

The structure of L. donovani pteridine reductase has been targeted to assist in a program of structure-based inhibitor research. Crystals that diffracted to 2.5 Å resolution were obtained and the structure has been solved. Unfortunately, the active site is disordered and this crystal form is unsuitable for use in characterizing enzyme–ligand interactions.
Keywords: antifolates, pteridine reductase, Leishmania, pterins, Trypanosoma
Abstract
Pteridine reductase (PTR1) is a potential target for drug development against parasitic Trypanosoma and Leishmania species, protozoa that are responsible for a range of serious diseases found in tropical and subtropical parts of the world. As part of a structure-based approach to inhibitor development, specifically targeting Leishmania species, well ordered crystals of L. donovani PTR1 were sought to support the characterization of complexes formed with inhibitors. An efficient system for recombinant protein production was prepared and the enzyme was purified and crystallized in an orthorhombic form with ammonium sulfate as the precipitant. Diffraction data were measured to 2.5 Å resolution and the structure was solved by molecular replacement. However, a sulfate occupies a phosphate-binding site used by NADPH and occludes cofactor binding. The nicotinamide moiety is a critical component of the active site and without it this part of the structure is disordered. The crystal form obtained under these conditions is therefore unsuitable for the characterization of inhibitor complexes.
1. Introduction
Leishmania are protozoan parasites (order Trypanosomatida, class Kinetoplastida) that cause a range of diseases and present a serious health risk to millions of people worldwide (Desjeux, 2004 ▶; Reithinger et al., 2007 ▶). The incidence of infection is primarily in tropical and subtropical regions of the world (Herwaldt, 1999 ▶). The different species of Leishmania are responsible for distinctive conditions (Reithinger et al., 2007 ▶). For example, L. donovani causes visceral leishmaniasis, a potentially fatal disease, while infection with L. major leads to cutaneous leishmaniasis. Several compounds are available to treat these infections, but increasing levels of drug resistance combined with the high cost and toxicity of antileishmanial drugs compromises the control of the diseases (Croft et al., 2006 ▶; Maltezou, 2010 ▶). These observations explain in part why the World Health Organization has identified the leishmaniases as neglected diseases and is urgently seeking novel therapeutic approaches (World Health Organization, 2007 ▶).
Our aim is to identify and characterize drug targets in these parasites and to apply structure-based approaches to develop potent inhibitors that possess the right chemical properties to underpin early-stage drug discovery (Hunter, 2009 ▶). A promising target with respect to infection with Leishmania sp. is the NADPH-dependent short-chain dehydrogenase/reductase pteridine reductase (PTR1; EC 1.5.1.33). This enzyme is unique to trypanosomatid parasites, where it supports the provision of reduced biopterins that are necessary for metacyclogenesis (Cunningham et al., 2001 ▶) and which are implicated in resistance to reactive oxygen and nitrogen species in Leishmania (Moreira et al., 2009 ▶). PTR1 catalyzes the reduction of biopterin to 7,8-dihydrobiopterin as well as its subsequent reduction to 5,6,7,8-tetrahydrobiopterin. Additionally, the enzyme catalyzes similar reactions in the salvage of unconjugated folates in Leishmania (Nare, Hardy et al., 1997 ▶; Nare, Luba et al., 1997 ▶). As Leishmania are auxotrophic for pteridines (folates and pterins) and are required to obtain these nutrients from their environment to maintain growth, disrupting this salvage process represents a potential therapeutic strategy.
We have previously studied the structure–mechanism–activity relationships for the enzymes from L. major (LmPTR1) and Trypanosoma brucei (TbPTR1) and determined the structures of a series of inhibitor complexes (Gourley et al., 2001 ▶; Dawson et al., 2006 ▶; Cavazzuti et al., 2008 ▶; Mpamhanga et al., 2009 ▶), whilst the structure of T. cruzi PTR2 has been reported by others (Schormann et al., 2005 ▶). We identified sequence and structural differences between LmPTR1 and TbPTR1 that explain why some inhibitors display a significant level of selectivity for one orthologue over the other (Gibson et al., 2009 ▶; Tulloch et al., 2010 ▶). Although we were able to routinely generate crystals of TbPTR1 that diffracted to between 2.0 and 1.0 Å resolution (Dawson et al., 2010 ▶), studies with LmPTR1 have been hampered by poor crystal quality and a lack of reproducibility. One crystal form of LmPTR1 diffracted to beyond 2.0 Å resolution but could only be obtained in the presence of NADPH and methotrexate (Gourley et al., 2001 ▶); when other ligands were present different crystal forms were obtained. The size of the asymmetric unit is increased from two to either four or eight subunits and the crystals are often mechanically twinned and diffract to lower resolution, with the diffraction pattern being anisotropic and highly mosaic (McLuskey et al., 2004 ▶; Schüttelkopf et al., 2005 ▶). An alternative source of Leishmania PTR1 was therefore sought for our investigations. Studies with L. tarentolae PTR1 resulted in a 2.8 Å resolution structure of the complex with NADPH, but despite its presence in the crystallization mixture the tight-binding ligand methotrexate was not observed in the electron-density maps (Zhao et al., 2003 ▶). This was not considered to be an improvement on the results that we had previously obtained, so we elected to initiate crystallographic studies of L. donovani PTR1 (LdPTR1), this also being the enzyme from the pathogen that causes the most serious form of leishmaniasis. There is a high level of conservation (91% sequence identity) between the L. major and L. donovani enzymes and homology modelling of the latter has suggested a close structural relationship in and around the active site (Kaur et al., 2010 ▶).
2. Methods
2.1. Expression and purification
The gene encoding LdPTR1 was cloned into the expression vector pET15b (Novagen) modified to encode a tobacco etch virus (TEV) protease-cleavable N-terminal hexahistidine tag. This plasmid was heat-shock transformed into Escherichia coli BL21 (DE3) GOLD cells (Stratagene) and selected on Luria–Bertani (LB) agar plates containing 50 mg l−1 carbenicillin. Cells were then cultured in LB medium containing the same antibiotic at 310 K with shaking. Gene expression was induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM when the cells reached the mid-log phase of growth (optical density at 600 nm of 0.6–0.8). After incubation at room temperature for 16 h, the cells were harvested by centrifugation (4000g, 277 K, 30 min), washed with fresh medium and the centrifugation was repeated. Cell pellets were frozen at 253 K until required.
For purification, the frozen cell pellet from 1 l culture was thawed on ice and resuspended in lysis buffer (50 mM Tris–HCl pH 7.7, 200 mM KCl, 20 mM imidazole) supplemented with an EDTA-free protease-inhibitor cocktail tablet (Roche) and 100 µg DNAse I (Sigma–Aldrich). Cells were lysed using a French cell press and the lysate was clarified by centrifugation (37 000g, 277 K, 30 min). The supernatant was filtered (0.2 µm) and applied onto a 5 ml HisTrap metal-chelating column (GE Healthcare) preloaded with Ni2+ and equilibrated with lysis buffer. A linear imidazole gradient was applied and the protein eluted at a concentration of approximately 160 mM imidazole. Fractions containing LdPTR1 were pooled and the histidine tag was cleaved by incubation with TEV protease at 303 K for 3 h. Imidazole was removed from the buffer by dialysis and cleavage continued at 277 K overnight before a second Ni2+-affinity step was performed, isolating the pure and cleaved LdPTR1. The protein was concentrated to approximately 10 mg ml−1 using a centrifugal filter unit with a molecular-weight cutoff of 3500 Da (Millipore) and the buffer was exchanged to 20 mM Tris–HCl pH 7.7. The high level of purity (>95%) and the molecular weight of the protein were confirmed by SDS–PAGE and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (data not shown). The yield of purified protein was relatively low at approximately 3 mg per litre of bacterial culture and we note that most of the material produced was insoluble.
2.2. Crystallization and data collection
Sitting-drop vapour diffusion was used to test a range of commercially available screens. Crystals were only obtained in the presence of both cofactor and an inhibitor and these were optimized in hanging drops. A solution of 5 mg ml−1 LdPTR1, 1 mM NADP+, 20 mM dithiothreitol and 1 mM methotrexate was incubated on ice for 1 h before crystallization. The protein solution (1 µl) was mixed with reservoir solution (1 µl) and stored at room temperature. Small orthorhombic plates with approximate dimensions of 0.1 × 0.1 × <0.05 mm formed within days above reservoirs containing 0.1 M MES pH 6.5, 10%(v/v) dioxane and 1.6 M ammonium sulfate (Fig. 1 ▶).
Figure 1.

A crystal of LdPTR1. The approximate dimensions of this sample were 0.1 × 0.1 × 0.03 mm.
Crystals were placed in a nylon loop and flash-cooled to 100 K in a stream of nitrogen gas after first being cryoprotected by passing them through a solution of 40% PEG 400. Initial in-house X-ray experiments using a Rigaku MicroMax-007 rotating-anode X-ray generator and an R-AXIS IV++ image-plate detector produced only smeared low-resolution diffraction images. The quality of the diffraction was improved by an annealing step in which the cryostream was manually diverted and reintroduced after 5–10 s (data not shown). Diffraction data were then collected on beamline I04 at the Diamond Light Source synchrotron.
2.3. Structure solution and refinement
The data were processed and scaled using MOSFLM (Leslie, 2006 ▶) and SCALA (Evans, 2006 ▶; Collaborative Computational Project, Number 4, 1994 ▶), respectively. The space group is C2221 and the unit-cell parameters are a = 107.51, b = 126.44, c = 87.51 Å. The Matthews coefficient (Matthews, 1968 ▶) of 2.5 Å3 Da−1, which corresponds to approximately 50% bulk solvent, suggested that two subunits occupy the asymmetric unit.
The LdPTR1 structure was solved by molecular replacement using Phaser (McCoy et al., 2007 ▶). The search model was a single LmPTR1 subunit (PDB code 1e7w; Gourley et al., 2001 ▶) in which divergent residues were truncated to Cβ and all ligands were removed. Two subunits were positioned and rigid-body refinement and all subsequent refinements were carried out using REFMAC5 (Murshudov et al., 1997 ▶). The graphics program Coot (Emsley & Cowtan, 2004 ▶) was used to inspect difference and electron-density maps for model fitting, solvent and ligand searching. Sulfate and water molecules were added after the protein model was completed and further rounds of refinement were performed. Noncrystallographic symmetry restraints were not employed during the analysis. Crystallographic statistics are shown in Table 1 ▶. Figures were prepared with PyMOL (DeLano, 2002 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 107.5, b = 126.4, c = 87.5 |
| Resolution range (Å) | 29.9–2.5 (2.6–2.5) |
| Wavelength (Å) | 0.973 |
| No. of measurements | 144524 (21138) |
| No. of unique reflections | 21004 (3022) |
| Multiplicity | 6.9 (7.0) |
| Completeness (%) | 99.9 (100.0) |
| Mean I/σ(I) | 11.9 (3.8) |
| Wilson B factor (Å2) | 46.9 |
| R merge † (%) | 9.6 (42.4) |
| R work ‡ (%) | 22.7 (28.0) |
| R free § (%) | 28.5 (33.0) |
| R.m.s.d. bonds (Å) | 0.019 |
| R.m.s.d. bond angles (°) | 1.802 |
| Ramachandran analysis | |
| Favoured (%) | 95.2 |
| Allowed (%) | 4.6 |
| Outliers (%) | 0.2 |
| Protein residues (total) | 432 |
| Atoms (total) | 3201 |
| Overall B factor (Å2) | 43.5 |
| Additional groups | |
| Waters | |
| No. | 24 |
| Average B factor (Å2) | 38.0 |
| Sulfates | |
| No. | 2 |
| Average B factor (Å2) | 53.4 |
R
merge = 
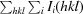 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
R
work = 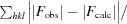
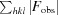 , where F
obs is the observed structure-factor amplitude and F
calc is the structure-factor amplitude calculated from the model.
, where F
obs is the observed structure-factor amplitude and F
calc is the structure-factor amplitude calculated from the model.
R free is the same as R work except calculated with a subset (5%) of data that were excluded from refinement calculations.
3. Results and discussion
3.1. Overall structure
PTR1 is a tetrameric enzyme and this crystal form of LdPTR1 has two subunits (labelled A and B) in the asymmetric unit; a 21 screw axis parallel to c generates the tetramer (Fig. 2 ▶ a). Each monomer is formed by a seven-stranded central β-sheet flanked by a set of three α-helices on either side (Fig. 2 ▶ b): a Rossmann-fold repeat (Gourley et al., 2001 ▶). The structures of subunits A and B are highly conserved, with an r.m.s.d. of 0.51 Å when 212 Cα atoms are matched; therefore, unless otherwise stated all descriptions refer to subunit A of LdPTR1.
Figure 2.

(a) Ribbon diagram of the LdPTR1 tetramer. Subunits A and B constitute the asymmetric unit. (b) Ribbon diagram of an LdPTR1 subunit. Seven red β-strands (numbered) are sandwiched between the blue α-helices; loop regions are coloured yellow. A sulfate is depicted as sticks in the active site with S coloured orange and O red; the N- and C-termini of the protein are labelled. Blue, red and black asterisks mark residues either side of the missing β3–α3, β4–α4 and β6–α6 loops, respectively.
3.2. A disordered active site
Sequence comparisons between LdPTR1 and LmPTR1 (data not shown) indicate that the residues involved in the construction of the active site and that are essential for catalysis in binding cofactor, substrates, products and inhibitors are strictly conserved and are also highly conserved in TbPTR1 (Dawson et al., 2006 ▶). However, the residues near the catalytic site were poorly ordered in contrast to the same regions of LmPTR1 and TbPTR1, with no electron density corresponding to NADP+ or methotrexate, ligands that were present in the crystallization conditions in a fivefold molar excess. The K i for methotrexate inhibition of LmPTR1 is 39 ± 19 nM and that with respect to TbPTR1 is 152 ± 16 nM (Dawson et al., 2006 ▶).
Ammonium sulfate was the precipitant for crystal growth and a sulfate ion binds in a polar cavity formed by the β1–α1 turn and the loop between β2 and α2, accepting hydrogen bonds donated from three main-chain amides (His38, Arg39 and Ser40) and Ser40 OG (Fig. 3 ▶). This polar cavity is the binding site for the adenine 2′-phosphate group of the cofactor (Gourley et al., 2001 ▶; Dawson et al., 2006 ▶).
Figure 3.
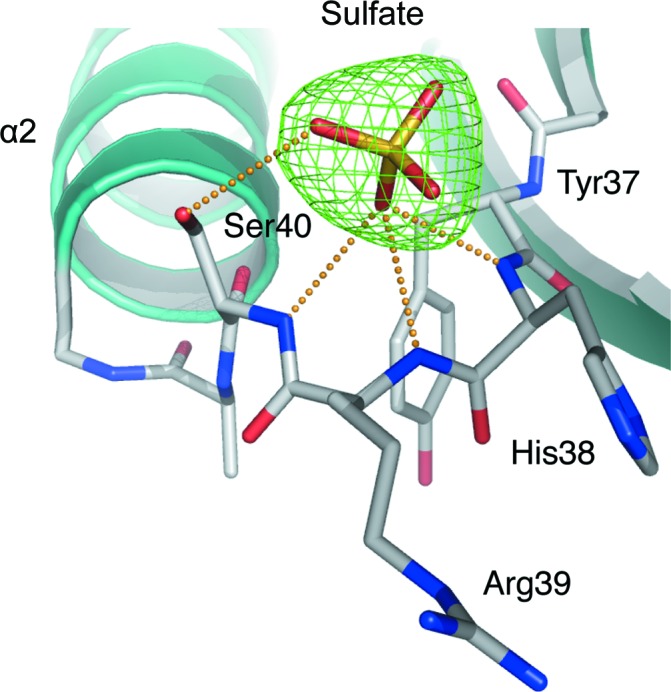
The F o − F c difference density OMIT map for sulfate bound in the LdPTR1 NADP+-binding site (green mesh contoured at 3.5σ). The orange dotted lines represent potential hydrogen bonds formed between the ion and the enzyme. These interactions are in the range 2.7–3.1 Å.
The core structure of the subunit is preserved between the LdPTR1 and LmPTR1 structures (Fig. 4 ▶) and an overlay of 202 Cα atoms common to both subunits resulted in an r.m.s.d. of 1.3 Å (calculated using Coot; Emsley & Cowtan, 2004 ▶). Owing to the absence of well defined electron density, the LdPTR1 model contains fewer residues compared with those of LmPTR1 or TbPTR1. Missing segments include residues 70–80 (the loop linking β3 and α3), 112–132 (the β4–α4 loop) and 227–254 (the β6–α6 loop). The β3–α3 loop is also poorly ordered in LmPTR1 (Schüttelkopf et al., 2005 ▶). However, the β4–α4 loop is well ordered in LmPTR1 but is missing in LdPTR1. This means that in LdPTR1, Phe113, which is a critical residue, is disordered. The phenylalanine, together with the nicotinamide, forms π-stacking interactions that stabilize ligand binding in the catalytic site (Gourley et al., 2001 ▶; Dawson et al., 2006 ▶). The largest break in the electron density involves residues 227–254, which form what is termed the substrate-binding loop linking β6 to α6 (Tulloch et al., 2010 ▶). Residues in this loop bind parts of folate substrates, products and some inhibitors. The absence of the β4–α4 loop vacates an area in the active-site cleft, allowing the β5–α5 loop of LdPTR1 to adopt a different position to fill the gap. Here, the Cα atoms of several residues relocate by between 10 and 16 Å.
Figure 4.
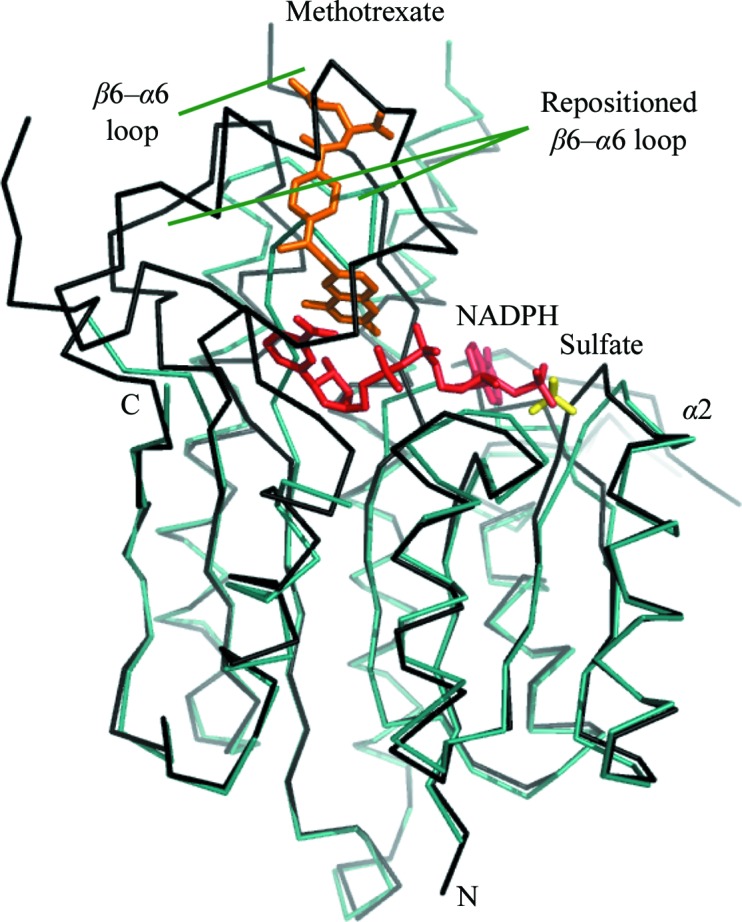
A Cα trace of one LdPTR1 subunit (cyan) overlaid with an LmPTR1 subunit (PDB code 1e7w; Gourley et al., 2001 ▶; black). The sulfate ion bound to LdPTR1 is shown as yellow sticks, while the NADPH and methotrexate binding to LmPTR1 are depicted as red and orange sticks, respectively. The β6–α6 loop of the LmPTR1 model is marked. This loop is absent from LdPTR1. The β5–α5 substrate-binding loop adopts different positions in the two structures.
In LdPTR1 Arg17 is disordered. This residue is important in binding the cofactor pyrophosphate (McLuskey et al., 2004 ▶), which in turn interacts with and positions the nicotinamide. Asp181, Tyr194 and Lys198 are the key catalytic residues. Asp181 is located within the link between β5 and α5, but this loop has been built into weak electron density relative to the structure as a whole. The orientation of Tyr194 is similar to that observed in LmPTR1. However, only the Cα atoms of Lys198 and its closest neighbours agree reasonably well with the identical LmPTR1 residues, while the side chain extends into a position that is inappropriate to form the stabilizing interaction formed with the nicotinamide ribose in LmPTR1 (Gourley et al., 2001 ▶; data not shown).
PTR1 displays a sequential ordered mechanism, with first cofactor binding and then substrate; following reduction the product leaves, followed by oxidized cofactor (Luba et al., 1998 ▶; Gourley et al., 2001 ▶). Substrate or inhibitors can only bind after the binary protein–cofactor complex has formed. Since sulfate binding blocks the 2′-phosphate-binding site, NADP+ is not present and this leads to disorder in the substrate-binding part of the active site.
4. Conclusions
The structure of apo LdPTR1 has been solved to 2.5 Å resolution. Attempts to improve the LdPTR1 crystal quality by attempting to crystallize the apo form and the binary complex with cofactor and by introducing other ligands in combination with oxidized and reduced cofactor failed. Even though we see no evidence for these ligands in the electron-density maps, the presence of both methotrexate and NADP+ was essential to obtain this crystal form. Despite their presence in the crystallization mixture, there was no electron density corresponding to these molecules. Instead, the high ammonium sulfate concentration in the crystallization conditions resulted in the replacement of an NADP+ phosphate by sulfate ions. The formation of the catalytic site of PTR1 is dependent on the presence of nicotinamide and in the absence of NADPH(+) this part of the active site is disordered. We conclude that owing to the disorder this crystal form is not suitable for characterizing the interactions of LdPTR1 with inhibitors.
Supplementary Material
PDB reference: pteridine reductase, 2xox
Acknowledgments
This research was funded by The Wellcome Trust (WT082596 and WT083481). We thank the Diamond Light Source for synchrotron beam time and excellent staff support.
References
- Cavazzuti, A., Paglietti, G., Hunter, W. N., Gamarro, F., Piras, S., Loriga, M., Allecca, S., Corona, P., McLuskey, K., Tulloch, L., Gibellini, F., Ferrari, S. & Costi, M. P. (2008). Proc. Natl Acad. Sci. USA, 105, 1448–1453. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Croft, S. L., Sundar, S. & Fairlamb, A. H. (2006). Clin. Microbiol. Rev. 19, 111–126. [DOI] [PMC free article] [PubMed]
- Cunningham, M. L., Titus, R. G., Turco, S. J. & Beverley, S. M. (2001). Science, 292, 285–287. [DOI] [PubMed]
- Dawson, A., Gibellini, F., Sienkiewicz, N., Tulloch, L. B., Fyfe, P. K., McLuskey, K., Fairlamb, A. H. & Hunter, W. N. (2006). Mol. Microbiol. 61, 1457–1468. [DOI] [PMC free article] [PubMed]
- Dawson, A., Tulloch, L. B., Barrack, K. L. & Hunter, W. N. (2010). Acta Cryst. D66, 1334–1340. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL. http://www.pymol.org.
- Desjeux, P. (2004). Comp. Immunol. Microbiol. Infect. Dis. 27, 305–318. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gibson, C. L., Huggan, J. K., Kennedy, A., Kiefer, L., Lee, J. H., Suckling, C. J., Clements, C., Harvey, A. L., Hunter, W. N. & Tulloch, L. B. (2009). Org. Biomol. Chem. 7, 1829–1842. [DOI] [PubMed]
- Gourley, D. G., Schüttelkopf, A., Leonard, G., Luba, J., Hardy, L., Beverley, S. & Hunter, W. N. (2001). Nature Struct. Biol. 8, 521–525. [DOI] [PubMed]
- Herwaldt, B. L. (1999). Lancet, 354, 1191–1199. [DOI] [PubMed]
- Hunter, W. N. (2009). J. Biol. Chem. 284, 11749–11753. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Luba, J., Nare, B., Liang, P.-H., Anderson, K. S., Beverley, S. M. & Hardy, L. W. (1998). Biochemistry, 37, 4093–4104. [DOI] [PubMed]
- Kaur, J., Sundar, S. & Singh, N. (2010). J. Antimicrob. Chemother. 65, 1742–1748. [DOI] [PubMed]
- Maltezou, H. C. (2010). J. Biomed. Biotechnol. 2010, 617521. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McLuskey, K., Gibellini, F., Carvalho, P., Avery, M. A. & Hunter, W. N. (2004). Acta Cryst. D60, 1780–1785. [DOI] [PubMed]
- Moreira, W., Leblanc, E. & Ouellette, M. (2009). Free Radic. Biol. Med. 46, 367–375. [DOI] [PubMed]
- Mpamhanga, C. P., Spinks, D., Tulloch, L. B., Shanks, E. J., Robinson, D. A., Collie, I. T., Fairlamb, A. H., Wyatt, P. G., Frearson, J. A., Hunter, W. N., Gilbert, I. H. & Brenk, R. (2009). J. Med. Chem. 52, 4454–4465. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nare, B., Hardy, L. W. & Beverley, S. M. (1997). J. Biol. Chem. 272, 13883–13891. [DOI] [PubMed]
- Nare, B., Luba, J., Hardy, L. W. & Beverley, S. (1997). Parasitology, 114, 101–110. [PubMed]
- Reithinger, R., Dujardin, J. C., Louzir, H., Pirmez, C., Alexander, B. & Brooker, S. (2007). Lancet Infect. Dis. 7, 581–596. [DOI] [PubMed]
- Schormann, N., Pal, B., Senkovich, O., Carson, M., Howard, A., Smith, C., DeLucas, L. & Chattopadhyay, D. (2005). J. Struct. Biol. 152, 64–75. [DOI] [PubMed]
- Schüttelkopf, A. W., Hardy, L. W., Beverley, S. M. & Hunter, W. N. (2005). J. Mol. Biol. 352, 105–116. [DOI] [PubMed]
- Tulloch, L. B., Martini, V. P., Iulek, J., Huggan, J. K., Lee, J. H., Gibson, C. L., Smith, T. K., Suckling, C. J. & Hunter, W. N. (2010). J. Med. Chem. 53, 221–229. [DOI] [PMC free article] [PubMed]
- World Health Organization. (2007). Global Plan to Combat Neglected Tropical Diseases 2008–2015. http://whqlibdoc.who.int/hq/2007/WHO_CDS_NTD_2007.3_eng.pdf.
- Zhao, H., Bray, T., Ouellette, M., Zhao, M., Ferre, R. A., Matthews, D., Whiteley, J. M. & Varughese, K. I. (2003). Acta Cryst. D59, 1539–1544. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: pteridine reductase, 2xox