

Abstract
γ-Glutamylcysteine (γ-GC) is an intermediate molecule of the glutathione (GSH) synthesis pathway. In the present study, we tested the hypothesis that γ-GC pretreatment in cultured astrocytes and neurons protects against hydrogen peroxide (H2O2)-induced oxidative injury. We demonstrate that pretreatment with γ-GC increases the ratio of reduced:oxidized GSH levels in both neurons and astrocytes and increases total GSH levels in neurons. In addition, γ-GC pretreatment decreases isoprostane formation both in neurons and astrocytes, as well as nuclear factor erythroid 2-related factor 2 (Nrf2) nuclear translocation in astrocytes in response to H2O2-induced oxidative stress. Furthermore, GSH and isoprostane levels significantly correlate with increased neuron and astrocyte viability in cells pretreated with γ-GC. Finally, we demonstrate that administration of a single intravenous injection of γ-GC to mice significantly increases GSH levels in the brain, heart, lungs, liver, and in muscle tissues in vivo. These results support a potential therapeutic role for γ-GC in the reduction of oxidant stress-induced damage in tissues including the brain.
INTRODUCTION
The ability of cells to withstand oxidative stress and maintain vitality in an aerobic environment is dependent upon antioxidant defense mechanisms that scavenge reactive oxygen species (ROS) before they inflict cellular damage. A delicate balance exists between cell death and survival; oxidative stress pushes the system toward death, while antioxidant defense mechanisms promote survival (Schulz, Lindenau et al. 2000). When intracellular ROS levels exceed the redox capacity, cells are subjected to oxidative stress, and oxidative damage ensues. Excessive production of ROS induces the oxidation of membrane polyunsaturated fatty acids, yielding a multitude of lipid peroxidation products, such as the F2-isoprostanes (F2-IsoPs), prostaglandin-like molecules produced by the free radical-mediated peroxidation of arachidonic acid (Morrow and Roberts 1999).
Many antioxidant defense mechanisms are activated by the translocation of nuclear factor erythroid 2-related factor 2 (Nrf2) from the cytoplasm to the nucleus. When bound to its inhibitory protein, Kelch-like ECH-associating protein 1 (Keap1, also known as INrf2), Nrf2 is targeted for ubiquitination and thus has relatively low protein stability. Upon exposure to oxidative stressors or electrophilic compounds, Nrf2 protein is liberated from Keap1, translocates into the nucleus and up-regulates cytoprotective and antioxidant genes that attenuate tissue injury (Wang, Fields et al. 2007). In addition, direct reactions between ROS and nonenzymatic antioxidant defenses, such as tocopherol, carotenoids, ascorbate and glutathione (GSH) break oxidative chain reactions and remove cellular ROS (Mead 1976).
GSH has a number of key cellular functions. It is a cofactor for several enzyme reactions, including: (1) transhydrogenases (where GSH serves to reduce oxidized centers on DNA, proteins and other biomolecules); (2) peroxidase enzymes (where GSH detoxifies peroxides generated from oxygen radical attacks on biological molecules); and (3) glutathione S-transferases (where GSH conjugates with endogenous substances, exogenous electrophiles and diverse xenobiotics) (Meister and Anderson 1983). In addition, GSH also regulates protein and gene expression via thiol:disulfide exchange reactions (Townsend, Tew et al. 2003). However, GSH’s primary role is as a cellular “redox buffer”, enabling cells to maintain the optimal electrostatic charge of proteins and cellular ion balance (Chance, Sies et al. 1979; Jewell, Bellomo et al. 1982; Orrenius, Ormstad et al. 1983). It is estimated that GSH accounts for approximately 90% of all cellular reducing equivalents, and its importance to biological reactions and the maintenance of cellular redox status is evident by its widespread phylogenetic expression in plants, mammals, fungi and some prokaryotic organisms (Townsend, Tew et al. 2003).
The central nervous system (CNS) is particularly sensitive to ROS-induced damage (Halliwell and Gutteridge 1985; LeBel and Bondy 1991). Neuronal membrane lipids are exquisitely enriched in polyunsaturated fatty acid side chains, which are especially sensitive to free radical attack. Ischemia and other conditions may lead to rapid membrane breakdown, resulting in increased free fatty acids within the CNS (Traystman, Kirsch et al. 1991). Secondly, the CNS has relatively low catalase activity and contains only moderate amounts of superoxide dismutase and glutathione peroxidase (Cohen and Wei 1988). Finally, several highly metabolic brain regions, such as the globus pallidus and substantia nigra, are rich in iron (Youdim 1988), which, via Fenton reactions, can readily generate ROS. Indeed, a variety of neurodegenerative diseases manifest abnormally low GSH (Benzi and Moretti 1995). For example, in Parkinson’s disease, ROS levels are increased (Youdim 1988); Alzheimer’s disease is characterized by decreased brain glutathione S-transferase activity; and experimental models of epilepsy are associated with altered GSH levels (Lovell, Xie et al. 1998; Candelario-Jalil, Al-Dalain et al. 2001). Abnormal respiratory chain function and iron accumulation leading to a progressive increase in oxidative damage have also been implicated in Friedreich’s ataxia (Bradley, Homayoun et al. 2004). The pathophysiology of prion diseases and the increased vulnerability for perturbation in normal prion protein function have also been shown to correlate with increased peroxidative stress (White, Collins et al. 1999).
Since oxidative stress and GSH dyshomeostasis are important events related to the above-mentioned neuropathological conditions, methods to increase endogenous GSH levels in oxidative stress-associated neurodegeneration hold considerable therapeutic interest (Pocernich, La Fontaine et al. 2000; Paintlia, Paintlia et al. 2004). Our previous studies demonstrate that increasing intracellular GSH levels by overexpression of glutamate cysteine ligase (GCL), an enzyme in the glutathione biosynthesis pathway, decreases cellular susceptibility to apoptosis (Le, Willis et al. 2010). In these experiments, we also noted that extracellular levels of γ-glutamylcysteine (γ-GC), the enzymatic product of GCL, were 50 – 200 μM. γ-GC is the immediate precursor to GSH and is directly transported into mammalian cells where it acts as a substrate of GSH synthetase (Anderson and Meister 1983), generating GSH in the absence of cysteine formation. Notably, studies on the neuroprotective effects of γ-GC are scarce.
Based on the promising role of γ-GC supplementation as a means for increasing intracellular GSH, and given that free radical production and oxidative injury have been implicated in neurodegeneration and that GSH is a scavenger of free radicals and plays an important role in reducing oxidative injury to cells, we hypothesized that γ-GC increases GSH content in neonatal rat cultures of primary astrocytes and neurons and protects them from oxidative damage in an experimental model of H2O2-induced injury. The efficacy of γ-GC in attenuating ROS-induced damage was measured with assays on cell viability, F2- isoprostane generation and the expression and nuclear translocation of Nrf2. Additional studies were carried out in vivo in mice to ascertain the efficacy of γ-GC in increasing GSH levels in the brain, heart, lung, liver and in muscle tissues.
METHODS
Materials
γ-Glutamylcysteine was purchased from Bachem Bioscience (Torrance, CA). Minimal essential medium (MEM) with Earle’s salts, heat-inactivated horse serum, penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA). All other chemicals were purchased from Sigma-Aldrich Chemicals (St. Louis, MO) unless otherwise specified. Rabbit anti-Nrf2 antibody was purchased from (Abcam, Cambridge, MA); FITC-conjugated goat anti-rabbit IgG antibody was purchased from Millipore (Billerica, MA); and goat anti-rabbit IgG was purchased from Pierce (Rockford, IL).
Primary astrocyte cultures
Astrocytic cultures from the cerebral cortices of newborn (1-day-old) Sprague– Dawley rats were established as previously described (Aschner, Mullaney et al. 1994). Briefly, rat pups were decapitated and the cerebral cortices removed. After removal of the meninges, the cerebral cortices were digested with bacterial dispase, a neutral protease (Invitrogen, Carlsbad, CA), and astrocytes were recovered by repeated removal of dissociated cells from brain tissues. Twenty-four hours after the initial plating, the media was changed to preserve the adhering astrocytes and to remove neurons, microglia and oligodendrocytes. The cultures were maintained at 37°C in a 95% air/5% CO2 incubator for 3 – 4 weeks in minimal essential medium (MEM) with Earle’s salts supplemented with 10% heat-inactivated horse serum, 100 U/ml of penicillin and 100 μg/ml of streptomycin (Invitrogen, Carlsbad, CA). The media was changed twice per week. The surface-adhering monolayer cultures were >95% positive for the astrocytic marker, glial fibrillary acidic protein (GFAP).
Primary neuronal cultures
Neuronal cortical cultures were prepared as previously described (Higgins and Banker 1998). Briefly, cortical cells from fetal (gestational day 17) rats were incubated with dispase, and clusters of cells were dissociated by gentle trituration with two flame-polished glass pipettes, followed by seeding the cells in 6-well or 96-well plates with Dulbecco’s modified eagle’s medium supplemented with 19.5% Ham’s F-12 nutrient mixture, 10% horse serum, 10% fetal bovine serum and 1% L-glutamine (200 mM). After 48 hours in culture, half of the medium was replaced by a feeding medium, comprised of 98% neurobasal medium supplemented with 2 % B-27, 0.25 % L-glutamine (200 mM), 25 mM KCl, and 0.1% β-mercaptoethanol. At 72 hours after plating, half the medium was again replaced with a feeding medium, and cultures were treated with 0.75 mM cytosine arabinoside (Ara C) to inhibit the proliferation of glial cells. The culture medium was then changed every 3 days for the duration of the experiment. The purity of the neuronal cultures was > 90%.
Glutathione Assays
Astrocytic and neuronal GSH was measured using a previously published high performance liquid chromatography (HPLC) protocol (Aschner, Mullaney et al. 1994). Briefly, cells were resuspended in saline and protein was removed by perchloric acid precipitation. Cellular GSH was measured by derivatization with iodoacetic acid and l-fluoro- 2,4-dinitrobenzene by the HPLC methods described by Fariss and Reed (Fariss and Reed 1987). Separations were achieved with a μ-Bondapak amine 10 μm cartridge (8 mm x 10 cm; Waters Assoc., Milford, MA) with a Waters Model 6OOE multisolvent delivery system using a methanol-acetate mobile phase and gradient elution. Derivatives were detected at 365 nm with a Waters Model 490 variable wavelength detector and were quantified with respect to standards using a Waters Model 745 data module (Lash and Tokarz 1990; Lash and Woods 1991). For tissue assays, a set of GSH standards and tissue samples were diluted in reaction buffer (100 mM NaPO4, 1mM EDTA, pH 7.5). These samples were then mixed with an equal volume of a reaction mixture containing 67 mM NaPO4, 0.67 mM EDTA pH 7.5, 0.67 mM dithiobis (2-nitrobenzoic acid), 500 μg/mL NADPH, and 1:200 dilution of glutathione reductase. GSH levels were measured at 412 nm using a Bio-Rad Model 680 Microplate Reader spectrophotometer.
Cell Viability
Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, which is based on the conversion of the tetrazolium salt, MTT, to a formazan product by mitochondrial dehydrogenase from live cells. Cells (2 × 105 cells/ml) were incubated with the designated drugs or vehicle control for the indicated times. Three hours before the end of the incubation period, 10 μl of phosphate-buffered saline (PBS) containing MTT (5 mg/ml) was added to each well, followed by the addition of 100 μl of isopropanol containing 1.0 N HCl to each well to dissolve the crystals formed by the reduction of the MTT reagent. Absorbance was measured using a plate reader (Molecular Devices, Sunnyvale, CA) at 570 nm with a 690 nm reference.
Trypan blue exclusion assays
Diluted cell suspensions in PBS were mixed with 0.4% trypan blue (1:1) and incubated for 3 minutes at room temperature. A drop of the trypan blue/cell mixture was placed in a hemacytometer, and cells were counted for the unstained (viable) vs. stained (dead) on the stage of a binocular microscope.
Measurement of F2-Isoprostanes
Total F2-IsoPs were measured in primary astrocyte and neuronal cultures exposed to H2O2 and/or γ-GC using gas chromatography/mass spectrometry (GC/MS) with selective ion monitoring (Morrow and Roberts 1999). Briefly, cells were resuspended in 0.5 ml of methanol containing 0.005% butylated hydroxytoluene, sonicated and then subjected to chemical saponification using 15% KOH to hydrolyze bound F2-IsoPs. The cell lysates were adjusted to a pH of 3, followed by the addition of 0.1 ng of 4H2-labeled 15-F2-IsoP internal standard. F2-IsoPs were subsequently purified by C18 and silica Sep-Pak extraction and by thin layer chromatography. They were then analyzed by pentafluorobenzyl ester, a trimethylsilyl ether derivative, via gas chromatography-mass spectrometry/negative ion chemical ionization. Cellular levels of F2-IsoPs were corrected for protein content. The protein content was determined following the manufacturer’s instructions for a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford IL).
Nuclear Translocation of Nrf2 by H2O2
Primary cerebral cortical astrocytes were grown on microscope cover glasses (Glaswarenfabrik Kari Hecht KG, Germany) and incubated in serum-deprived medium for 6 hours. The immunocytochemical method was used as described previously with minor modifications (Nancy, Wolthuis et al. 1999). For immunostaining, the cells were fixed in 100% methanol for 15 min and washed three times with PBS. After blocking in 10% bovine serum albumin in PBS for 1 hour at room temperature, the cells were incubated for 1 hour with polyclonal rabbit anti-Nrf2 antibody (1:100) in PBS containing 0.5% bovine serum albumin. The cells were incubated with FITC-conjugated goat anti-rabbit IgG antibody (1:100, Millipore, Billerica, MA) after serial washings with PBS. Counter-staining with propidium iodide verified the location and integrity of nuclei. Stained cells were washed and examined using a laser scanning confocal microscope (Leica TCS NT; Leica Microsystems, Wetzlar, Germany).
Western blotting for Nrf2
After the whole cell lysates were prepared using RIPA buffer (Sigma, St. Louis, MO), equal amounts of protein were loaded and separated by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes, and immunoblotting was performed using primary antibodies:polyclonal rabbit anti-Nrf2 antibody (1:1000, Abcam, Cambridge, MA) for Nrf2 and monoclonal mouse anti β-actin (1:5000, Sigma, St. Louis, MO) for β-actin. After three washes with PBS, goat anti-rabbit IgG (1:5000, Pierce, Rockford, IL) for Nrf2 and goat anti-mouse IgG (1:5000, Pierce, Rockford, IL) for β-actin were incubated, followed by washing with PBS and detection by enhanced chemiluminescence technique (Pierce, Rockford, IL).
Mouse γ-glutamylcysteine supplementation
Adult C57BL6 mice were injected intravenously with 400mg/kg γ-glutamylcysteine reconstituted in sterile PBS via tail vein injection. Five mice were used to generate values at each time point. At the specified time, mice were euthanized using carbon dioxide, and tissues were immediately harvested and frozen in liquid nitrogen. Tissues were carefully weighed, placed in 5x volume of lysis buffer (50 mM Tris pH 7.8, 1 mM EDTA, 150 mM NaCl, 1% Igepal CA-630) and homogenized. An equal volume of 5% sulfosalicylic acid was then added for protein precipitation, and samples were incubated on ice for 30 minutes. Samples were then centrifuged, and the supernatant was collected. The supernatants were then diluted 1:5 with neutralizing solution (200 mM NaPO4, 2 mM EDTA, pH 7.5) and assayed for glutathione concentration (see above).
Statistical analysis
Measurements of F2-IsoPs, cellular viability and MMT reduction were conducted in duplicate or triplicate wells/experiment, and the mean from three to four independent experiments was used for statistical analysis. All data were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test. Statistical significance was placed at p < 0.05 for all tests.
RESULTS
γ-Glutamylcysteine Pretreatment Restores Intracellular Reduced Glutathione Stores After Oxidative Stress In Vitro
As an in vitro model of oxidant injury, primary astrocyte and neuron cell cultures were incubated with 100 μM H2O2 for 4 hours (Fig. 1). After this incubation period, the cell cultures were harvested, and intracellular reduced and oxidized GSH levels were measured. As shown in Fig. 1A, treatment with H2O2 did not significantly decrease astrocyte total GSH levels compared to untreated controls (20.1±1.7nmol/mg protein). Similarly, pretreatment of astrocytes with γ-GC for 2 hours prior to treatment with H2O2 did not significantly increase total intracellular GSH levels (Fig. 1A). However, 2 hours of pretreatment with 100 μM or 500 μM γ-GC prior to H2O2 treatment in astrocytes significantly increased the reduced:oxidized ratio of intracellular GSH compared to control astrocytes (p < 0.05) (Fig. 1B).
Fig. 1.
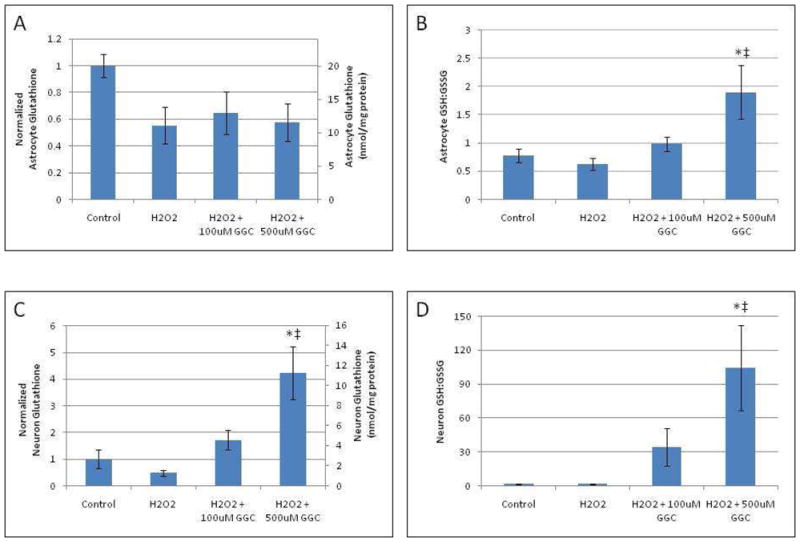
A-D. In vitro pretreatment with γ-GC reverses the decreased GSH levels in astrocytes and neurons after exposure to H2O2. Intracellular GSH levels were measured in astrocytes (A) and neurons (C) after exposure to H2O2 for 4 hours with/without pretreatment with γ-GC for 2 hours and normalized to the untreated control cells. Reduced:oxidized GSH ratio was determined in astrocytes (B) and neurons (D) after exposure to H2O2 for 4 hours with/without a 2 hour pretreatment with γ-GC. Asterisks indicate statistical significance (p < 0.05) as compared to untreated controls. ‡ indicates statistical significance (p < 0.05) as compared to H2O2-only treated groups.
Treatment of neurons with 100 μM H2O2 caused a trend similar to the one noted in astrocytes (Fig. 1A), with total neuronal GSH levels decreasing (albeit not significantly) from a control concentration of 2.66±0.94nmol/mg protein (Fig. 1C). Pretreatment of neurons with 100 μM or 500 μM γ-GC for 2 hours prior to the addition of H2O2 significantly (p < 0.05) increased total neuronal GSH levels compared to cells treated with H2O2 alone (Fig. 1C). Pretreatment of neurons with 100 μM or 500 μM γ-GC prior to H2O2 treatment also significantly (p < 0.05) increased the neuronal reduced:oxidized GSH ratio compared to neurons treated with H2O2 alone (Fig. 1D). Simultaneous introduction of of γ-GC with H2O2 with 4 hour incubation did not alter either total GSH levels or the reduced:oxidized GSH ratio in astrocytes or neurons (data not shown).
γ-Glutamylcysteine Pretreatment Decreases Lipid Peroxidation and Cellular Response to Oxidant Injury
Since pretreatment with γ-GC significantly increased the reduced:oxidized ratio of intracellular GSH and reversed the decrease in GSH levels in both H2O2-treated astrocytes and neurons, we studied whether pretreatment with γ-GC also decreases cellular markers of oxidant injury. F2-IsoP2, an oxidative biomarker produced via lipid peroxidation, was analyzed to assess whether γ-GC efficiently attenuates F2-IsoP2 generation in response to oxidant challenge with H2O2. In astrocytes, pretreatment with 100 μM or 500 μM γ-GC for 2 hours followed by the addition of H2O2 significantly (p < 0.05) reduced the H2O2-induced increase in F2-IsoP2 levels to levels indistinguishable from controls (Fig. 2A). H2O2 also induced a significant (p < 0.05) increase in neuronal F2-IsoP2 levels. Pretreatment of neurons with 500 μM γ-GC decreased IsoP2 formation to levels similar to untreated controls (Fig. 2B). In contrast, pretreatment with 100 μM γ-GC was ineffective in reducing neuronal IsoP2 levels, and these levels remained indistinguishable from those in neurons treated with H2O2 alone.
Fig. 2.

Pretreatment with γ-GC attenuates isoprostane formation in H2O2-exposed astrocytes and neurons. After cells were treated with the designated drugs, cell homogenates were prepared to measure IsoP2 as described in the Methods section. F2- IsoP2 formation in astrocytes (A) and neurons (B) after exposure to H2O2 for 4 hours with/without pretreatment with γ-GC for 2 hours and normalized to untreated control cells. Asterisks indicate statistical significance (p < 0.05) as compared to untreated controls. ‡ indicates statistical significance (p < 0.05) as compared to H2O2-only treated groups.
Nrf2 expression and Nuclear Translocation
We also studied Nrf2 expression and nuclear translocation as a marker of cellular response to oxidative injury. In response to oxidative stress, Nrf2 translocates to the nucleus where it acts as a transcriptional activator for genes containing antioxidant response elements (AREs). To further understand the effect of γ-GC pretreatment on astrocytic response to oxidant injury, we compared Nrf2 expression and subcellular localization with and without γ-GC. Treatment of astrocytes with H2O2 increased Nrf2 protein levels in whole lysates (Fig. 3A). Pretreatment of astrocytes with 500 μM γ-glutamylcysteine for 2 hours prior to the addition of H2O2, however, attenuated this Nrf2 response (Fig. 3A). To corroborate these findings, we detected by immunocytochemistry the subcellular localization of Nrf2 in astrocytes treated with H2O2. As shown in Fig. 3B, astrocytes treated with H2O2 revealed a marked increase in the nuclear expression of Nrf2 compared to the control astrocytes, indicating dissociation of the Nrf2-Keap1 complex. In contrast, astrocytes pretreated with γ-GC prior to H2O2 exposure showed very low nuclear Nrf2 expression, similar to control astrocytes.
Fig. 3.

Pretreatment of astrocytes with γ-GC attenuates H2O2-induced Nrf2 expression and nuclear translocation. (A) Nrf2 expression was measured by western blotting of the whole cell lysates of astrocytes after exposure to H2O2 for 4 hours or pretreated with γ-GC for 2 hours prior to H2O2 exposure. (B) The nuclear translocation of Nrf2 in astrocytes after exposure to H2O2 for 4 hours or pretreated with γ-GC for 2 hours prior to H2O2 exposure.
γ-Glutamylcysteine Pretreatment Increases Cell Viability After Oxidant Injury
Pretreatment of astrocytes and neurons with γ-GC increased intracellular GSH levels and decreased oxidative injury, as measured both by F2-IsoP2 production and nuclear Nrf2 translocation (see above). Since astrocytes and neurons pretreated with γ-GC have reduced oxidative damage, we hypothesized that these cells would also have increased survival rates after exposure to H2O2. Indeed, as shown in Fig. 4A, pretreatment of astrocytes with either 100 μM or 500 μM γ-GC for 2 hours prior to H2O2 treatment fully reversed the H2O2- induced (p < 0.05) decrease in cell viability to a level indistinguishable from controls. Similar results were observed in neurons, in which the H2O2-induced decrease in cell viability was significantly (p < 0.05) attenuated by 500 μM, but not 100 μM γ-GC pretreatment (Fig. 4A).
Fig. 4.

Pretreatment of astrocytes and neurons with γ-GC prior to exposure to H2O2 increases the viability of astrocytes and neurons, respectively. (A) Cell viability was measured by Trypan blue exclusion assay as described in the Methods section. (B) Cell viability was assessed by MTT assay in astrocytes and neurons exposed to H2O2 with/without γ-GC pretreatment and normalized to untreated control cells. Asterisks indicate statistical significance (p < 0.05) as compared to untreated controls. ‡ indicates statistical significance (p < 0.05) as compared to groups treated with H2O2 alone.
We also studied astrocyte and neuron viability following oxidative injury using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay. Metabolically active cells are able to reduce MTT to a colored derivative that is easily quantified. Astrocytes treated with H2O2 showed a significant decrease in cell viability compared to controls, and this effect was reversed with 500 μM γ-GC pretreatment. In neurons pretreated with 500 μM, γ-GC significantly (p < 0.05) reversed the H2O2-induced decrease in cell viability (Fig. 4B). Neurons pretreated with 100μM γ-GC demonstrated a significant increase in MTT absorbance, indicating a reduction in mitochondrial dysfunction. However, this dosage was not sufficient to significantly alter overall neuronal viability after exposure to H2O2.
Intravenous γ-Glutamylcysteine Administration Increases Tissue Glutathione Levels In Vivo
Since γ-GC treatment increased GSH levels in vitro, we studied whether in vivo γ-GC administration increases total tissue GSH levels. Mice were administered 400mg/kg γ-GC intravenously, and tissues of various organs were subsequently harvested at different time points and assayed for total GSH levels. As shown in Fig. 5, GSH levels in the red blood cells were significantly increased at 30 min post-injection and increased to a peak level at 90 min post-injection of 1.88 ± 0.11 times the normalized control level (p < 0.05). Brain tissue exhibited significantly increased GSH levels at 90 min post-injection, whereas lung, heart, liver and muscle tissues had elevated GSH levels 60 min post-injection. At 180 min post-injection, brain tissue continued to demonstrate a sustained increase in GSH levels.
Fig. 5.

Intravenous bolus injection of γ-GC increases mouse tissue glutathione levels in vivo. A 400 mg/kg bolus of γ-GC was administered intravenously via tail vein injection. Various organ tissues were harvested at serial time points and assayed for tissue glutathione levels. Asterisks indicate statistical significance (p < 0.05) as compared to control groups.
DISCUSSION
In the present study, we demonstrate that in vitro pretreatment with γ-GC in primary neuronal and astrocyte cultures attenuates cellular injury following oxidative stress. Pretreatment with γ-GC reverses a H2O2-induced reduction of total intracellular GSH levels in neurons, increases the reduced:oxidized ratio of intracellular GSH in both neurons and astrocytes, decreases astrocytic oxidative stress (as measured by IsoP2) and increases both neuronal and astrocytic cell viability. Finally, we demonstrate that in vivo intravenous injection of γ-GC in mice induces a sustained and significant increase in total tissue GSH levels in various organs including the brain, heart, lung, liver and in muscle tissues. These results support the paradigm of γ-GC as a therapeutic candidate, which increases GSH levels in vivo and in vitro and modulates the intracellular thiol status both under in vitro conditions. The observed increase of brain GSH content in γ-GC-treated mice is particularly important due to the need to increase antioxidant defenses in oxidative stress-related neuropathological conditions (Halliwell and Gutteridge 1985; LeBel and Bondy 1991) (Traystman, Kirsch et al. 1991); (Youdim 1988) (Lovell, Xie et al. 1998; White, Collins et al. 1999; Candelario-Jalil, Al-Dalain et al. 2001; Bradley, Homayoun et al. 2004).
There have been numerous reports addressing the use of NAC as an antioxidant and demonstrating its efficacy in reducing oxidant injury in human neurodegenerative disorders (Banaclocha 2001). Although the main effects of NAC are thought to arise from its deacetylation into cysteine, the rate-limiting substrate for GSH synthesis (Lauterburg, Corcoran et al. 1983), both of these GSH precursors have potential neurotoxic side effects (Puka-Sundvall, Eriksson et al. 1995). Indeed, both NAC and cysteine potentiate glutamate toxicity in cultured cerebellar granule cells (Eimerl and Schramm 1992). Similarly, cysteine elevates the influx of Ca2+ in cultured granule cells and causes neurotoxicity in the neonatal rat brain under in vivo conditions (Janaky, Varga et al. 1993; Lehmann, Hagberg et al. 1993). The Na+-dependent system, XAG−, is the major transporter for cysteine (as well as cystine, glutamate and aspartate) both in neurons and astrocytes (Shanker, Allen et al. 2001). Due to its ability to increase the GSH level without the formation of cysteine, γ-GC supplementation presents a strategy for ameliorating oxidant injury without the potential of neurotoxicity. Further, because of its co-transport via the Na+-dependent system, XAG− also likely attenuates extracellular glutamate concentrations, thus preventing the “side effects” inherent to the cellular augmentation of GSH levels by means of NAC. In addition, the use of NAC to increase GSH levels is subject to the feedback inhibition of the glutamate cysteine ligase enzyme, which is the rate-limiting enzyme in de novo GSH synthesis. Because γ-GC is the enzymatic product of glutamate cysteine ligase, the conversion of γ-GC to GSH is not subject to feedback inhibition and is theoretically only limited by γ-GC, glycine, and glutathione synthetase concentrations.
Astrocytes play an important role in providing cysteine for neuronal GSH production, a process which requires an extracellular cysteine source for glutathione production (Wang and Cynader 2000). Our results demonstrate that neurons can readily take up extracellular γ-GC to increase GSH production. In contrast to neurons, the addition of γ-GC to astrocytes did not increase the total intracellular GSH level. In keeping with previous reports, our control astrocytes contained intracellular GSH levels approximately 10-fold higher than in neurons (Kranich, Dringen et al. 1998). Therefore, our level of γ-GC supplementation may not be sufficient to detect a significant increase in intracellular GSH levels in astrocytes. Alternatively, the dissimilarity between astrocytes and neurons may reflect different uptake systems, export functions, or glutathione synthetase activity between neurons and astrocytes.
The addition of γ-GC, though, increased the ratio of reduced:oxidized GSH in both neurons and astrocytes, representing a major shift in cellular oxidative status. This effect is likely attributable to the increased GSH production in neurons. However, since total GSH levels did not increase in astrocytes, γ-GC may exert some effects due to its inherent reducing capacity. Indeed, previous studies have demonstrated that γ-GC can exert antioxidant effects in yeast incapable of GSH synthesis (Grant, MacIver et al. 1997). In both neurons and astrocytes, the described effects were only observed after a 2-hour pre-incubation, as simultaneous addition of γ-GC with H2O2 did not alter the intracellular GSH or reduced:oxidized GSH ratios. This finding implies that γ-GC is not a direct free radical scavenger, and the exact mechanism of its GSH-independent effects remains unknown. In addition, the need for pre-incubation limits the use of γ-GC to situations in which γ-GC can be used as a preventative measure, rather than a treatment to be used after oxidative injury has occurred.
Our data also establish, for the first time, the efficacy of γ-GC in attenuating H2O2- induced ROS generation and cell death in the primary cultures of astrocytes. Our studies support the concept that administration of γ-GC increases intracellular reduced GSH content, thereby enhancing the ability of cells to withstand the consequences of oxidative stress and preserve their redox capacity, resulting in decreased markers of oxidative injury as evidenced by decreased IsoP2 production and Nrf2 nuclear translocation. The in vitro studies reported herein establish strong support for the efficacy of γ-GC in attenuating oxidative stress in cultured astrocytes.
In neurons, pretreatment with γ-GC increased GSH levels and increased cellular viability after exposure to H2O2. In contrast, γ-GC pretreatment did not significantly decrease isoprostane levels as compared to H2O2-treated neurons. However, neurons pretreated with 500 μM γ-GC did not have a significant increase in isoprostane formation after exposure to H2O2 as compared to control neurons. In addition, comparing neurons pretreated with 100μM and 500μM γ-GC demonstrates a non-significant trend towards returning to baseline.
In this study, we used a single intravenous injection of γ-glutamylcysteine to study the immediate effects of γ-GC pretreatment. After a bolus injection of γ-GC, the GSH levels were significantly increased in the blood as early as 30 min after injection, with a peak level occurring at 90 min post-injection. GSH levels in the brain, heart, lung, and in muscle tissues also increased significantly, peaking at 90-120 min post-injection. These pharmacokinetic results are consistent with a multi-compartment model for GSH redistribution. Of note, unlike GSH, γ-GC appears capable of crossing the blood-brain barrier and thus affecting neuronal levels of GSH. Previous studies have also indicated that intraperitoneal injection of an esterified γ-GC increases GSH levels slowly (76 hours) compared to the present drug administration regime (up to 3 hours) (Joshi, Hardas et al. 2007). Interestingly, in the liver, GSH levels are significantly increased 60 min post-injection and decrease precipitously thereafter. Since the liver is the major organ of GSH production, we hypothesize that γ-GC undergoes rapid uptake and conversion to GSH in the liver, after which it is converted to an intermediary and redistributed to other tissues throughout the body.
In summary, these studies indicate that γ-GC rapidly increases GSH levels in a number of major organs, decreases markers for oxidant injury in in vitro experiments and increases cell viability after oxidant injury. This evidence supports the role of γ-GC in modulating the intracellular thiol status both under in vitro and in vivo conditions, supporting the potential role of γ-GC for GSH supplementation in a variety of clinical settings involving acute and chronic oxidant damage. The observed increase of brain GSH content in γ-GC-treated mice is of particular importance due to the need to increase antioxidant defenses in oxidative stress-related neuropathological conditions, such as epilepsy, Friedreich’s ataxia, Parkinson’s and Alzheimer’s diseases.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health Grant 5T32HL007256, Clinical Nutrition Research Unit (National Institutes of Diabetes and Digestive and Kidney Diseases) and divisional funding from the Division of Pediatric Critical Care Medicine, Vanderbilt Children’s Hospital. MA was supported by grants from the National Institutes of Health NIEHS 07331 and 10563. MS was supported by The Margaret O’Malley Chair in Molecular Genetics.
Non-Standard Abbreviations
- ROS
Reactive oxygen species
- GSH
Glutathione
- γ-GC
γ-Glutamylcysteine
- F2-IsoP
F2-isoprostane
- CNS
Central nervous system
- NAC
N-acetylcysteine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson ME, Meister A. Transport and direct utilization of gamma-glutamylcyst(e)ine for glutathione synthesis. Proc Natl Acad Sci U S A. 1983;80(3):707–11. doi: 10.1073/pnas.80.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Mullaney KJ, et al. Intracellular glutathione (GSH) levels modulate mercuric chloride (MC)- and methylmercuric chloride (MeHgCl)-induced amino acid release from neonatal rat primary astrocytes cultures. Brain Res. 1994;664(1–2):133–40. doi: 10.1016/0006-8993(94)91963-1. [DOI] [PubMed] [Google Scholar]
- Banaclocha MM. Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med Hypotheses. 2001;56(4):472–7. doi: 10.1054/mehy.2000.1194. [DOI] [PubMed] [Google Scholar]
- Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol Aging. 1995;16(4):661–74. doi: 10.1016/0197-4580(95)00066-n. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Homayoun S, et al. Role of oxidative damage in Friedreich’s ataxia. Neurochem Res. 2004;29(3):561–7. doi: 10.1023/b:nere.0000014826.00881.c3. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Al-Dalain SM, et al. Selective vulnerability to kainate-induced oxidative damage in different rat brain regions. J Appl Toxicol. 2001;21(5):403–7. doi: 10.1002/jat.768. [DOI] [PubMed] [Google Scholar]
- Chance B, Sies H, et al. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59(3):527– 605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Cohen MD, Wei CI. Effects of ammonium metavanadate treatment upon macrophage glutathione redox cycle activity, superoxide production, and intracellular glutathione status. J Leukoc Biol. 1988;44(2):122–9. doi: 10.1002/jlb.44.2.122. [DOI] [PubMed] [Google Scholar]
- Eimerl S, Schramm M. An endogenous metal appears to regulate NMDA receptor mediated 45Ca influx and toxicity in cultured cerebellar granule cells. Neurosci Lett. 1992;137(2):198–202. doi: 10.1016/0304-3940(92)90403-t. [DOI] [PubMed] [Google Scholar]
- Fariss MW, Reed DJ. High-performance liquid chromatography of thiols and disulfides: dinitrophenol derivatives. Methods Enzymol. 1987;143:101–9. doi: 10.1016/0076-6879(87)43018-8. [DOI] [PubMed] [Google Scholar]
- Grant CM, MacIver FH, et al. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine [In Process Citation] Mol Biol Cell. 1997;8(9):1699–1707. doi: 10.1091/mbc.8.9.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. The importance of free radicals and catalytic metal ions in human diseases. Mol Aspects Med. 1985;8(2):89–193. doi: 10.1016/0098-2997(85)90001-9. [DOI] [PubMed] [Google Scholar]
- Higgins D, Banker G. Primary dissociated cell cultures. In: Banker G, Goslin K, editors. Culturing Nerve Cells. Cambridge MA: MIT Press; 1998. pp. 37–78. [Google Scholar]
- Janaky R, Varga V, et al. Glutathione modulates the N-methyl-D-aspartate receptor-activated calcium influx into cultured rat cerebellar granule cells. Neurosci Lett. 1993;156(1–2):153–7. doi: 10.1016/0304-3940(93)90461-s. [DOI] [PubMed] [Google Scholar]
- Jewell SA, Bellomo G, et al. Bleb formation in hepatocytes during drug metabolism is caused by disturbances in thiol and calcium ion homeostasis. Science. 1982;217(4566):1257–9. doi: 10.1126/science.7112127. [DOI] [PubMed] [Google Scholar]
- Joshi G, Hardas S, et al. Glutathione elevation by gamma-glutamyl cysteine ethyl ester as a potential therapeutic strategy for preventing oxidative stress in brain mediated by in vivo administration of adriamycin: Implication for chemobrain. J Neurosci Res. 2007;85(3):497–503. doi: 10.1002/jnr.21158. [DOI] [PubMed] [Google Scholar]
- Kranich O, Dringen R, et al. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: preference for cystine. Glia. 1998;22(1):11–8. [PubMed] [Google Scholar]
- Lash LH, Tokarz JJ. Oxidative stress in isolated rat renal proximal and distal tubular cells. Am J Physiol. 1990;259(2 Pt 2):F338–47. doi: 10.1152/ajprenal.1990.259.2.F338. [DOI] [PubMed] [Google Scholar]
- Lash LH, Woods EB. Cytotoxicity of alkylating agents in isolated rat kidney proximal tubular and distal tubular cells. Arch Biochem Biophys. 1991;286(1):46–56. doi: 10.1016/0003-9861(91)90007-6. [DOI] [PubMed] [Google Scholar]
- Lauterburg BH, Corcoran GB, et al. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest. 1983;71(4):980–91. doi: 10.1172/JCI110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TM, Willis AS, et al. An ethnic-specific polymorphism in the catalytic subunit of glutamate-cysteine ligase impairs the production of glutathione intermediates in vitro. Mol Genet Metab. 2010;101(1):55–61. doi: 10.1016/j.ymgme.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBel CP, Bondy SC. Oxygen radicals: common mediators of neurotoxicity. Neurotoxicol Teratol. 1991;13(3):341–6. doi: 10.1016/0892-0362(91)90081-7. [DOI] [PubMed] [Google Scholar]
- Lehmann A, Hagberg H, et al. Cysteine sulphinate and cysteate: mediators of cysteine toxicity in the neonatal rat brain? Eur J Neurosci. 1993;5(10):1398–412. doi: 10.1111/j.1460-9568.1993.tb00926.x. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, et al. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology. 1998;51(6):1562–6. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- Mead J. Free radical mechanisms of lipid damage and consequences for cellular membranes. In: Pryor WA, editor. Free Radicals in Biology. Vol. 1. New York: Academic Press; 1976. pp. 51–68. [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–60. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Nancy V, Wolthuis RM, et al. Identification and characterization of potential effector molecules of the Ras-related GTPase Rap2. J Biol Chem. 1999;274(13):8737–45. doi: 10.1074/jbc.274.13.8737. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Ormstad K, et al. Turnover and functions of glutathione studied with isolated hepatic and renal cells. Fed Proc. 1983;42(15):3177–88. [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, et al. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78(3):347– 61. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- Pocernich CB, La Fontaine M, et al. In-vivo glutathione elevation protects against hydroxyl free radical-induced protein oxidation in rat brain. Neurochem Int. 2000;36(3):185–91. doi: 10.1016/s0197-0186(99)00126-6. [DOI] [PubMed] [Google Scholar]
- Puka-Sundvall M, Eriksson P, et al. Neurotoxicity of cysteine: interaction with glutamate. Brain Res. 1995;705(1–2):65–70. doi: 10.1016/0006-8993(95)01139-0. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, et al. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267(16):4904–11. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Shanker G, Allen JW, et al. Methylmercury inhibits cysteine uptake in cultured primary astrocytes, but not in neurons. Brain Research. 2001;914(1–2):159–165. doi: 10.1016/s0006-8993(01)02791-3. [DOI] [PubMed] [Google Scholar]
- Townsend DM, Tew KD, et al. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57(3–4):145–55. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traystman RJ, Kirsch JR, et al. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol. 1991;71(4):1185–95. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- Wang J, Fields J, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43(3):408–14. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74(4):1434–42. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- White AR, Collins SJ, et al. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol. 1999;155(5):1723–30. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB. Iron in the brain: implications for Parkinson’s and Alzheimer’s diseases. Mt Sinai J Med. 1988;55(1):97–101. [PubMed] [Google Scholar]