cells and/or the vascular compartment would be a promising strategy to counteract the CD36 dependent component of the brain damage produced by post-ischemic inflammation.
Supplementary Material
S1. CD36 is expressed in the hippocampus of neonatal mice used for slice preparation. Total RNA was isolated from the hippocampus of an adult mouse (8 weeks, lane 1), two 6-day old pups of CD36 −/− (lane 2 and 3) and two WT controls (lane 4 and 5). CD36 expression was examined by PCR analysis with 25 cycles. MW: molecular weight marker is 100 basepair ladder with size indicated.
Acknowledgments
Supported by NIH grants NS34179 (CI) and HL87062 (SSG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, Iadecola C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41:898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20:420–425. doi: 10.1016/j.coi.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Austin PE, McCulloch EA, Till JE. Characterization of the factor in L-cell conditioned medium capable of stimulating colony formation by mouse marrow cells in culture. J Cell Physiol. 1971;77:121–134. doi: 10.1002/jcp.1040770202. [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol. 2010;41:242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- Duport S, Garthwaite J. Pathological consequences of inducible nitric oxide synthase expression in hippocampal slice cultures. Neuroscience. 2005;135:1155–1166. doi: 10.1016/j.neuroscience.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Felger JC, Abe T, Kaunzner UW, Gottfried-Blackmore A, Gal-Toth J, McEwen BS, Iadecola C, Bulloch K. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun. 2010;24:724–737. doi: 10.1016/j.bbi.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya K, Takeda H, Azhar S, McCarron RM, Chen Y, Ruetzler CA, Wolcott KM, DeGraba TJ, Rothlein R, Hugli TE, del Zoppo GJ, Hallenbeck JM. Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody: a bedside-to-bench study. Stroke. 2001;32:2665–2674. doi: 10.1161/hs3211.098535. [DOI] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Grisham MB, Johnson GG, Lancaster JR., Jr Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol. 1996;268:237–246. doi: 10.1016/s0076-6879(96)68026-4. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- Hooper C, Fry VA, Sevastou IG, Pocock JM. Scavenger receptor control of chromogranin A-induced microglial stress and neurotoxic cascades. FEBS Lett. 2009;583:3461–3466. doi: 10.1016/j.febslet.2009.09.049. [DOI] [PubMed] [Google Scholar]
- Hyman MC, Petrovic-Djergovic D, Visovatti SH, Liao H, Yanamadala S, Bouis D, Su EJ, Lawrence DA, Broekman MJ, Marcus AJ, Pinsky DJ. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J Clin Invest. 2009;119:1136–1149. doi: 10.1172/JCI36433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallenbeck J, Del Zoppo G, Jacobs T, Hakim A, Goldman S, Utz U, Hasan A. Immunomodulation strategies for preventing vascular disease of the brain and heart: workshop summary. Stroke. 2006;37:3035–3042. doi: 10.1161/01.STR.0000248836.82538.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Bone marrow spawns brain killers. Nat Med. 2004;10:1044–1045. doi: 10.1038/nm1004-1044. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Cho S, Feuerstein GZ, Hallenbeck J. Cerebral Ischemia and Inflammation. Churchill Livingstone; New York: 2004. pp. 883–894. [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C, Iadecola C, Nathan C, Ding A. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97–108. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T, Hori M, Matsumoto M. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke. 2000;31:1735–1743. doi: 10.1161/01.str.31.7.1735. [DOI] [PubMed] [Google Scholar]
- Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation. 2005;2:20–35. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs P, Parisi L, Stoppini L. Interface organotypic hippocamlpal slice cultures. In: Fedoroff S, Richardson A, editors. Protocols for Neural Cell Culture. 3. Humana Press; 2001. pp. 13–27. [Google Scholar]
- Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Nuriel T, Deeb RS, Hajjar DP, Gross SS. Protein 3-nitrotyrosine in complex biological samples: quantification by high-pressure liquid chromatography/electrochemical detection and emergence of proteomic approaches for unbiased identification of modification sites. Methods Enzymol. 2008;441:1–17. doi: 10.1016/S0076-6879(08)01201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sughrue ME, Mehra A, Connolly ES, Jr, D’Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;14:14.1.1. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. CD36 is expressed in the hippocampus of neonatal mice used for slice preparation. Total RNA was isolated from the hippocampus of an adult mouse (8 weeks, lane 1), two 6-day old pups of CD36 −/− (lane 2 and 3) and two WT controls (lane 4 and 5). CD36 expression was examined by PCR analysis with 25 cycles. MW: molecular weight marker is 100 basepair ladder with size indicated.