

LR11/sorLA contains in its extracellular region a large (∼700-residue) Vps10p domain that is implicated in its intracellular protein-trafficking function. Here, the expression, purification, crystallization and preliminary crystallographic characterization of this domain are described.
Keywords: LR11, Vps10p domain, deglycosylation, CHO lec 3.2.8.1 cells
Abstract
Low-density lipoprotein receptor (LDLR) relative with 11 binding repeats (LR11; also known as sorLA) is genetically associated with late-onset Alzheimer’s disease and is thought to be involved in neurodegenerative processes. LR11 contains a vacuolar protein-sorting 10 protein (Vps10p) domain. As this domain has been implicated in protein–protein interaction in other receptors, its structure and function are of great biological interest. Human LR11 Vps10p domain was expressed in mammalian cells and the purified protein was crystallized using the hanging-drop vapour-diffusion method. Enzymatic deglycosylation of the sample was critical to obtaining diffraction-quality crystals. Deglycosylated LR11 Vps10p-domain crystals belonged to the hexagonal space group P6122. A diffraction data set was collected to 2.4 Å resolution and a clear molecular-replacement solution was obtained.
1. Introduction
LDLR relative with 11 binding repeats (LR11) is a type 1 membrane protein that is expressed abundantly in the central as well as the peripheral nervous system and to a lesser extent in other organs (Hermans-Borgmeyer et al., 1998 ▶; Yamazaki et al., 1996 ▶; Motoi et al., 1999 ▶). LR11 expression is selectively reduced in the brains of Alzheimer’s disease (AD) patients (Scherzer et al., 2004 ▶). Genetic studies have also revealed a strong association of LR11 genetic variants with the risk of AD in several populations (Rogaeva et al., 2007 ▶; Lee et al., 2007 ▶; Bettens et al., 2008 ▶). Although the exact molecular mechanism underlying these phenomena is still unclear, LR11 has been hypothesized to be involved in the intracellular trafficking of amyloid precursor protein (APP) between the trans-Golgi network and early endosomes (Andersen et al., 2005 ▶; Willnow et al., 2008 ▶), reducing the chance of APP processing in the late endosomes. In fact, overexpression of LR11 in HEK293 cells reduced the levels of extracellular amyloid β peptide produced (Offe et al., 2006 ▶). In addition to direct interaction with APP during this trafficking (Andersen et al., 2006 ▶), LR11 may also directly interact with β-secretase, blocking the β-secretase–APP interaction (Spoelgen et al., 2006 ▶).
Uniquely among the LDLR gene family proteins, LR11 possesses an ∼700-amino-acid domain that was initially identified in the vacuolar protein-sorting 10 protein (Vps10p), a sorting protein in yeast that transports carboxypeptidase Y from the Golgi to the vacuole (Marcusson et al., 1994 ▶). Mammalian receptors that contain this domain constitute another family of proteins that are highly expressed in neuronal tissues (Willnow et al., 2008 ▶). Sortilin is the most extensively studied member of this family and is thought to be involved in the intracellular sorting of brain-derived neurotrophic factor (Chen et al., 2005 ▶). The crystal structure of the sortilin Vps10p domain in complex with the neuronal peptide neurotensin has recently been reported (Quistgaard et al., 2009 ▶), revealing that the Vps10p domain is comprised of a unique ten-bladed β-propeller fold followed by two small cysteine-rich domains (10CC-a and 10CC-b domains) that make intimate contacts with the bottom face of the propeller. It was also revealed that the ligand peptide is deeply buried in the tunnel of the ten-bladed β-propeller fold. There is no precedent for β-propeller domains with a blade number greater than eight and the Vps10p domain is probably the largest of the known extracellular modules that fold as a single structural unit (Bork et al., 1996 ▶). Given the potential role of the Vps10p domain in the intracellular trafficking exerted by LR11, it is of great biological as well as medical importance to gain insight into the three-dimensional structure of the LR11 Vps10p domain. To this end, here we report the expression, purification, crystallization and preliminary crystallographic analysis of LR11 Vps10p domain.
2. Methods
2.1. Expression and purification of LR11 Vps10p domain
Human LR11 cDNA (gene accession No. NM_003105) was used to amplify the Vps10p domain, corresponding to the N-terminal 753-residue portion (Ohwaki et al., 2007 ▶). The primer sequences were 5′-CCGGAATTCGCCACCATGGCGACACGGAGCAG-3′ (the EcoRI site is shown in bold) and 5′-CGATCTAGAGGGACAGGGGACCAGCTCTCCTTCC-3′ (the XbaI site is shown in bold). The resultant PCR product was cloned into the EcoRI/XbaI site of pcDNA3.1/Myc-His (Invitrogen) that had been modified to include tag sequences at the C-terminus, SRLENLYFQ^GGHHHHHH HHIEQKLISEEDLNMHTGHHHHHH, containing a tobacco etch virus (TEV) protease recognition sequence (shown in italics with the cleavage site indicated by a caret) and tandem His-tag (bold) and Myc-tag (underlined) sequences. Using this plasmid, CHO lec 3.2.8.1 cells (Stanley, 1989 ▶) were transfected as described previously (Nogi et al., 2006 ▶). The transfected cells were plated in 96-well plates and selected for resistance against 1.5 mg ml−1 G418. Confluent culture supernatants from the single-colony wells were subjected to immunoblotting using anti-Myc antibody (Invitrogen) and the clone with the highest secretion level of LR11 Vps10p domain was chosen for production-scale culture in roller bottles. The recombinant protein was initially fractionated from the culture medium by ammonium sulfate precipitation (50% saturation), dissolved in wash buffer (20 mM Tris pH 8.0, 0.3 M NaCl, 50 mM imidazole) and applied onto an Ni–NTA agarose column (Qiagen). The column was washed with wash buffer and eluted with elution buffer (20 mM Tris pH 8.0, 0.3 M NaCl, 0.25 M imidazole). The eluted material was dialyzed in buffer A (20 mM Tris pH 8.0, 50 mM NaCl) and incubated with hexahistidine-tagged TEV protease at an enzyme:substrate ratio of 1:15(w:w) for 16 h at 277 K to remove the C-terminal tag. For deglycosylation, endoglycosidase H (Endo H; New England Biolabs) was added at a ratio of 5 U enzyme per 1 µg substrate protein and was incubated together with TEV protease under the same conditions as described above. The samples were then reapplied onto the Ni–NTA agarose column to remove the cleaved tags and the protease. The flowthrough fractions were collected and applied onto a Mono Q 5/50 GL column (GE Healthcare). The column was equilibrated with buffer A and eluted with a linear gradient of NaCl (50–300 mM). LR11 Vps10p domain eluted as a single peak at around 150 mM NaCl. The purified protein was concentrated to 6 mg ml−1 using an Ultrafree-0.5 centrifugal filter (30 kDa molecular-weight cutoff; Millipore). Typical yields ranged between 0.5 and 1.3 mg per litre of culture supernatant.
2.2. Crystallization
Initial screening for crystallization conditions was carried out using Index, Crystal Screen, Crystal Screen 2 and SaltRx from Hampton Research and Wizard Screens I and II from Emerald BioSystems. In these screens, a Mosquito crystallization robot (TTP Labtech) was used to dispense a mixture of 0.1 µl protein solution (LR11 Vps10p domain in 20 mM Tris pH 8.0, 150 mM NaCl) and 0.1 µl reservoir solution. Drops were equilibrated against 100 µl reservoir solution using the sitting-drop vapour-diffusion method at 293 K. The initial crystallization condition (solution No. 11 of Crystal Screen; 0.1 M sodium citrate tribasic pH 5.6, 1.0 M ammonium phosphate monobasic) was further optimized using a 24-well crystallization plate with the hanging-drop vapour-diffusion method. Each well contained 350 µl reservoir solution and the drop consisted of a mixture of 0.5 µl protein solution and 0.5 µl reservoir solution.
2.3. Data collection and phasing
For X-ray diffraction experiments, crystals were soaked in reservoir solution containing 20% glycerol or ethylene glycol and flash-cooled in liquid nitrogen. X-ray diffraction data for the crystal grown from the fully glycosylated sample were collected at 100 K on beamline BL44XU at SPring-8 (Harima, Japan) using a wavelength of 0.900 Å and a DIP-6040 imaging-plate detector (Bruker). An X-ray diffraction data set for the crystal grown from the Endo H-treated protein was collected at 95 K on beamline BL-17A at the Photon Factory (Tsukuba, Japan) using a wavelength of 0.980 Å and an ADSC Quantum 270 CCD detector. For the latter, data collection was performed with a total oscillation range of 130° and each diffraction image was obtained with an oscillation angle of 0.5° and an exposure time of 5 s. The diffraction data were processed and scaled using the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶). Data-collection statistics are listed in Table 1 ▶. Phase determination was performed by the molecular-replacement method using the program MOLREP (Vagin & Teplyakov, 2010 ▶). The initial model was refined with REFMAC5 (Murshudov et al., 1997 ▶) and a weighted 2F obs − F calc electron-density map was calculated using the coefficients produced by REFMAC5. Inspection of the crystal packing and the electron-density map was performed with Coot (Emsley & Cowtan, 2004 ▶).
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| No. of crystals | 1 |
| Beamline | Photon Factory BL-17A |
| Wavelength (Å) | 0.980 |
| Detector | ADSC Quantum 270 |
| Crystal-to-detector distance (mm) | 279.6 |
| Rotation range per image (°) | 0.5 |
| Total rotation range (°) | 130 |
| Exposure time per image (s) | 5 |
| Resolution range (Å) | 50–2.4 (2.44–2.40) |
| Space group | P6122 |
| Unit-cell parameters (Å) | a = b = 126.4, c = 290.3 |
| Mosaicity (°) | 0.251 |
| Total No. of measured intensities | 678636 |
| Unique reflections | 54346 (2670) |
| Multiplicity | 12.5 (10.2) |
| Mean I/σ(I) | 15.9 |
| Completeness (%) | 99.6 (100.0) |
| Rmerge† (%) | 5.5 (46.9) |
| Rmeas or Rr.i.m. (%) | 5.8 (43.8) |
| Overall B factor from Wilson plot (Å2) | 58.3 |
R
merge = 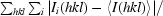
 , where I
i(hkl) is the ith intensity of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith intensity of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
3. Results and discussion
LR11 Vps10p domain contains six potential N-linked glycosylation sites and seven disulfide bonds, demanding production in a mammalian-cell expression system. We used CHO lec 3.2.8.1 cells as the production host because they are known to produce glycoproteins with homogeneous glycoforms, which is ideal for crystallization (Davis et al., 1993 ▶). The relative molecular mass of the purified protein was estimated to be 87 kDa on an SDS–PAGE gel (Fig. 1 ▶). The difference from the calculated molecular weight (76 kDa) is consistent with the presence of glycosylation. Initial crystallization trials were performed using fully glycosylated sample. Crystals were obtained under the condition 0.1 M sodium acetate pH 4.5, 1.2 M sodium dihydrogen phosphate at 293 K and grew to dimensions of approximately 30 × 30 × 100 µm within two months (Fig. 2 ▶ a). Processing of the X-ray diffraction images indicated that the unit-cell parameters of the crystal were a = b = 125.8, c = 292.7 Å (Fig. 2 ▶ a). However, the resolution limit of X-ray diffraction was very low (below 6 Å) and we could not determine the atomic resolution structure using these crystals. We reasoned that the presence of highly flexible glycan chains in the crystal prevented ordered tight packing of the LR11 Vps10p domain. Therefore, we trimmed the glycan chains from the purified protein using Endo H. Endo H cleaves between the two GlcNAc residues at the base of N-glycans, leaving a single GlcNAc residue at each glycosylation sequon that maintains overall protein solubility. Although Endo H exhibits varying degrees of cleavage efficiency towards different glycan chains, it is able to efficiently cleave the Man5GlcNAc2 chain, which is the predominant glycoform present on proteins expressed in CHO lec 3.2.8.1 cells (Davis et al., 1993 ▶). Endo H treatment of LR11 Vps10p domain resulted in a reduction of the relative molecular mass by ∼6 kDa, which is consistent with the complete removal of six glycan chains (Fig. 1 ▶). The deglycosylated sample could be crystallized under the same conditions and with the same incubation period (∼2 months) as the untreated sample (Fig. 2 ▶ b). Remarkably, the quality of the crystals was dramatically improved and the crystals diffracted to 2.4 Å resolution (Fig. 2 ▶ b), while the unit-cell parameters (a = b = 126.4, c = 290.3 Å) remained essentially unchanged from the original crystal of fully glycosylated sample (Table 1 ▶).
Figure 1.

SDS–PAGE analysis of the purified LR11 Vps10p domain before and after Endo H treatment. 2 µg of the purified protein was either treated with 10 U Endo H (+) or left untreated (−) and subjected to SDS–PAGE using 8% gel under reducing conditions followed by staining with Coomassie Brilliant Blue. Lane M contains molecular-weight markers (labelled in kDa).
Figure 2.

Crystals and diffraction patterns obtained using samples before (a) and after (b) Endo H treatment. 6.0 Å (a) and 2.4 Å (b) resolution circles are shown. Expanded views of the regions marked with a rectangle are shown in the insets.
The symmetry and systematic absences of the diffraction intensities indicated that the new crystal of deglycosylated LR11 Vps10p domain belonged to either space group P6122 or its enantiomer P6522. In an attempt to determine the initial phases, the molecular-replacement method was performed using the atomic coordinates of the sortilin Vps10p domain (PDB code 3f6k; Quistgaard et al., 2009 ▶) as a search model. Specifically, the β-propeller module was extracted from the coordinates. In addition, residues and atoms that do not map onto the sequence of the LR11 Vps10p domain were deleted from the model with the ‘FILE_SEQUENCE’ option of MOLREP. While the sortilin Vps10p domain shows only 25% sequence identity to the LR11 Vps10p domain, it gave a clear solution in P6122 for which the peak-to-noise ratios of the rotation and translation functions were 5.46 and 24.85, respectively. One monomer was located in the asymmetric unit, showing an R factor of 62.8%. The resulting model was subjected to initial rigid-body and subsequent restrained refinement, which resulted in a reduction of the R and R free factors to 55.5% and 55.9%, respectively. Preliminary model building revealed that all of the potential N-linked glycosylation sites are located on the surface of the protein and we could identify clear electron density that is likely to represent the remaining GlcNAc moiety at five of the six sites. Furthermore, three of these sites are very close to the neighbouring molecules. Although the three N-glycans are not directly involved in crystal packing, it is possible that deglycosylation with Endo H stabilized the packing by lowering the disorder arising from the flexibility of the carbohydrates. In fact, the Matthews coefficient (Matthews, 1968 ▶) and solvent content (4.4 Å3 Da−1 and 72.2%, respectively) suggested a loose crystal packing (Kantardjieff & Rupp, 2003 ▶), indicating the presence of a large space that can accommodate flexible N-glycans. Further model building and structure refinement are now in progress and the mechanism by which the deglycosylation improved the crystal quality may be clarified after completion of the model assignment, including the carbohydrates.
Acknowledgments
We would like to thank Drs L. M. G. Chavas, Y. Yamada, N. Matsugaki and N. Igarashi of the Photon Factory and E. Yamashita, M. Suzuki and A. Nakagawa of SPring-8 BL44-XU for providing data-collection facilities and for support. We also thank K. Tamura-Kawakami, E. Mihara and M. Nampo for their excellent technical support and M. Nakano for preparation of the manuscript. This work was partly supported by a Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), by a Grant-in-Aid for Scientific Research on Priority Areas from MEXT and by a Protein 3000 Project grant from MEXT.
References
- Andersen, O. M. et al. (2005). Proc. Natl Acad. Sci. USA, 102, 13461–13466.
- Andersen, O. M., Schmidt, V., Spoelgen, R., Gliemann, J., Behlke, J., Galatis, D., McKinstry, W. J., Parker, M. W., Masters, C. L., Hyman, B. T., Cappai, R. & Willnow, T. E. (2006). Biochemistry, 45, 2618–2628. [DOI] [PubMed]
- Bettens, K., Brouwers, N., Engelborghs, S., De Deyn, P. P., Van Broeckhoven, C. & Sleegers, K. (2008). Hum. Mutat. 29, 769–770. [DOI] [PubMed]
- Bork, P., Downing, A. K., Kieffer, B. & Campbell, I. D. (1996). Q. Rev. Biophys. 29, 119–167. [DOI] [PubMed]
- Chen, Z.-Y., Ieraci, A., Teng, H., Dall, H., Meng, C.-X., Herrera, D. G., Nykjaer, A., Hempstead, B. L. & Lee, F. S. (2005). J. Neurosci. 25, 6156–6166. [DOI] [PMC free article] [PubMed]
- Davis, S. J., Puklavec, M. J., Ashford, D. A., Harlos, K., Jones, E. Y., Stuart, D. I. & Williams, A. F. (1993). Protein Eng. 6, 229–232. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hermans-Borgmeyer, I., Hampe, W., Schinke, B., Methner, A., Nykjaer, A., Süsens, U., Fenger, U., Herbarth, B. & Schaller, H. C. (1998). Mech. Dev. 70, 65–76. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Lee, J. H., Cheng, R., Schupf, N., Manly, J., Lantigua, R., Stern, Y., Rogaeva, E., Wakutani, Y., Farrer, L., St George-Hyslop, P. & Mayeux, R. (2007). Arch. Neurol. 64, 501–506. [DOI] [PMC free article] [PubMed]
- Marcusson, E. G., Horazdovsky, B. F., Cereghino, J. L., Gharakhanian, E. & Emr, S. D. (1994). Cell, 77, 579–586. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Motoi, Y., Aizawa, T., Haga, S., Nakamura, S., Namba, Y. & Ikeda, K. (1999). Brain Res. 833, 209–215. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nogi, T., Yasui, N., Hattori, M., Iwasaki, K. & Takagi, J. (2006). EMBO J. 25, 3675–3683. [DOI] [PMC free article] [PubMed]
- Offe, K., Dodson, S. E., Shoemaker, J. T., Fritz, J. J., Gearing, M., Levey, A. I. & Lah, J. J. (2006). J. Neurosci. 26, 1596–1603. [DOI] [PMC free article] [PubMed]
- Ohwaki, K., Bujo, H., Jiang, M., Yamazaki, H., Schneider, W. J. & Saito, Y. (2007). Arterioscler. Thromb. Vasc. Biol. 27, 1050–1056. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Quistgaard, E. M., Madsen, P., Groftehauge, M. K., Nissen, P., Petersen, C. M. & Thirup, S. S. (2009). Nature Struct. Mol. Biol. 16, 96–98. [DOI] [PubMed]
- Rogaeva, E. et al. (2007). Nature Genet. 39, 168–177. [DOI] [PMC free article] [PubMed]
- Scherzer, C. R., Offe, K., Gearing, M., Rees, H. D., Fang, G., Heilman, C. J., Schaller, C., Bujo, H., Levey, A. I. & Lah, J. J. (2004). Arch. Neurol. 61, 1200–1205. [DOI] [PubMed]
- Spoelgen, R., von Arnim, C. A., Thomas, A. V., Peltan, I. D., Koker, M., Deng, A., Irizarry, M. C., Andersen, O. M., Willnow, T. E. & Hyman, B. T. (2006). J. Neurosci. 26, 418–428. [DOI] [PMC free article] [PubMed]
- Stanley, P. (1989). Mol. Cell. Biol. 9, 377–383. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Willnow, T. E., Petersen, C. M. & Nykjaer, A. (2008). Nature Rev. Neurosci. 9, 899–909. [DOI] [PubMed]
- Yamazaki, H., Bujo, H., Kusunoki, J., Seimiya, K., Kanaki, T., Morisaki, N., Schneider, W. J. & Saito, Y. (1996). J. Biol. Chem. 271, 24761–24768. [DOI] [PubMed]