

The small terminase subunit of bacteriophage P22 was overexpressed in E. coli, purified under native conditions and crystallized as a nonamer. High-quality diffraction data were collected to 1.75 Å resolution using synchrotron radiation.
Keywords: small terminase, large terminase, DNA packaging, bacteriophage P22, nonamers
Abstract
The packaging of viral genomes into preformed empty procapsids is powered by an ATP-dependent genome-translocating motor. This molecular machine is formed by a heterodimer consisting of large terminase (L-terminase) and small terminase (S-terminase) subunits, which is assembled into a complex of unknown stoichiometry, and a dodecameric portal protein. There is considerable confusion in the literature regarding the biologically relevant oligomeric state of terminases, which, like portal proteins, form ring-like structures. The number of subunits in a hollow oligomeric protein defines the internal diameter of the central channel and the ability to fit DNA inside. Thus, knowledge of the exact stoichiometry of terminases is critical to decipher the mechanisms of terminase-dependent DNA translocation. Here, the gene encoding bacteriophage P22 S-terminase in Escherichia coli has been overexpressed and the protein purified under native conditions. In the absence of detergents and/or denaturants that may cause disassembly of the native oligomer and formation of aberrant rings, it was found that P22 S-terminase assembles into a concentration-independent nonamer of ∼168 kDa. Nonameric S-terminase was crystallized in two different crystal forms at neutral pH. Crystal form I belonged to space group P21212, with unit-cell parameters a = 144.2, b = 144.2, c = 145.3 Å, and diffracted to 3.0 Å resolution. Crystal form II belonged to space group P21, with unit-cell parameters a = 76.48, b = 100.9, c = 89.95 Å, β = 93.73°, and diffracted to 1.75 Å resolution. Preliminary crystallographic analysis of crystal form II confirms that the S-terminase crystals contain a nonamer in the asymmetric unit and are suitable for high-resolution structure determination.
1. Introduction
P22 is a prototypical member of the Podoviridae family of double-stranded DNA (dsDNA) bacteriophages that infects Salmonella enterica (Teschke & Parent, 2010 ▶). The infectious virion consists of an icosahedral T = 7 capsid ∼650 Å in diameter interrupted at a unique vertex by an ∼2.8 MDa tail apparatus (Lander et al., 2006 ▶; Chang et al., 2006 ▶). The tail complex, which is also known as the ‘portal vertex structure’, is formed by five proteins repeated in different stoichiometries (Lander et al., 2009 ▶). This includes a dodecamer of the portal protein gp1 (Bazinet et al., 1988 ▶), 12 copies of gp4 (Olia et al., 2006 ▶), a hexamer of gp10 (Olia, Bhardwaj et al., 2007 ▶), a trimer of the tail needle gp26 (Andrews et al., 2005 ▶; Olia, Casjens et al., 2007 ▶; Bhardwaj et al., 2007 ▶) and six copies of the trimeric tailspike gp9 (Goldenberg & King, 1982 ▶).
The morphogenesis of P22 has been well characterized and most if not all of the factors involved in virus maturation have been characterized (Teschke & Parent, 2010 ▶). The virus first assembles as a spherical metastable procapsid of ∼600 Å in diameter that undergoes exothermic expansion (to ∼650 Å diameter) during packaging (Zhang et al., 2000 ▶; Thuman-Commike et al., 1996 ▶). Packaging of the P22 genome (∼43 kbp) into the procapsid is powered by an ATP-dependent motor, which consists of a complex of small and large terminase subunits (abbreviated as S-terminase and L-terminase, respectively; Casjens & Huang, 1982 ▶; Jackson et al., 1982 ▶; Poteete et al., 1979 ▶, 1983 ▶) bound to a dodecameric portal protein. DNA packaging proceeds by the ‘headful packaging’ mechanism, which means that the P22 procapsid is filled to capacity with DNA, followed by a nucleolytic cleavage catalyzed by the L-terminase subunit that frees the virion genome from the concatemeric DNA (Casjens & Weigele, 2005 ▶). After packaging of a single copy of the P22 genome (∼43 kbp), the portal channel is closed by the sequential addition of gp4, gp10 and gp26 followed by gp9 (Strauss & King, 1984 ▶).
A wealth of genetic and biochemical evidence (Rao & Feiss, 2008 ▶) from well characterized phages (e.g. SPP1, T3, T4, T7 and λ) has suggested that L-terminase and S-terminase play distinct roles in DNA packaging. L-terminase possesses the ATPase and nuclease activities that are required to translocate the viral genome inside the procapsid. In contrast, S-terminase recognizes the packaging-initiation sites (known as pac in P22) in preparation of packaging and stimulates the ATPase activity of the large subunit during packaging. Unfortunately, the oligomeric states of S-terminases in different viruses remain controversial. In P22, the S-terminase subunit (gp3) has been reported to assemble into a nonamer (Nemecek et al., 2007 ▶, 2008 ▶). In contrast, it was found that the SPP1 terminase small subunit (G1P) assembles into a decameric ring (Camacho et al., 2003 ▶), while both the T4 (gp16) and T7 (gp18) S-terminase subunits are thought to assemble into octamers (Lin et al., 1997 ▶; White & Richardson, 1987 ▶). In Sf6, a phage closely related to P22, a recent crystal structure of S-terminase revealed an octameric ring (Zhao et al., 2010 ▶) with an internal channel that was too small to accommodate dsDNA. This structure raises concerns over the ability of S-terminases to ‘thread’ DNA inside the central channel and instead lends support to a model in which DNA ‘wraps’ around the perimeter of S-terminase (Nemecek et al., 2008 ▶). In this paper, we report the crystallization of bacteriophage P22 S-terminase. We have determined that recombinant P22 S-terminase purified under native conditions folds into a nonameric ring both in solution and in the crystal. We hypothesize that the internal channel of this ring is large enough to accommodate hydrated dsDNA and thus S-terminase binds DNA like a portal protein.
2. Materials and methods
2.1. Cloning, expression and purification of native S-terminase
The gene encoding the S-terminase protein was PCR-amplified and ligated into the BamHI/HindIII sites of vector pMAL c2E (New England Biolabs), which yields S-terminase fused to an N-terminal maltose-binding protein (MBP). MBP-S-terminase fusion protein was expressed in Escherichia coli strain BL21 (DE3) pLysS cells in Luria–Bertani medium supplemented with 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol. Cells were grown at 310 K to an optical density at 600 nm of 0.6 and S-terminase expression was induced by the addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 14 h at 289 K. Cell pellets expressing MBP-S-terminase fusion protein were lysed by sonication in lysis buffer consisting of 20 mM Tris–HCl pH 7.5, 300 mM NaCl, 1 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM phenylmethanesulfonyl fluoride (PMSF). MBP-S-terminase was affinity-purified from a soluble E. coli lysate using amylose–agarose resin (New England Biolabs) and eluted with 10 mM maltose. The MBP tag was cleaved off by overnight incubation with PreScission protease (2 units per 100 µg fusion protein) and the S-terminase was further purified on a Superdex 200 size-exclusion column (GE Life Sciences). For limited proteolysis experiments, purified S-terminase at 1 mg ml−1 was mixed with chymotrypsin (at 5 µg ml−1) in digestion buffer (20 mM Tris–HCl, 200 mM NaCl, 5 mM β-mercaptoethanol, 5% glycerol and 1 mM MgCl2 pH 7.5). The protease:substrate ratio used for digestion was 1:200(w:w) and the reaction was incubated on ice for 30 min. 10 µl aliquots of digestion mixture were taken at time intervals and the reaction was stopped by adding 1 mM PMSF followed by boiling for 5 min in 10 µl 2× SDS sample buffer. Samples were then analyzed by SDS–PAGE (12.5%) and stained with Coomassie Brilliant Blue G-250.
2.2. Analytical ultracentrifugation
Analytical ultracentrifugation experiments were carried out using a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge equipped with absorbance optics and an eight-hole An-50 Ti analytical rotor at the Kimmel Cancer Center X-ray Crystallography and Molecular Characterization shared-resource facility at Thomas Jefferson University. Sedimentation-velocity experiments were carried out at 283 K and 72 500g using standard Epon two-channel centerpieces with quartz windows. P22 S-terminase at a concentration of 15 µM was analyzed in crystallization buffer (20 mM Tris–HCl, 250 mM NaCl, 5 mM β-mercaptoethanol, 5% glycerol and 1 mM MgCl2 pH 7.5). Sedimentation boundaries were analyzed by the continuous distribution [c(s)] method using the program SEDFIT (Schuck, 2000 ▶). The program SEDNTERP v.1.09 (Laue et al., 1992 ▶) was used to correct the experimental s value (s*) to standard conditions at 293 K in water (s 20,w) and to calculate the partial specific volume of S-terminase.
2.3. Crystallization of oligomeric S-terminase
S-terminase at 10 mg ml−1 in crystallization buffer was crystallized by the microbatch-under-oil method in Crystal Quick 96-well plates (Hampton Research) using a Hydra II crystallization robot at the Kimmel Cancer Center X-ray Crystallography and Molecular Characterization shared-resource facility at Thomas Jefferson University. Conditions initially screened included the Index and the PEG/Ion screens (Hampton Research). All crystallization hits were reproduced using the hanging-drop vapor-diffusion method in Linbro 24-well plates (Hampton Research) by mixing 2 µl protein solution (at 7–10 mg ml−1) with an equal volume of reservoir solution and equilibrating against 600 µl reservoir solution. Two different conditions from the PEG/Ion screen yielded diffracting crystals: condition No. 10 [20%(w/v) PEG 3350, 0.2 M sodium iodide pH 7.0] and condition No. 14 [20%(w/v) PEG 3350, 0.2 M potassium thiocyanate pH 7.0]. The PEG/Ion screen hits were reproduced and optimized using the volumetric method without any additional pH adjustment. In both cases S-terminase crystals grew at 293 K within one week.
2.4. X-ray data collection and analysis
Single crystals of native S-terminase were cryocooled after adding 27.5% ethylene glycol to the reservoir solution. Several data sets were collected on beamline F-1 at Cornell High Energy Synchrotron Source (CHESS; Ithaca, New York, USA) and beamline X6A at the National Synchrotron Light Source (NSLS; Upton, New York, USA). Diffraction data were reduced to hkl intensities using the programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶) from the HKL-2000 package and were further analyzed with CCP4 programs (Collaborative Computational Project, Number 4, 1994 ▶). Self-rotation functions were computed with GLRF (Tong & Rossmann, 1997 ▶). A complete summary of diffraction statistics is given in Table 1 ▶.
Table 1. Summary of diffraction data statistics for native P22 S-terminase crystal forms I and II.
Values in parentheses are for the highest resolution shell.
| Crystal form I | Crystal form II | |
|---|---|---|
| Crystallization condition | 20%(w/v) PEG 3350, 0.2 M sodium iodide pH 7.0 | 20%(w/v) PEG 3350, 0.2 M potassium thiocyanate pH 7.0 |
| Beamline | NSLS X6A | NSLS X6A |
| Wavelength (Å) | 0.98 | 0.98 |
| Space group | P21212 | P21 |
| Reflections (total/unique) | 686006/58407 | 2483158/126076 |
| Unit-cell parameters (Å, °) | a = 144.18, b = 144.24, c = 145.37, α = β = γ = 90.0 | a = 76.48, b = 100.90, c = 89.85, α = γ = 90.0, β = 93.73 |
| Resolution (Å) | 30–3.0 (3.1–3.0) | 30–1.75 (1.8–1.75) |
| Completeness (%) | 95.3 (96.2) | 93.5 (62.5) |
| Multiplicity | 3.3 (3.1) | 4.4 (3.3) |
| Rmerge† (%) | 8.8 (43.1) | 8.8 (67.4) |
| 〈I〉/〈σ(I)〉 | 21.0 (3.9) | 28.2 (1.9) |
R
merge = 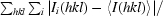
 , where Ii(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
, where Ii(hkl) is the ith intensity measurement of reflection hkl, including symmetry-related reflections, and 〈I(hkl)〉 is its average.
3. Results and discussion
3.1. P22 S-terminase purified under native conditions forms nonamers in solution
The S-terminase subunit of bacteriophage P22 consists of 162 residues and has a molecular mass of ∼18.5 kDa. An initial attempt to express recombinant untagged (or His-tagged) S-terminase in E. coli revealed poor solubility. Although a fraction of the protein could be solubilized using 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPs; Nemecek et al., 2007 ▶), we decided to avoid the use of detergents, which can perturb the native oligomeric structure of S-terminase. As an alternative strategy, the gene coding P22 S-terminase was fused to maltose-binding protein (MBP) for overexpression in E. coli and affinity-purified under native conditions on amylose beads. The MBP-S-terminase fusion protein was perfectly soluble; the S-terminase remained soluble even after cleavage of the affinity tag (MBP) and could be concentrated to ∼10 mg ml−1. The digestion mixture containing S-terminase and MBP was further purified on a Superdex 200 gel-filtration column, which gave three distinct peaks (Fig. 1 ▶ a). The first peak (peak a) contained a large and likely aggregated population of S-terminase migrating in the void volume. The second peak (peak b) also contained an oligomeric species of S-terminase of ∼200 kDa, as determined using molecular-mass calibration markers (data not shown). The third peak (peak c) contained exclusively cleaved MBP, which migrated at the expected mass of ∼42 kDa.
Figure 1.

P22 S-terminase assembles into a nonamer in solution. (a) Size-exclusion chromatogram of overexpressed P22 S-terminase. The Superdex 200 gel-filtration column was calibrated using molecular-mass markers (data not shown). S-terminase elutes as two oligomeric species corresponding to a large aggregate migrating in the void volume (molecular mass >600 kDa; peak a) and a smaller species of ∼200 kDa (peak b). The third peak (peak c) contains free MBP. (b) Sedimentation-velocity profile of P22 S-terminase measured at 15 µM concentration in 0.25 M sodium chloride at 283 K. Top, raw absorbance at 278 nm plotted as a function of the radial position. Data at intervals of 14 min are shown as dots for sedimentation at 30 000 rev min−1. The monophasic sedimentation boundary suggests that S-terminase exists as a monodisperse single species in solution. Middle, the residuals between the fitted curve and the raw data plotted as a function of radius. Bottom, the diffusion-free sedimentation-coefficient distribution c(s) derived from sedimentation velocity of P22 S-terminase corresponds to an estimated molecular mass of ∼167.8 kDa. Given the size of monomeric S-terminase (∼18.5 kDa), this molecular mass is consistent with a nonamer.
We used sedimentation-velocity analysis to determine the oligomerization of S-terminase in peak b. Fig. 1 ▶(b) shows a typical sedimentation profile of S-terminase at 15 µM final concentration in 20 mM HEPES, 0.25 M sodium chloride pH 7.5. Interestingly, the sedimentation boundary exhibits monophasic behavior, which is indicative of a single major (>96%) component in solution migrating with a sedimentation coefficient of 5.09 S. Conversion of the distribution of the apparent sedimentation coefficient to molecular mass for three independent runs revealed a molecular mass of ∼167.8 kDa. This value agrees with a nonamer of S-terminase, which has an expected molecular mass of 9 × 18.5 = 166.5 kDa. In addition, the S-terminase nonameric quaternary structure was independent of concentration under the range of concentrations tested (1–15 µM), suggesting a low oligomerization constant. These findings agree well with recent reports by Nemecek et al. (2007 ▶, 2008 ▶). Using native mass spectrometry and negative-stain electron microscopy, these authors also found that P22 S-terminase assembles into a nonamer. However, while they also observed monomeric S-terminase in equilibrium with the nonamer, our preparation of S-terminase was always fully oligomeric and we failed to observe monomeric S-terminase even at concentrations as low as 1 µM. This discrepancy is likely to reflect the fact that Nemecek and coworkers expressed His-tagged S-terminase, which is poorly soluble, in E. coli and used 10 mM CHAPs to solubilize it from inclusion bodies. Instead, our S-terminase was perfectly soluble before and after removing the MBP affinity tag and did not require solubilization with detergents.
3.2. Crystallization of nonameric S-terminase
We subjected purified P22 S-terminase to microbatch crystallization under oil using a Hydra II crystallization robot. Standard Index and PEG/Ion crystallization screens (Hampton Research) gave at least 12 distinct crystallization hits at pH values between 6.5 and 7.5. Most crystallization conditions contained highly concentrated solutions of various PEGs and yielded elongated crystals. All hits were repeated in-house using the hanging-drop vapor-diffusion method. The largest crystals were obtained using 20%(w/v) PEG 3350 pH 7.0 as a crystallization agent, in the presence of either 0.2 M sodium iodide (Fig. 2 ▶ a) or 0.2 M potassium thiocyanate (Fig. 2 ▶ b). These two crystal forms (which are referred to as crystal forms I and II, respectively) were morphologically very different, crystal form I being elongated and crystal form II shaped like a plate. Regardless of the crystallization condition used, all crystals grew within 5–7 d.
Figure 2.

Crystallization of S-terminase. (a) P22 S-terminase crystal form I obtained by the hanging-drop vapor-diffusion method using 0.2 M sodium iodide, 20%(w/v) PEG 3350 pH 7.0 as the crystallization agent. Individual crystals have average dimensions of 80 × 100 × 350 µm. (b) Crystals of form II with a plate-like morphology were also obtained by the hanging-drop vapor-diffusion method, using 0.2 M potassium thiocyanate, 20%(w/v) PEG 3350 pH 7.0 as the crystallization agent. The largest plates have average dimensions of 50 × 500 × 800 µm. (c) 12.5% SDS–PAGE analysis of the purified S-terminase used for crystallization (lane 2) and of a dissolved S-terminase crystal (lane 3). Lane 1 contains molecular-weight markers (kDa). (d) Limited proteolysis analysis of S-terminase in the presence of chymotrypsin. Aliquots of the digestion mixture were collected after 1 min (lane 3), 15 min (lane 4) and 30 min (lane 5); the reaction was blocked with 1 mM PMSF and resolved by 12.5% SDS–PAGE. Single and double asterisks indicate the positions of full-length and C-terminally truncated S-terminase, respectively. Lane 1 contains molecular-weight markers (kDa).
To confirm that the S-terminase crystals contained protein and not salt, a single crystal was isolated from a two-week-old crystallization drop, carefully washed in PBS and analyzed by SDS–PAGE. As shown in Fig. 2 ▶(c), the crystal was found to contain a slightly degraded version of S-terminase. Using MALDI mass spectrometry, we found that the crystallized protein contained a C-terminal deletion of the last ∼15–20 residues (data not shown). Inspection of the protein sequence revealed a cluster of positively charged residues at positions 140–151 (140-KGDRDKRRSRIK-151) which is a possible target for a protease with trypsin-like specificity that is likely to have copurified from E. coli. Attentive analysis of freshly purified S-terminase suggested that the C-terminal degradation occurred within 5–7 d of purification. Interestingly, this was also the approximate time necessary to obtain crystals. Intuitively, we thought that heterogeneity in the C-terminus of S-terminase could potentially result in partially disordered crystals and hence poor diffraction quality. To avoid this potential problem, freshly purified S-terminase was subjected to limited proteolysis in the presence of chymotrypsin. In a time course of digestion using a protease:S-terminase ratio of 1:200, we found that the degradation seen in solution after 5–7 d could be reproduced in only 15–30 min (Fig. 2 ▶ d, lanes 4–5). Once again, mass-spectrometric analysis of chymotrypsin-treated S-terminase confirmed that the C-terminal cleavage was as homogeneous as that observed in the crystallization drop (data not shown). Accordingly, crystallization attempts using chymotrypsin-treated S-terminase yielded crystals in only 4–5 h; however, these crystals failed to grow larger than 50 µm in the longest dimension, likely as a consequence of the excessively fast nucleation kinetics. We tried to slow the rate of crystallization by adding 10%(v/v) glycerol to the protein solution to increase its solubility and by reducing the protein concentration and lowering the temperature of crystallization. Despite all efforts, chymotrypsin-treated S-terminase failed to give crystals as large as those obtained by slow in-drop proteolysis.
3.3. Crystallographic analysis reveals a nonameric quaternary structure
All crystals were screened for diffraction using synchrotron radiation. Diffraction analysis confirmed that of the 12 crystal forms initially obtained, only two (Figs. 2 ▶ a and 2 ▶ b) reproducibly diffracted X-rays to better than 3.5 Å resolution. The first crystal form (Fig. 2 ▶ a) had an orthorhombic (P21212) pseudo-cubic unit cell with unit-cell parameters a = 144.2, b = 144.2, c = 145 Å and diffracted weakly to ∼3 Å resolution (Table 1 ▶). In contrast, crystal form II (Fig. 2 ▶ b) belonged to a primitive monoclinic space group (P21) with unit-cell parameters a = 76.48, b = 100.90, c = 89.85 Å, β = 93.73° and diffracted to 1.75 Å resolution. A characteristic 0.5° oscillation diffraction pattern of this second crystal form is shown in Fig. 3 ▶(a). The diffraction data for crystal form II were of excellent quality, with a cumulative R merge and 〈I〉/〈σ(I)〉 of 7.1% and 28.2, respectively (Table 1 ▶).
Figure 3.

Diffraction analysis confirms that S-terminase is a nonamer. (a) Diffraction image recorded from S-terminase crystal form II oscillated 0.5° with an exposure time of 30 s. The image was measured on NSLS beamline X6A using an X-ray wavelength of 0.98 Å. Distinct and intense diffraction spots are observed to 1.75 Å resolution (Table 1 ▶). (b, c) Stereographic projections of the κ = 180° (b) and κ = 40° (c) sections of the P22 S-terminase self-rotation function, showing the directions of twofold and ninefold NCS axes. The function was computed using the program GLRF (Tong & Rossmann, 1997 ▶) and the maps are contoured at 3.5σ (b) and 10.0σ (c) in steps of 0.25σ. The polar angles ψ, ϕ, κ follow the Rossmann convention (Rossmann & Blow, 1962 ▶), with the orthogonal y axis aligned with the crystal b axis; the x axis lies in the crystal ab plane and the z axis coincides with the c axis. The polar angle ψ denotes the inclination of the rotation axis from the y axis, which in space group P21 corresponds to the crystallographic screw axis. The polar angle ϕ denotes the inclination of the rotation axis from the x axis. (d) One-dimensional κ plot showing the intensity of the self-rotation function at ϕ = 120°, ψ = 90°, varying κ from 0° to 360°. The peak at the origin (0° = 360°) has been partially omitted for clarity (the self-rotation has a maximum value at the origin) and the plot is only shown between 10° and 350°. The presence of a ninefold NCS axis is revealed by a maximum of the self-rotation function every 40°.
To determine the oligomeric state of S-terminase, we focused on crystal form II, for which higher resolution and superior quality data were obtained. We investigated the local symmetry of the form II S-terminase crystal by self-rotation functions using polar angles (Rossmann & Blow, 1962 ▶). Self-rotation functions were computed with GLRF (Tong & Rossmann, 1997 ▶) using approximately 2000 reflections with an 〈I〉/〈σ(I)〉 higher than 5 in the resolution range 10.0–3.5 Å and a radius of integration of 25 Å. In a spherical polar coordinate system, the self-rotation function is defined by three angles, ϕ, ψ and κ, which describe a rotation of angle κ about an axis defined by ϕ and ψ in a suitable orthogonal system (Rossmann & Blow, 1962 ▶). The angle ψ denotes the inclination of the rotation axis from the y axis, ϕ denotes the angle between the projection of the rotation-function axis on the xz plane and the x axis, and κ is the rotation of the self-rotation vector around itself. To investigate the existence of local twofold noncrystallographic symmetry (NCS) axes, we computed a self-rotation function in space group P21 for κ = 180° (a search for twofold NCS rotation axes). As shown in Fig. 3 ▶(b), the stereographic projections at κ = 180° revealed a constellation of nine distinct peaks of ∼3.5σ, which correspond to nine copies of the S-terminase subunit per ring. The stereographic projections also allowed us to determine the orientation of the ninefold NCS axis, which gives a maximum of the rotation function at angles ϕ = 120°, ψ = 90° and κ = 40° (Fig. 3 ▶ c). Finally, we computed a one-dimensional search by fixing ϕ = 120° and ψ = 90° and searching κ in the entire polar space 1–360° (Fig. 3 ▶ d). This one-dimensional κ plot revealed a regular periodicity of a maximum of the rotation function every 40°, thereby confirming that S-terminase is a nonamer. Thus, crystal form II contains a nonamer of P22 S-terminase in the asymmetric unit, which results in approximately 41% solvent content and a Matthews coefficient V M of ∼2.07 Å3 Da−1.
4. Conclusions
In this paper, we report the crystallization and preliminary crystallographic analysis of P22 S-terminase overexpressed in E. coli. Using analytical ultracentrifugation and self-rotation analysis, we demonstrated that P22 S-terminase purified under native conditions assembles into a nonamer of ∼168 kDa. We found that S-terminase is susceptible to proteolytic cleavage in the C-terminal basic stretch 140–151, which has previously been implicated in DNA binding (Nemecek et al., 2008 ▶). Proteolysis of S-terminase is likely to be carried out by trypsin-like proteases copurified from E. coli. Despite the use of protease inhibitors during the initial steps of purification, the proteolytic cleavage seen in the drop (Fig. 2 ▶ c) goes to completion within 5–7 d. Counterintuitively, the slow degradation of the S-terminase C-terminus in the crystallization drop was essential to obtain large and well ordered crystals. When the degradation was accelerated in vitro by digesting the protein with chymotrypsin, homogeneous C-terminally truncated S-terminase nucleated and crystallized in hanging drops within only 4–5 h, but failed to give large crystals. This is consistent with the high tendency of cleaved S-terminase to crystallize, which could not be slowed down by working at 277 K or by adding glycerol. Thus, the presence of a flexible moiety in the C-terminus of S-terminase together with the fortuitous presence of trypsin-like protease activity in the purified sample were both essential to obtain high-quality crystals of P22 S-terminase. Evidently, the slow rate at which the full-length protein is cleaved in the drop results in an optimal concentration of truncated S-terminase that forms well ordered crystals within approximately one week.
The oligomeric state of P22 S-terminase presented in this paper agrees well with recent reports by Nemecek et al. (2007 ▶, 2008 ▶), but is surprisingly different from the recently described structure of the bacteriophage Sf6 terminase, which was crystallized as an octamer (Zhao et al., 2010 ▶). The latter has an internal diameter that is too small to accommodate dsDNA. This indirectly lends support to a model in which DNA is bound on the outside of the terminase (like a ‘gear’) as opposed to being ‘threaded’ inside the central channel like in a portal protein. Intuitively, the number of subunits building up a hollow oligomeric protein such as S-terminase dictates the diameter of the central channel and thus the ability to ‘thread’ DNA. This suggests that knowing the exact stoichiometry of S-terminase is essential to study the mechanism of DNA packaging. A similar controversy on the biologically relevant oligomeric conformation of portal proteins has been well documented in the literature (Trus et al., 2004 ▶). For instance, overexpressed SPP1 portal protein assembles into a 13-fold symmetric oligomer in vitro (Orlova et al., 1999 ▶; Lebedev et al., 2007 ▶), but is dodecameric in the mature virion (Lurz et al., 2001 ▶) or in the as-isolated connector bound to tail factors gp6 and gp15 (Orlova et al., 2003 ▶). While it is possible that the oligomeric state of S-terminase varies in different viruses, it appears unlikely that the oligomeric state changes between two bacteriophages as similar as Sf6 and P22, which belong to the same family (Podoviridae) and genus (‘P22-like’). Thus, the work presented in this paper provides the necessary prerequisites to solve the atomic structure of a prototypical viral S-terminase in a nonameric quaternary structure. This will hopefully shed light on how S-terminase recognizes the packaging-initiation site in phages such as P22 that package DNA using the ‘headful packaging’ mechanism (Casjens & Weigele, 2005 ▶).
Acknowledgments
We thank Kaylen Lott for critical reading of the manuscript. We are also grateful to Vivian Stojanoff and the staff at NSLS beamline X6A and macCHESS for beam time and assistance in data collection. This work was supported by NIH grant 1R56 AI076509-01A1 to GC.
References
- Andrews, D., Butler, J. S., Al-Bassam, J., Joss, L., Winn-Stapley, D. A., Casjens, S. & Cingolani, G. (2005). J. Biol. Chem. 280, 5929–5933. [DOI] [PubMed]
- Bazinet, C., Benbasat, J., King, J., Carazo, J. M. & Carrascosa, J. L. (1988). Biochemistry, 27, 1849–1856. [DOI] [PubMed]
- Bhardwaj, A., Olia, A. S., Walker-Kopp, N. & Cingolani, G. (2007). J. Mol. Biol. 371, 374–387. [DOI] [PubMed]
- Camacho, A. G., Gual, A., Lurz, R., Tavares, P. & Alonso, J. C. (2003). J. Biol. Chem. 278, 23251–23259. [DOI] [PubMed]
- Casjens, S. & Huang, W. M. (1982). J. Mol. Biol. 157, 287–298. [DOI] [PubMed]
- Casjens, S. & Weigele, P. (2005). Viral Genome Packaging Machines: Genetics, Structure and Mechanism, edited by C. Catalano, pp. 80–88. Georgetown: Landes Publishing.
- Chang, J., Weigele, P., King, J., Chiu, W. & Jiang, W. (2006). Structure, 14, 1073–1082. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Goldenberg, D. & King, J. (1982). Proc. Natl Acad. Sci. USA, 79, 3403–3407. [DOI] [PMC free article] [PubMed]
- Jackson, E. N., Laski, F. & Andres, C. (1982). J. Mol. Biol. 154, 551–563. [DOI] [PubMed]
- Lander, G. C., Khayat, R., Li, R., Prevelige, P. E., Potter, C. S., Carragher, B. & Johnson, J. E. (2009). Structure, 17, 789–799. [DOI] [PMC free article] [PubMed]
- Lander, G. C., Tang, L., Casjens, S. R., Gilcrease, E. B., Prevelige, P., Poliakov, A., Potter, C. S., Carragher, B. & Johnson, J. E. (2006). Science, 312, 1791–1795. [DOI] [PubMed]
- Laue, T. M., Shah, B., Ridgeway, T. M. & Pelletier, S. L. (1992). Analytical Ultracentrifugation in Biochemistry and Polymer Science, edited by S. E. Harding, A. J. Rowe & J. Horton, pp. 90–125. Cambridge: Royal Society of Chemistry.
- Lebedev, A. A., Krause, M. H., Isidro, A. L., Vagin, A. A., Orlova, E. V., Turner, J., Dodson, E. J., Tavares, P. & Antson, A. A. (2007). EMBO J. 26, 1984–1994. [DOI] [PMC free article] [PubMed]
- Lin, H., Simon, M. N. & Black, L. W. (1997). J. Biol. Chem. 272, 3495–3501. [DOI] [PubMed]
- Lurz, R., Orlova, E. V., Gunther, D., Dube, P., Droge, A., Weise, F., van Heel, M. & Tavares, P. (2001). J. Mol. Biol. 310, 1027–1037. [DOI] [PubMed]
- Nemecek, D., Gilcrease, E. B., Kang, S., Prevelige, P. E. Jr, Casjens, S. & Thomas, G. J. Jr (2007). J. Mol. Biol. 374, 817–836. [DOI] [PMC free article] [PubMed]
- Nemecek, D., Lander, G. C., Johnson, J. E., Casjens, S. R. & Thomas, G. J. Jr (2008). J. Mol. Biol. 383, 494–501. [DOI] [PMC free article] [PubMed]
- Olia, A. S., Al-Bassam, J., Winn-Stapley, D. A., Joss, L., Casjens, S. R. & Cingolani, G. (2006). J. Mol. Biol. 363, 558–576. [DOI] [PubMed]
- Olia, A. S., Bhardwaj, A., Joss, L., Casjens, S. & Cingolani, G. (2007). Biochemistry, 46, 8776–8784. [DOI] [PubMed]
- Olia, A. S., Casjens, S. & Cingolani, G. (2007). Nature Struct. Mol. Biol. 14, 1221–1226. [DOI] [PubMed]
- Orlova, E. V., Dube, P., Beckmann, E., Zemlin, F., Lurz, R., Trautner, T. A., Tavares, P. & van Heel, M. (1999). Nature Struct. Biol. 6, 842–846. [DOI] [PubMed]
- Orlova, E. V., Gowen, B., Droge, A., Stiege, A., Weise, F., Lurz, R., van Heel, M. & Tavares, P. (2003). EMBO J. 22, 1255–1262. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Poteete, A. R., Jarvik, V. & Botstein, D. (1979). Virology, 95, 550–564. [DOI] [PubMed]
- Poteete, A. R., Sauer, R. T. & Hendrix, R. W. (1983). J. Mol. Biol. 171, 401–418. [DOI] [PubMed]
- Rao, V. B. & Feiss, M. (2008). Annu. Rev. Genet. 42, 647–681. [DOI] [PubMed]
- Rossmann, M. G. & Blow, D. M. (1962). Acta Cryst. 15, 24–31.
- Schuck, P. (2000). Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed]
- Strauss, H. & King, J. (1984). J. Mol. Biol. 172, 523–543. [DOI] [PubMed]
- Teschke, C. M. & Parent, K. N. (2010). Virology, 401, 119–130. [DOI] [PMC free article] [PubMed]
- Thuman-Commike, P. A., Greene, B., Jakana, J., Prasad, B. V., King, J., Prevelige, P. E. Jr & Chiu, W. (1996). J. Mol. Biol. 260, 85–98. [DOI] [PubMed]
- Tong, L. & Rossmann, M. G. (1997). Methods Enzymol. 276, 594–611. [PubMed]
- Trus, B. L., Cheng, N., Newcomb, W. W., Homa, F. L., Brown, J. C. & Steven, A. C. (2004). J. Virol. 78, 12668–12671. [DOI] [PMC free article] [PubMed]
- White, J. H. & Richardson, C. C. (1987). J. Biol. Chem. 262, 8845–8850. [PubMed]
- Zhang, Z., Greene, B., Thuman-Commike, P. A., Jakana, J., Prevelige, P. E. Jr, King, J. & Chiu, W. (2000). J. Mol. Biol. 297, 615–626. [DOI] [PubMed]
- Zhao, H., Finch, C. J., Sequeira, R. D., Johnson, B. A., Johnson, J. E., Casjens, S. R. & Tang, L. (2010). Proc. Natl Acad. Sci. USA, 107, 1971–1976. [DOI] [PMC free article] [PubMed]