

The structure of the small laccase from S. coelicolor is reported at improved resolution and in a different space group. The soaked ligand is bound between laccase molecules.
Keywords: laccases, ligand soaking, oxidoreductases, multicopper blue proteins
Abstract
The paper reports the structure of the small laccase from Streptomyces coelicolor determined from a crystal soaked with potassium hexacyanoferrate [K4Fe(CN)6]. The decolorization of the natively blue crystal observed upon soaking indicates the reduction of the enzyme in the crystal. The ligand binds between laccase molecules and stabilizes the crystal. The increased diffraction limit of the diffraction data collected from this crystal enabled the refinement of the small laccase structure at 2.3 Å resolution, which is the highest resolution obtained to date.
1. Introduction
Laccases (EC 1.10.3.2) are multicopper oxidoreductases that oxidize diphenols and related substances (Solomon et al., 2001 ▶; Mayer & Staples, 2002 ▶; Giardina et al., 2010 ▶). The reaction cycle is completed by the reduction of dioxygen to water. Laccases usually consist of three cupredoxin-like domains, but several two-domain enzymes with laccase activity have also been described (DiSpirito et al., 1985 ▶; Endo et al., 2003 ▶; Machczynski et al., 2004 ▶).
Four copper ions are necessary for laccase function. The type 1 copper (the nomenclature is according to spectroscopic properties) is an acceptor of electrons from substrates. Electrons (four electrons from four sequential substrates) are then transferred to a trinuclear copper cluster at a distance of ∼13 Å, where dioxygen is reduced to water. The trinuclear copper cluster consists of a type 2 copper ion and two type 3 copper ions. The two type 3 copper ions have slightly different binding distances to the surrounding histidines. Distinct roles for the two type 3 copper ions have recently been determined for the multicopper oxidase Fet3p (Augustine et al., 2010 ▶).
The crystal structures of three two-domain oxidoreductases have been published: laccase from Streptomyces coelicolor (SLAC; Skálová et al., 2009 ▶; PDB code 3cg8; molecular weight 30 kDa), a metagenomic laccase (Komori et al., 2009 ▶; PDB code 2zwn) and a multicopper oxidase from Nitrosomonas europaea (Lawton et al., 2009 ▶; PDB code 3g5w). These enzymes were found to form trimers, with three type 1 copper ions and three trinuclear copper clusters (which are always placed between two neighbouring chains of the trimer).
According to an evolutionary theory for multicopper blue proteins proposed by Nakamura & Go (2005 ▶), there are three types (A, B and C) of two-domain multicopper oxidoreductases. Type A is the common ancestor of types B and C. Type B (for which the only three-dimensional structure known is that of SLAC) is a common ancestor of three-domain laccases, three-domain ascorbate oxidases and six-domain ceruloplasmins. Type C (for which three-dimensional structures have been determined for the metagenomic laccase and the oxidase from N. europaea) is the ancestor of two-domain trimeric nitrite reductases.
There are significant structural differences between enzymes of types B and C. The position of the type 1 copper in the type B enzyme is in domain 2, close to the centre and on the surface of the trimer. It resembles the typical position of this metal in laccases according to secondary-structure matching and also with respect to the trinuclear copper cluster. In type C enzymes it is placed in domain 1, as in nitrite reductase, farther away from the centre of the trimer compared with the type B enzyme. The position of the type 1 copper in the type B enzyme can be transformed onto that in type C enzymes by a rotation of approximately 180°. The axis of rotation lies in the plane of the trinuclear cluster and runs through the type 2 copper ion and between the type 3 copper ions. In this way, not only does the location of the type 1 copper ion change, but the roles of the type 3 copper ions in the enzyme mechanism also exchange.
The level of sequence identity between the two type C enzymes (3g5w and 2zwn) is 62%, with an r.m.s.d. of Cα coordinates of the corresponding residues of 0.8 Å. In relation to the type B laccase SLAC, they show sequence identities of 19–20% and r.m.s.d.s of 2.6–2.7 Å (all equivalent Cα atoms according to secondary-structure matching).
After the native structure of SLAC was solved at 2.7 Å resolution, an effort was made to obtain a structure of a complex with a ligand. Potassium hexacyanoferrate was known to reduce type 1 copper ions. Here, we present the structure of SLAC after soaking with potassium hexacyanoferrate [K4Fe(CN)6]. The ligand is found between the SLAC trimers in the crystal, participating in stabilization of the contacts. In the context of our previous work, this study provides a structure of SLAC at a higher resolution, in a higher symmetry space group and with more precise structural details.
2. Materials and methods
2.1. Crystallization and data collection
Details of the expression, purification and crystallization of SLAC have been published previously (Skálová et al., 2007 ▶). A crystal (rhombic dodecahedron) of diameter 180 µm was soaked in buffer solution [40%(v/v) polyethylene glycol 550 monomethyl ether (PEG 550 MME), 0.1 M NaCl, 0.1 M glycine pH 9.0] with potassium hexacyanoferrate added to a concentration of 50 mM. The crystal changed colour from blue to colourless after 10 s in the solution. It was then vitrified in liquid nitrogen. Single-crystal diffraction data were collected on beamline BL14.1 at the BESSY II synchrotron-radiation source of the Helmholtz Zentrum Berlin at a temperature of 100 K using a MAR Mosaic 225 CCD detector.
2.2. Data processing and refinement
The data were indexed, integrated and scaled in HKL-2000 (Otwinowski & Minor, 1997 ▶) and imported into the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶). The data-collection and processing parameters are given in Table 1 ▶. The enzyme crystallized in a cubic system, with unit-cell parameters a = b = c = 176.7 Å. The unit-cell parameters differed by several angstroms from the tetragonal unit-cell parameters (a = b = 180.9, c = 177.1 Å) of the native structure (PDB code 3cg8) and the crystal belonged to the higher symmetry space group P4332, leading to a reduction in the content of the asymmetric unit from one trimer (in the native structure) to only one chain of the trimer. The individual chains of the functional trimer of SLAC are related by crystal symmetry in this case.
Table 1. Data-collection statistics and structure-refinement parameters.
Values in parentheses are for the highest resolution shell.
| Data-collection statistics | |
| Wavelength (Å) | 0.91841 |
| No. of oscillation images | 200 |
| Crystal-to-detector distance (mm) | 245 |
| Exposure time per image (s) | 10 |
| Oscillation width (°) | 0.5 |
| Space group | P4332 |
| Unit-cell parameters (Å) | a = b = c = 176.7 |
| Resolution range (Å) | 50.0–2.3 (2.34–2.30) |
| No. of observations | 3078811 |
| No. of unique reflections | 42751 (2077) |
| Data completeness (%) | 100 (100) |
| Overall multiplicity (I+ = I−) | 24.1 (24.6) |
| Friedel pair completeness (%) | 99 |
| Mosaicity (°) | 0.3 |
| Average I/σ(I) | 33.9 (6.3) |
| Solvent content (%) | 83.8 |
| Matthews coefficient (Å3 Da−1) | 7.59 |
| Rmerge† | 0.114 (0.605) |
| Rp.i.m.‡ | 0.025 (0.116) |
| Wilson B factor (Å2) | 43.6 |
| Structure-refinement parameters | |
| Rwork§ | 0.161 |
| Rfree§ | 0.178 |
| Rall§ | 0.162 |
| Average B factor (Å2) | 25.6 |
| R.m.s.d. of bond lengths from ideal (Å) | 0.016 |
| R.m.s.d. of bond angles from ideal (°) | 1.483 |
| No. of non-H atoms | 2440 |
| No. of protein monomers per asymmetric unit | 1 |
| No. of water molecules | 234 |
| Other localized moieties | 4 Cu, 1 Fe (part of potassium hexacyanoferrate), 2 Na, 5 fragments of PEG 550 MME |
R
merge = 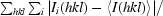
 (Diederichs & Karplus, 1997 ▶), where Ii(hkl) and 〈I(hkl)〉 are the observed individual and mean intensities of a reflection with indices hkl, respectively,
(Diederichs & Karplus, 1997 ▶), where Ii(hkl) and 〈I(hkl)〉 are the observed individual and mean intensities of a reflection with indices hkl, respectively, 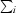 is the sum over the individual measurements of a reflection with indices hkl and
is the sum over the individual measurements of a reflection with indices hkl and  is the sum over all reflections.
is the sum over all reflections.
R
p.i.m. = 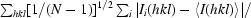 (Weiss, 2001 ▶), where N is the multiplicity.
(Weiss, 2001 ▶), where N is the multiplicity.
R
work = 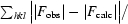
 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes for the working set of reflections. R
free is the same as R
work but for 3% of the data that were omitted from refinement. R
all sums over all reflections.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes for the working set of reflections. R
free is the same as R
work but for 3% of the data that were omitted from refinement. R
all sums over all reflections.
Initial phases were assigned by molecular replacement in MOLREP (Vagin & Teplyakov, 2010 ▶) using chain A of the previous SLAC structure (3cg8). The structure was refined using REFMAC5.5 (Murshudov et al., 1997 ▶) and manual modifications were carried out in XtalView (McRee, 1999 ▶) and Coot (Emsley & Cowtan, 2004 ▶). 3% of reflections were used as test reflections for the R free statistic. The occupancy factors of the copper and iron ions were set according to anomalous occupancy refinement (as implemented in REFMAC5.5). The quality of the geometry, the contacts and the consistency with maps were assessed using the validation tools available in Coot 0.1-PRE-1 and the PDB validation server ADIT. According to the MolProbity Ramachandran plot (Chen et al., 2010 ▶), 98.2% of residues lie in favoured regions and all residues lie in allowed regions of the plot. The final structure parameters are summarized in Table 1 ▶.
2.3. Experiments with other ligands
Only the structural results for SLAC with potassium hexacyanoferrate (PDB code 3kw8) are presented and discussed in this paper. A short summary of trials with two other ligands follows.
2.3.1. 2,5-Xylidine
A native SLAC crystal of diameter 260 µm was soaked in reservoir solution [40%(v/v) PEG 550 MME, 0.1 M NaCl, 0.1 M glycine pH 9.0] with 2,5-xylidine added to a concentration of 10 mM. Diffraction data were measured at the same station as in the case of the potassium hexacyanoferrate complex and were processed to 2.7 Å resolution in space group P43212. A significant positive difference peak was observed in the expected substrate-binding site between Tyr229 and Tyr230. It was not possible to interpret the peak unambiguously as 2,5-xylidine and the structure was not refined.
2.3.2. Cocrystallization with ABTS
A crystal of SLAC of diameter 130 µm was grown in the presence of 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS). A blue crystal was obtained (as in the case of the native crystals), unlike the experience of Enguita et al. (2004 ▶) in which a colour change was observed from light blue to dark magenta during soaking with ABTS. Crystallization was performed by the hanging-drop vapour-diffusion technique (using drops consisting of 1 µl protein solution and 1 µl precipitant) with a reservoir solution consisting of 40%(v/v) PEG 550 MME, 0.1 M NaCl, 0.1 M glycine pH 9.0 and 0.05 M ABTS at a temperature of 298 K. Diffraction data were measured at the same station as in the above cases and were processed to 3.25 Å resolution in space group P43212. As in the case of 2,5-xylidine, a significant positive peak in a difference Fourier map was observed in the expected substrate-binding site between Tyr229 and Tyr230. It was not possible to interpret the peak unambiguously as ABTS and the structure was not refined any further.
3. Results
3.1. Overall structure
The overall structure of the SLAC trimer does not deviate from the previous results (Fig. 1 ▶). The type 1 copper is placed close to the surface near the centre of the triangular-shaped SLAC. The reported structure (PDB code 3kw8) confirms the basic features of the previous structure (PDB code 3cg8) solved at 2.7 Å resolution and new structural details are apparent.
Figure 1.

SLAC: a trimeric two-domain laccase from S. coelicolor. Copper ions are shown in magenta and the iron ions of potassium hexacyanoferrate are depicted as brown spheres.
The major differences between the previous and the currently reported structures are observed at the termini and are connected with different crystal packing. Superposition of the main-chain atoms of the complete trimer (by secondary-structure matching in CCP4) produces a root-mean-square deviation of 0.33 Å.
3.2. Crystal packing
The trimers of SLAC in the reported structure are formed around a crystallographic threefold axis and therefore each chain participates in identical interactions with a neighbouring enzyme trimer. In the previously determined structure 3cg8 one trimer in the asymmetric unit participates in contacts with three other trimers, with two of the interactions being identical to those in the cubic form (3kw8) and one being significantly different (Fig. 2 ▶). The latter interaction in the tetrameric form is stronger, with 12 hydrogen bonds connecting the trimers, while only one direct and several water-mediated contacts exist in the cubic form. The distinct interface in the tetragonal form lies on a crystallographic twofold axis between the interacting trimers. Thus, the difference in packing in the cases of 3cg8 (tetragonal) and 3kw8 (cubic) is caused by a preference for a different type of intermolecular interaction. Hexacyanoferrate ions mediate intermolecular interactions in the case of the cubic form and their binding would not be possible within the interface specific to the tetragonal crystal form.
Figure 2.

Crystal packing of laccase from S. coelicolor in the reported structure 3kw8 (green, space group P4332) and in the previous structure 3cg8 (blue, space group P43212). The central trimers of the two structures (black) were superimposed using the SSM algorithm (Krissinel & Henrick, 2004 ▶). The occurrence of alternative packing (top) distinguishes the tetragonal and cubic forms.
In the tetragonal form there are differences between the conformations of several terminal residues of individual SLAC chains. In the cubic form the crystallographic threefold symmetry of the trimer excludes this possibility and the terminal residues of the three chains are identical.
3.3. Type 1 copper and substrate-binding site
The occupancy of the type 1 copper (residue 401; occupancy 1.0; binding to His231, His293 and Cys288) is 100% according to anomalous occupancy refinement in REFMAC5. Based on a superposition of SLAC with complexes of three-domain laccases with ligands, the expected substrate-binding site of SLAC is located between residues Tyr229 and Tyr230 near the threefold axis of the trimer. Only water molecules (703, 823, 923 and 924) were localized here in this structure.
There is no sign of the presence of a hexacyanoferrate ion in the vicinity of the expected substrate-binding site. According to the Protein Data Bank, at least four ligands have been found in the substrate-binding site of a laccase: 2,5-xylidine (an arylamine; Bertrand et al., 2002 ▶; PDB code 1kya), ABTS (Enguita et al., 2004 ▶; 1uvw and 1of0), p-toluate (Matera et al., 2008 ▶; 2hrg) and 2,6-dimethoxyphenol (Kallio et al., 2009 ▶; 3fu7, 3fu8 and 3fu9). Unlike three-domain laccases, SLAC lacks a well shaped binding pocket. This decreases the probability of the observation of a ligand localized in the reaction site in a crystal structure.
No significant variation of the type 1 copper coordination sphere was observed in comparison to the native structure, despite the fact that reduction of the type 1 copper sites was confirmed visually by crystal decolourization during soaking with potassium hexacyanoferrate.
3.4. Trinuclear copper site
The occupancy factors of the copper ions, based on anomalous occupancy refinement in REFMAC5, are as follows: 1.0 for type 3 Cu (residue 402; binding to His289 at 2.1 Å, His104′ at 2.0 Å and His156′ at 2.0 Å), 0.7 for type 3 Cu (residue 403; binding to His287 at 2.4 Å, His236 at 2.0 Å and His158′ at 2.4 Å) and 0.3 for type 2 Cu (residue 404; binding to His234 at 2.0 Å and His102′ at 1.8 Å). Fig. 3 ▶(a) shows the positions of copper ions in the trinuclear copper cluster (modelled with isotropic atomic displacement parameters) and surrounding water molecules as deposited. Oxygen A701 corresponds to an OH− ion bound between the two type 3 copper ions Cu402 and Cu403.
Figure 3.

Alternative interpretation of the trinuclear copper cluster site. (a) Situation in the deposited structure. Distances between copper ions and water molecules, which commonly accompany the copper cluster, are shown in angstroms. (b) Two additional positive F o − F c peaks (5.5σ) which can be interpreted as waters or O atoms participating in the reaction cycle; the coordinates of these two atoms are not present in the deposited entry. The F o − F c map is contoured at the 4σ level (green) and electron density based on anomalous differences is contoured at the 6σ level (violet). The varying level of anomalous signal corresponds to the occupancy of the copper ions. (c, d) Difference OMIT map: two perpendicular views. For calculation of this F o − F c map, contoured at the 4σ level in green, Cu404 and water molecules 928, 929, 701, 930 and 736 were omitted from the model.
Two additional positive F o − F c maxima at the 5.5σ level are present at the trinuclear copper cluster (Fig. 3 ▶ b). The first peak lies between Cu402 (type 3) and Cu404 (type 2) and may correspond to the OH− expected in the resting state (Solomon et al., 2001 ▶). The second peak is found close to a line connecting Cu402 (type 3) and Cu403 and may be an alternative position of a water molecule in the solvent channel; this water may possibly alternate with Cu403 with partial occupancy.
Figs. 3 ▶(c) and 3 ▶(d) show positive peaks of an F o − F c electron-density map which appear when the localized water molecules near the trinuclear copper site and Cu404 are omitted. This suggests a partially disordered arrangement of water molecules and/or O atoms in the direction of the expected water channel. In spite of this, the localized F o − F c maxima for individual water molecules and Cu404 are unambiguous and reach levels in the range 11.8–17.4σ.
To examine changes in the trinuclear copper cluster, subsets of data from the beginning (the first 30 images of the total 200; unit-cell parameter a = b = c = 176.5 Å) and the end of data collection (the last 30 images; unit-cell parameter = 177.2 Å) were processed independently for this purpose. The data completeness of each of the subsets was 97%. The distance Cu402—Cu403 increased by 0.2 Å between these two sets of data (from 4.8 to 5.0 Å, comparing refined positions of both copper ions), which may indicate progressive reduction of the trinuclear copper site. The phenomenon of an increase in the type 3 copper distance of 0.1 Å when comparing a short and a long X-ray-exposed structure with a parallel spectroscopic confirmation of the reduction of the type 1 copper caused by X-ray radiation has been described by Hakulinen et al. (2006 ▶).
3.5. Iron and other ligands
An iron with occupancy 0.4, which is part of a hexacyanoferrate ion, is located between residues Arg203 and Arg203′ on a twofold axis between two laccase molecules in the crystal, 4.3 Å distant from Arg203 NH2 (Fig. 4 ▶). There is sufficient space for the whole hexacyanoferrate ion to be modelled and positive F o − F c peaks are present at the 5σ level, but the electron density in this region is not so unambiguously interpretable as to allow inclusion of the whole ion in the model. An anomalous difference peak at the 8σ level coincides with the position of the iron ion. A similar binding of hexacyanoferrate ions has been experimentally observed in organic and inorganic crystal structures, such as in the structure of bis[diaqua-bis(ethylene-1,2-diamine-N,N′)-copper(II)]hexacyanoferrate(II) tetrahydrate (Kuchár et al., 2004 ▶; Cambridge Structural Database code EYIQUI).
Figure 4.

(a) Binding of a hexacyanoferrate ion (brown) on a twofold symmetry axis between two molecules of laccase. Only iron is built in the deposited structure. The ion binds between the Arg203 residues of two symmetrically related molecules; the 2F o − F c map is shown at the 1σ level in blue with contact distances in angstroms. (b) Overall view of the laccase trimer with three bound hexacyanoferrate ions (brown). Copper ions are shown in magenta.
PEG 550 MME binds on the enzyme surface in a similar manner as observed in the first SLAC structure, but more details can be observed. In two cases, a ring-like structure of part of the polymer chain is found in contact with an ion (most likely sodium, which is present at high concentration in the mother liquor) near Glu80 (Fig. 5 ▶) and near Tyr314. In the near vicinity of the hexacyanoferrate ion bound in the intermolecular interface, a PEG 550 MME chain is localized similarly as in the previous structure (near Arg203). However, in this case it very likely bears an ion (modelled as water 932) which mediates the formation of a ‘polymer–ion–[Fe(CN)6]4−–ion–polymer’ complex across the crystallographic twofold axis. At the same time, the hexacyanoferrate ion is held in position by Arg203 and Arg203′.
Figure 5.

A fragment of PEG 550 MME (C atoms in yellow) with a sodium ion (blue) bound on the protein surface to residue Glu80. The 2F o − F c map is contoured at 1.6σ (blue mesh); contact distances are given in angstroms.
The structure contains 234 localized water molecules, which surround the protein and fill its cavity.
4. Conclusions
The small laccase from S. coelicolor crystallizes in a second form that is closely related to the previously reported form. The functional trimer is organized around the threefold axis of the cubic space group. Despite the overall similarity of the unit-cell parameters and the high solvent content, one of the crystal-packing interfaces in the trimer changes significantly between the two forms.
Potassium hexacyanoferrate apparently reduced the type 1 copper, but no significant structural changes were observed. A hexacyanoferrate ion binds far from the substrate-binding site in the trimer–trimer interface in a complex interaction with surface arginine residues, polymer chains and positive ions.
The asymmetry of the type 3 copper ions is clearly confirmed by this higher resolution structure. One type 3 copper ion is present with an occupancy factor of 1.0, whereas the other is present with an occupancy of 0.7. The type 2 copper ion is again observed with a significantly decreased occupancy of 0.3. Partial disorder of atomic coordinates is observed in the electron density along the trinuclear cluster, which could be associated with the functional dynamic exchange of atoms/ions at this location during the enzymatic cycle.
Supplementary Material
PDB reference: laccase, 3kw8
Acknowledgments
This work was supported by the Czech Science Foundation (305/07/1073), the Grant Agency of the Academy of Sciences of the Czech Republic (IAA500500701) and by the EC under ELISA grant agreement No. 226716 (synchrotron access funding, projects 09.1.81077 and 09.2.90262). The authors wish to thank Dr U. Müller of the Helmholtz-Zentrum Berlin, Albert-Einstein-Strasse 15 for support at beamline BL14.1 of BESSY II.
References
- Augustine, A. J., Kjaergaard, C., Qayyum, M., Ziegler, L., Kosman, D. J., Hodgson, K. O., Hedman, B. & Solomon, E. I. (2010). J. Am. Chem. Soc. 132, 6057–6067. [DOI] [PMC free article] [PubMed]
- Bertrand, T., Jolivalt, C., Briozzo, P., Caminade, E., Joly, N., Madzak, C. & Mougin, C. (2002). Biochemistry, 41, 7325–7333. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–274. [DOI] [PubMed]
- DiSpirito, A. A., Taaffe, L. R., Lipscomb, J. D. & Hooper, A. B. (1985). Biochim. Biophys. Acta, 827, 320–326.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Endo, K., Hayashi, Y., Hibi, T., Hosono, K., Beppu, T. & Ueda, K. (2003). J. Biochem. (Tokyo), 133, 671–677. [DOI] [PubMed]
- Enguita, F. J., Marcal, D., Martins, L. O., Grenha, R., Henriques, A. O., Lindley, P. F. & Carrondo, M. A. (2004). J. Biol. Chem. 279, 23472–23476. [DOI] [PubMed]
- Giardina, P., Faraco, V., Pezzella, C., Piscitelli, A., Vanhulle, S. & Sannia, G. (2010). Cell. Mol. Life Sci. 67, 369–385. [DOI] [PMC free article] [PubMed]
- Hakulinen, N., Kruus, K., Koivula, A. & Rouvinen, J. (2006). Biochem. Biophys. Res. Commun. 350, 929–934. [DOI] [PubMed]
- Kallio, J. P., Auer, S., Jänis, J., Andberg, M., Kruus, K., Rouvinen, J., Koivula, A. & Hakulinen, N. (2009). J. Mol. Biol. 392, 895–909. [DOI] [PubMed]
- Komori, H., Miyazaki, K. & Higuchi, Y. (2009). FEBS Lett. 583, 1189–1195. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Kuchár, J., Černák, J. & Massa, W. (2004). Acta Cryst. C60, m418–m420. [DOI] [PubMed]
- Lawton, T. J., Sayavedra-Soto, L. A., Arp, D. J. & Rosenzweig, A. C. (2009). J. Biol. Chem. 284, 10174–10180. [DOI] [PMC free article] [PubMed]
- Machczynski, M. C., Vijgenboom, E., Samyn, B. & Canters, G. W. (2004). Protein Sci. 13, 2388–2397. [DOI] [PMC free article] [PubMed]
- Matera, I., Gullotto, A., Tilli, S., Ferraroni, M., Scozzafava, A. & Briganti, F. (2008). Inorg. Chim. Acta, 361, 4129–4137.
- Mayer, A. M. & Staples, R. C. (2002). Phytochemistry, 60, 551–565. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nakamura, K. & Go, N. (2005). Cell. Mol. Life. Sci. 62, 2050–2066. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Skálová, T., Dohnálek, J., Østergaard, L. H., Østergaard, P. R., Kolenko, P., Dušková, J. & Hašek, J. (2007). Acta Cryst. F63, 1077–1079. [DOI] [PMC free article] [PubMed]
- Skálová, T., Dohnálek, J., Østergaard, L. H., Østergaard, P. R., Kolenko, P., Dušková, J., Štěpánková, A. & Hašek, J. (2009). J. Mol. Biol. 385, 1165–1178. [DOI] [PubMed]
- Solomon, E. I., Chen, P., Metz, M., Lee, S.-K. & Palmer, A. E. (2001). Angew. Chem. Int. Ed. 40, 4570–4590. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: laccase, 3kw8