

Abstract
We describe herein our progress toward the synthesis of halicyclamine A, which possesses very interesting biological activities and has never been synthesized. For this purpose, we proposed a stereoselective Diels-Alder reaction as a key step for the establishment of the stereogenic triad of the bis(piperidinyl) core of this molecule. A series of NMR studies was then conducted to establish the correct stereochemical assignment subsequent to the Diels-Alder reaction.
Keywords: Total synthesis, Marine alkaloid, Organotrifluoroborate, Stereoselective Diels-Alder reaction, Halicyclamine A
1. Introduction
Halicyclamine A is one member of a growing class of tetracyclic diamine alkaloids isolated from the marine sponge Haliclona sp. (Figure 1). Crews and coworkers first isolated halicyclamine A and determined its structure.1 Haliclona sp. inhibits inosine monophosphate dehydrogenase (IMPDH) at concentrations of 1 μg/mL. IMPDH is a major therapeutic target, and drugs that are directed at IMPDH have been approved or are currently being evaluated for antiproliferative, antiviral, and anticancer chemotherapies as well as immunosuppressive activity. Halicyclamine A is cytotoxic against P388 with an IC50 value of 0.45 μg/mL.2 Besides this activity, it was recently shown that this molecule has great potential against tuberculosis with MIC values of 1.0–5.0 μg/mL for Mycobacterium smegmatis, Mycobacterium bovis BCG, and M. tuberculosis H37Ra growth inhibition in both active and dormant states.3
Figure 1.

Sponge Haliclona Sp. and structure of halicyclamine A.
Although there is considerable evidence concerning its biogenetic origin, and efforts directed toward biomimetic syntheses have been recorded,4,5 this molecule has yet to succumb to total synthesis, and its absolute configuration has yet to be determined. Noteworthy, however, is the recent synthesis of the related (±)-haliclonacyclamine C by Sulikowski et al., who built the core of that molecule by Stille cross-coupling of the two piperidinyl moieties followed by a non-stereoselective hydrogenation of the resulting diene, with subsequent separation of the formed diastereomers.6 There are two fundamental challenges associated with the synthesis of halicyclamine A. The first is the establishment of the three contiguous stereocenters about the bis(piperidinyl) ring system. The second challenge is the formation of two macrocyclic ring systems in which the stereochemistry about the conjugated diene systems must be controlled. Herein, we report the assembly of the stereogenic triad in the central nucleus of halicyclamine A.
2. Results and discussion
The central concept for the synthesis was to install the three stereocenters at once using a diastereoselective intramolecular Diels-Alder reaction, taking advantage of an internal hydrogen bonding effect that was reported earlier on other substrates.7 We envisioned that a Suzuki-Miyaura cross-coupling reaction between a potassium alkenyltrifluoroborate and a 2-bromoallylic alcohol would lead to the diene, while a Wittig-Horner reaction would be utilized to construct the dienophile. The Diels-Alder adduct could be subjected to a reductive amination, with subsequent oxidative cleavage of the cyclohexene leading to an α-hydroxy ketone derivative. Appropriate manipulation of this intermediate and cyclization would yield the core of halicyclamine A (Scheme 1).
Scheme 1.
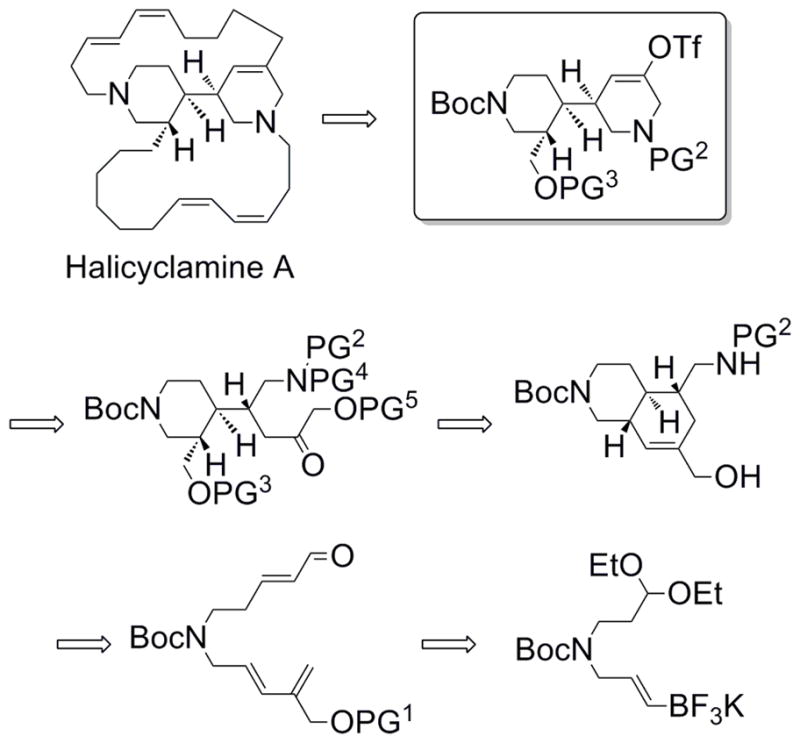
Proposed retrosynthesis.
Following the synthetic approach outlined in Scheme 2, the synthesis began with the Boc-protection of propargylamine (1) followed by N-alkylation using sodium hydride and 3-chloro-1,1-diethoxypropane. Amine 2 was then subjected to a Snieckus hydroboration.8 The resulting boronic acid was quenched with potassium hydrogen fluoride to access the corresponding potassium alkenyltrifluoroborate 3 as a waxy solid. Attempts at recrystallization failed because of the high solubility of 3 in most organic solvents. The volatile byproducts stemming from the reaction were removed by Kugelrohr distillation, and the resulting purified compound 3 was used in the next step without further purification. Suzuki-Miyaura cross-coupling9 of 3 with TBDPS-protected 2-bromo-2-propen-1-ol 4 provided the cross-coupled product 5 in good yield. Deprotection of the acetal using Amberlyst 15® led to the free aldehyde 6, which was subjected to a Wittig reaction with (triphenylphosphoranylidene)acetaldehyde in chloroform in moderate yield. Of note, only one isomer of enal 7 was observed. The latter was assigned as the E isomer based on the coupling constant of the vinylic protons (J = 15.1 Hz, see also Supporting Information), which is in agreement with the reported reactivity of (triphenylphosphoranylidene)acetaldehyde.10 The removal of the TBDPS group of compound 7 with a mixture of acetic acid and TBAF in DMF provided the desired product 8 in moderate yield.11
Scheme 2.

Synthesis of the Diels-Alder substrate.
With compound 8 in hand, the Diels-Alder reaction was attempted. The reaction worked well using the conditions described by Sherburn et al.,7 affording only one diastereoisomer of the bicyclic alcohol 9 (Scheme 3). A reductive amination with benzylamine in the presence of sodium triacetoxyborohydride afforded the corresponding amine 10 (Scheme 4), the stereochemistry of which was confirmed by NMR experiments (vide infra). The primary alcohol and the secondary amine were successively protected with a TBS group (11) and with a Cbz group (12), respectively. An ozonolysis was then performed using zinc and acetic acid as a reducing reagent, providing the keto-aldehyde 13 in moderate yield. The selective reduction of the aldehyde with sodium borohydride at low temperature gave the expected keto-alcohol 14. Owing to the relative instability of the intermediate keto-aldehyde, the sequence was conducted without any purification, which resulted in a significantly improved yield. The resulting alcohol was protected with TBDPS chloride to access the ketone 15. Selective deprotection of the TBS ether yielded the corresponding alcohol 16. Hydrogenolysis of the Cbz group of the amine required the use of palladium(II) hydroxide to give the secondary amine of compound 17. A Mitsunobu cyclization12 was attempted to access the core of Halicyclamine A 18 but initial attempts at this transformation have not been successful. The formation of an unidentified product was observed, perhaps due to side-reactions involving the carbonyl functional group or the low reactivity of the primary alcohol and benzylamine system.
Scheme 3.

Stereochemistry of the Diels-Alder reaction.
Scheme 4.

Diels-Alder reaction and oxidative cleavage.
3. Studies on the stereochemistry of the amine 10
The stereochemistry of the three contiguous stereocenters simultaneously formed by the Diels-Alder reaction was investigated. The study was conducted on the amine 10 instead of the aldehyde 9 because the NMR spectrum of the amine was more suitable for this purpose. The chemical shifts of each of the protons of the molecule were assigned by 2D NMR experiments (COSY, HMQC; See also Supporting Information).
The relative stereochemistry between protons 4a and 5 in the Diels-Alder adduct 9 or the amine 10 was assigned as trans owing to their stereochemical relationship in the enal 7. Consequently, it remained to assign the hydrogens at the ring junction (4a and 8a) as being either in a cis or in a trans relationship (Figure 2).
Figure 2.
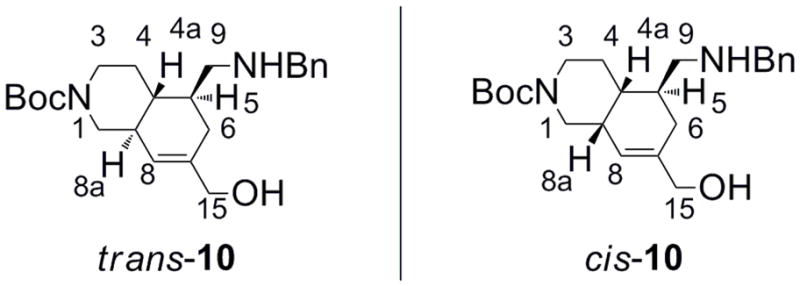
The two possible stereoisomers for amine 10.
The NOESY NMR experiment clearly showed a NOE effect between the two signals at 1.59–1.77 and 1.80–1.96 ppm (Figure 3), which correspond to protons 6/8a and 4′/5, respectively.
Figure 3.
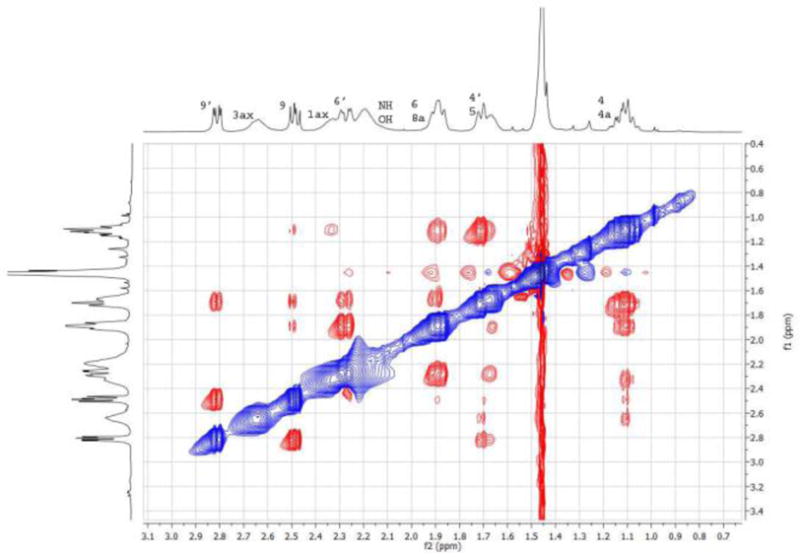
NOESY NMR 2D experiment for amine 10.
With the help of the NOESY spectrum and Chem3D model (Figure 4, see also Supporting Information for the discussion), we determined that this effect can be attributed to protons 5 and 8a, which demonstrated that trans-10 is the stereoisomer which was obtained, corresponding to the stereochemical relationship reported for halicyclamine A.
Figure 4.
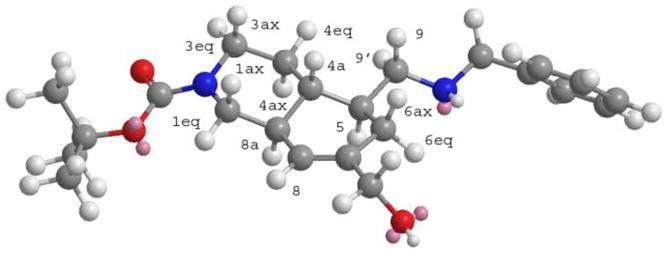
3D Structure of the amine 10 deduced from NMR analyses.
4. Conclusion
In summary, an advanced intermediate for the synthesis of the core of halicyclamine A was prepared with the correct stereochemistry, in 17 steps and a global yield of 1.27%. The original approach for this synthesis featured a diastereoselective intramolecular Diels-Alder reaction. Initial attempts at ring closure to form the second piperidinyl ring have failed but studies are still in progress for the completion of the synthesis by activating the α-hydroxy ketone or protecting the ketone to avoid side reactions.
Supplementary Material
Acknowledgments
This work was supported by a grant from the French Ministère des Affaires étrangères (BFE – Lavoisier) and the NIH (R01 GM-081376). We thank Dr. Rakesh Kohli (University of Pennsylvania) for the HRMS data. We are also grateful to Johnson-Matthey for their donation of palladium(II) acetate.
Footnotes
Experimental procedures and spectral characterization for all compounds are available free of charge.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jaspars M, Pasupathy V, Crews P. J Org Chem. 1994;59:3253–3255. [Google Scholar]
- 2.Matsunaga S, Miyata Y, van Soest RWM, Fusetani N. J Nat Prod. 2004;67:1758–1760. doi: 10.1021/np049824a. [DOI] [PubMed] [Google Scholar]
- 3.Arai M, Sobou M, Vilchéze C, Baughn A, Hashizume H, Pruksakorn P, Ishida S, Matsumoto M, Jacobs WR, Jr, Kobayashi M. Bioorg Med Chem. 2008;16:6732–6736. doi: 10.1016/j.bmc.2008.05.061. [DOI] [PubMed] [Google Scholar]
- 4.(a) Jakubowicz K, Abdeljelil KB, Herdemann M, Martin M-T, Gateau-Olesker A, Al Mourabit A, Marazano C, Das BC. J Org Chem. 1999;64:7381–7387. [Google Scholar]; (b) Sanchez-Salvatori MdR, Marazano C. J Org Chem. 2003;68:8883–8889. doi: 10.1021/jo034099e. [DOI] [PubMed] [Google Scholar]
- 5.Sinigaglia I, Nguyen TM, Wypych JC, Delpech B, Marazano C. Chem Eur J. 2010;16:3594–3597. doi: 10.1002/chem.201000142. [DOI] [PubMed] [Google Scholar]
- 6.Smith BJ, Sulikowski GA. Angew Chem Int Ed. 2010;49:1599–1602. doi: 10.1002/anie.200905732. [DOI] [PubMed] [Google Scholar]
- 7.Cayzer TN, Paddon-Row MN, Sherburn MS. Eur J Org Chem. 2003:4059–4068. [Google Scholar]
- 8.Kalinin AV, Scherer S, Snieckus V. Angew Chem Int Ed. 2003;42:3399–3404. doi: 10.1002/anie.200351312. [DOI] [PubMed] [Google Scholar]
- 9.Molander GA, Felix LA. J Org Chem. 2005;70:3950–3956. doi: 10.1021/jo050286w. [DOI] [PubMed] [Google Scholar]
- 10.Trippett S, Walker DM. J Chem Soc. 1961:1266–1272. [Google Scholar]
- 11.Higashibayashi S, Shinko K, Ishizu T, Hashimoto K, Shirahama H, Nakata M. Synlett. 2000:1306–1308. [Google Scholar]
- 12.Kim S, Lee T, Lee E, Lee J, Fan GJ, Lee SK, Kim D. J Org Chem. 2004;69:3144–3149. doi: 10.1021/jo049820a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.