

A variant of the diaminopimelate/lysine pathway has recently been defined following the discovery of the enzyme l,l-diaminopimelate aminotransferase (DapL). The cloning of the cDNA, recombinant expression, purification and preliminary diffraction analysis of DapL from the alga C. reinhardtii are presented.
Keywords: algaecides; Chlamydomonas reinhardtii; l,l-diaminopimelate aminotransferase; lysine biosynthesis
Abstract
In the anabolic synthesis of diaminopimelate and lysine in plants and in some bacteria, the enzyme l,l-diaminopimelate aminotransferase (DapL; EC 2.6.1.83) catalyzes the conversion of tetrahydrodipicolinic acid (THDPA) to l,l-diaminopimelate, bypassing the DapD, DapC and DapE enzymatic steps in the bacterial acyl pathways. Here, the cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of DapL from the alga Chlamydomonas reinhardtii are presented. Protein crystals were grown in conditions containing 25%(w/v) PEG 3350 and 200 mM lithium sulfate and initially diffracted to ∼1.35 Å resolution. They belonged to space group P212121, with unit-cell parameters a = 58.9, b = 91.8, c = 162.9 Å. The data were processed to 1.55 Å resolution with an R merge of 0.081, an R p.i.m. of 0.044, an R r.i.m of 0.093 and a V M of 2.28 Å3 Da−1.
1. Introduction
The essential amino acid lysine (Lys) is synthesized via two routes. One pathway uses the intermediate α-aminoadipic acid (AAA) and occurs in yeast, fungi and several archaeal species (Nishida et al., 1999 ▶; Velasco et al., 2002 ▶). The other pathway utilizes the intermediate meso-diaminopimelate (m-DAP). This pathway is present in most bacterial species and photosynthetic cohorts. We recently discovered a variant of the DAP pathway by the identification and characterization of the enzyme l,l-diaminopimelate aminotransferase (DapL; EC 2.6.1.83) from the organism Arabidopsis thaliana (Hudson et al., 2006 ▶) and subsequently from a number of bacterial species, including some that are deemed to be pathogenic (McCoy et al., 2006 ▶; Hudson et al., 2008 ▶; Liu et al., 2010 ▶).
There are four variants of the DAP/Lys pathway and all share the same enzymatic steps for the synthesis of tetrahydrodipicolinic acid (THDPA) from aspartate (Hudson et al., 2006 ▶). What makes these anabolic pathways unique is the synthesis of m-DAP from tetrahydrodipicolinic acid (THDPA). Most bacteria employ the use of acetylated or succinylated (acyl) intermediates to convert THDPA to l,l-DAP using the enzymes DapD, DapC and DapE (Fig. 1 ▶). Another variant of the pathways utilizes the enzyme m-DAP dehydrogenase (Ddh), which converts THDPA to m-DAP in a single step, bypassing the DapD, DapC, DapE and DapF enzymatic reactions that are present in the acyl pathways (Fig. 1 ▶). The recently discovered DapL pathway synthesizes l,l-DAP from THDPA using the amino acid glutamate as the amino donor and THDPA as the amino acceptor in one step, bypassing the DapD, DapC and DapE steps that are present in the bacterial acyl pathways (Fig. 1 ▶; Hudson et al., 2006 ▶). The conversion of m-DAP to lysine, catalyzed by the enzyme m-DAP decarboxylase (LysA), is the ultimate step in the pathway. This enzymatic reaction is common to all four variants. Given that animals do not possess the machinery necessary for the de novo biosynthesis of lysine, the enzymes associated with this pathway are attractive targets for the development of antibiotics, herbicides and algaecides. Accordingly, we have been engaged in study of the structure and function of enzymes of lysine biosynthesis from a variety of pathogens (Dobson et al., 2004 ▶, 2005 ▶; Boughton et al., 2008 ▶; Burgess, Dobson, Bailey et al., 2008 ▶; Griffin et al., 2008 ▶; Domigan et al., 2009 ▶).
Figure 1.

Lysine anabolic pathways as denoted by the acyl pathways (a), the dehydrogenase pathway (b) and the l,l-diaminopimelate aminotransferase pathway (c). The abbreviations of the enzymes are as follows: tetrahydrodipicolinate acylase, DapD; acyl-amino-ketopimelate aminotransferase, DapC; acyl-ketopimelate deacylase, DapF; diaminopimelate epimerase, DapF; diaminopimelate decarboxylase, LysA; m-diaminopimelate dehydrogenase, Ddh; l,l-diaminopimelate aminotransferase, DapL.
Recently, the macromolecular structure of DapL from Arabidopsis was solved via X-ray crystallography (Watanabe et al., 2007 ▶, 2008 ▶). These structures underpinned the rational design of a range of inhibitors of the DapL enzyme, which have been developed to bind the active site (Fan et al., 2010 ▶).
We are interested in studying the macromolecular structure of DapL from the alga Chlamydomonas reinhardtii, which is a critical step in our effort to rationally design novel algaecides. Here, we present the cloning, recombinant expression, purification, crystallization and preliminary crystallographic studies of DapL from C. reinhardtii.
2. Materials and methods
2.1. Chlamydomonas growth, total RNA isolation and cDNA synthesis
C. reinhardtii strain CC-1690 was obtained from the Chlamydomonas Culture Collection (http://www.chlamy.org/) and was grown in Tris–acetate–phosphate (TAP) medium. The strain was grown with a 16 h light and 8 h dark period for 7 d. The temperature was 297 K during the light period and 293 K during the dark period. The light intensity was approximately 120 µE m−2 s−1.
Total RNA was isolated from C. reinhardtii using the RNeasy Plant Mini Kit (Qiagen, Valencia, California, USA) using the manufacturer’s protocol. cDNA was synthesized in a reaction containing 1 µl oligo(dT)12–18 primer, 5 µg total RNA, 1 µl 10 mM dNTP mix and DEPC-treated water up to 13 µl. The mixture was incubated at 338 K for 5 min, followed by incubation on ice for 5 min. The Reverse Transcription System Kit (Promega, Madison, Wisconsin, USA) was used to synthesize cDNA following the manufacturer’s protocol.
2.2. Amplification and cloning of the C. reinhardtii dapL cDNA
The full-length cDNA annotated by the locus tag CHLREDRAFT_129557 was amplified by PCR. The cDNA was amplified using 12 pmol forward and reverse primers, 1 mM MgSO4, 0.5 mM of each of the four deoxynucleotide triphosphates, 2 µl cDNA product and 1 U Platinum Pfx DNA polymerase (Invitrogen Corporation, Carlsbad, California, USA) using the following PCR conditions: one cycle at 367 K for 2 min followed by 30 cycles of 367 K for 15 s, 333 K for 30 s and 345 K for 2 min. The forward and reverse primers used to amplify the gene were 5′-CCCCCGAATTC ATGCAGCTCAACGTGCGGTCCACCGCCAGC-3′ and 5′-CCCCCAAGCTT CTAGTTACGCTTGCCGTAGGCCTCCTTAAA-3′. The bold sequences represent the restriction-enzyme sites used in the cloning of the cDNA, while the italicized sequences represent initiation and termination codons. For cloning, the PCR fragment was digested with EcoRI and HindIII and ligated into the plasmid pET30a (EMD Biosciences, Gibbstown, New Jersey, USA) to produce the plasmid pET30a-Cr-DapL. The recombinant protein derived from this plasmid carries a hexahistidine and S-TAG epitope derived from the pET30a plasmid at the amino terminus. To confirm the fidelity of the PCR reaction, the PCR fragment was sequenced from the pET30a construct using the T7 promoter primer 5′-TAATACGACTCACTATAGGG-3′ and the T7 terminator primer 5′-TATGCTAGTTATTGCTCAG-3′ and was compared with the published sequence deposited in the public database (http://www.chlamy.org/cgi-bin/webblast.pl).
2.3. Expression and purification
The plasmid pET30a-Cr-DapL was transformed into Escherichia coli BL21-CodonPlus-RIPL strain (Agilent Technologies, La Jolla, California, USA). For protein expression and purification, the strain was grown in LB broth containing 50 µg ml−1 kanamycin and 34 µg ml−1 chloramphenicol at 310 K to an OD600 of 0.5. Protein expression was induced with 0.5 mM IPTG for 4 h at 298 K. The cells were lysed by sonication in a solution consisting of 50 mM sodium phosphate pH 8.0 and 300 mM NaCl. The soluble extract was incubated with 1.5 ml Talon Metal Affinity Resin (Clontech, Mountain View, California, USA) for 30 min at 277 K. The resin was washed three times with sonication buffer containing 10 mM imidazole pH 8.0 and the enzyme was eluted with sonication buffer containing 250 mM imidazole. The pure protein was concentrated in an Amicon Ultra 10 kDa molecular-weight cutoff spin-filter unit, replacing the elution buffer with 100 mM HEPES containing 1 mM DTT and 2 mM EDTA pH 7.6 for storage. Prior to crystallization, the purified protein was subjected to size-exclusion chromatography on an S200 column pre-equilibrated with 20 mM Tris–HCl, 5 mM DTT, 2 mM EDTA pH 7.8 to remove any precipitated protein. The protein was then concentrated with an Amicon Ultra 10 kDa molecular-weight cutoff spin-filter unit.
2.4. Crystallization
Crystallization screens were conducted as described previously (Atkinson et al., 2009 ▶; Burgess, Dobson, Dogovski et al., 2008 ▶; Dobson et al., 2008 ▶). Protein crystallization experiments were performed at the Bio21 Collaborative Crystallization Centre (C3; CSIRO, Parkville, Melbourne, Australia) using the PACT Suite and JCSG+ Suite crystal screens at 281 and 293 K (Newman et al., 2005 ▶, 2008 ▶). The screens were set up using the sitting-drop vapour-diffusion method. The crystals shown in Fig. 3(a) were obtained at 293 K from a 300 nl drop formed from 150 nl protein solution (8.6 mg ml−1 in 20 mM Tris–HCl, 5 mM DTT, 2 mM EDTA pH 7.8) and 150 nl reservoir solution [JCSG+ condition H9; 200 mM lithium sulfate, 25%(w/v) polyethylene glycol 3350, 100 mM bis-tris propane pH 5.5 including 0.02%(w/v) sodium azide].
2.5. Data collection and processing
For X-ray data collection, the crystal was briefly soaked in cryoprotectant solution consisting of reservoir solution made up to 20%(v/v) glycerol and flash-cooled in liquid nitrogen. Intensity data were collected at 110 K at the Australian Synchrotron (MX2 beamline). Data were collected in 0.5° rotations over 110° using an ADSC Q315r CCD positioned 120 mm from the crystal with an exposure time of 2.5 s and 80% attenuation. The diffraction data were processed using the programs MOSFLM (Leslie, 1992 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
C. reinhardtii DapL was successfully cloned, expressed and purified to homogeneity by a two-step purification protocol involving binding to Talon Metal Affinity Resin. The purity of the enzyme was assessed by SDS–PAGE (Fig. 2 ▶) and the enzyme activity was measured using the DapL quantitative forward and reverse assays (Hudson et al., 2006 ▶). The specific activity of the enzyme in the forward direction (THDPA to l,l-DAP) was 0.7 µmol min−1 mg−1 protein and that in the reverse direction (l,l-DAP to THDPA) was 12.0 µmol min−1 mg−1 protein.
Figure 2.

SDS–PAGE analysis of purified recombinant DapL. Lane 1, protein markers (kDa). Lane 2, 10 µg uninduced soluble protein extract. Lane 3, 10 µg induced soluble extract. Lane 4, 1.0 µg purified recombinant DapL protein. The proteins were resolved on an SDS–PAGE gel containing 10%(w/v) acrylamide and the gel was stained using Coomassie Blue.
Purified DapL protein was screened for crystallization using the JCSG+ and PACT screens, but only afforded crystals in the following condition: 200 mM lithium sulfate, 25%(w/v) polyethylene glycol 3350, 100 mM bis-tris propane pH 5.5 (JCSG+ condition H9). The crystals in Fig. 3 ▶(a) appeared after 5 d and continued to grow over a further 5 d. The larger crystals appeared to develop imperfections in crystal morphology.
Figure 3.

Crystals of DapL grown in JCSG+ condition H9. The scale bar indicates 0.2 mm.
The crystal showed excellent diffraction (to ∼1.35 Å resolution) and belonged to space group P212121, with unit-cell parameters a = 58.9, b = 91.8, c = 162.9 Å. However, the very intense beam on MX2 resulted in a signifant loss of resolution, even after 20° of data had been collected, presumably owing to radiation damage. Nonetheless, the data were scaled to 1.55 Å resolution with reasonable completeness (data-collection statistics are summarized in Table 1 ▶). The axial reflections showed systematic absences that were consistent with three twofold screw axes. The R merge was 0.081, R p.i.m. was 0.044, R r.i.m was 0.093, V M was 2.28 Å3 Da−1 and the estimated solvent content was 48% given two molecules in the asymmetric unit. The crystals shown in Fig. 3 ▶(a) could also be produced under the same conditions but at 281 K, although they grew noticeably more slowly. Diffraction data were also collected from these crystals, but the resolution was reduced: the best resolution was ∼1.9 Å. We anticipate that the structure of DapL from C. reinhardtii will provide insight into the design of novel algaecides.
Table 1. X-ray data-collection statistics.
Values in parentheses are for the highest resolution shell. The Matthews coefficient and the estimate of the solvent content are based on a molecular weight of 97 661 Da in the asymmetric unit (two molecules of DapL of 48 830 Da each).
| Wavelength (Å) | 0.95369 |
| No. of images | 220 |
| Oscillation (°) | 0.5 |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 58.9, b = 91.8, c = 162.9 |
| Resolution (Å) | 35.35–1.55 (1.63–1.55) |
| Average mosaicity (°) | 0.27 |
| Observed reflections | 492985 |
| Unique reflections | 124970 |
| Completeness (%) | 97.3 (94.3) |
| Rmerge† | 0.081 (0.494) |
| Rp.i.m.‡ | 0.044 (0.283) |
| Rr.i.m. | 0.093 (0.573) |
| Mean I/σ(I) | 10.6 (2.7) |
| Multiplicity | 3.9 (3.9) |
| Wilson B value (Å2) | 13.6 |
| Estimated molecules per asymmetric unit | 2 |
| VM (Å3 Da−1) | 2.28 |
| Solvent content (%) | 48 |
R
merge = 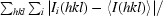
 .
.
R
p.i.m. = 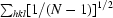
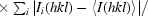 .
.
Acknowledgments
We wish to thank the School of Biological and Medical Sciences at RIT for the support of this work through a Faculty Evaluation and Development (FEAD) 2010 award to AOH. IG is currently enrolled in the Biotechnology program at RIT. The DAP isomers used for enzymatic assays were a gift provided by Dr John Vederas of the University of Alberta, Canada. We would also like to acknowledge the support and assistance of the friendly staff, especially Dr Janet Newman, at the Bio21 Collaborative Crystallographic Centre at CSIRO Molecular and Health Technologies, Parkville, Melbourne. This research was undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia. The views expressed herein are those of the authors and are not necessarily those of the owner or operator of the Australian Synchrotron. RCJD acknowledges the C. R. Roper Bequest for Fellowship support. The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Atkinson, S. C., Dobson, R. C. J., Newman, J. M., Gorman, M. A., Dogovski, C., Parker, M. W. & Perugini, M. A. (2009). Acta Cryst. F65, 253–255. [DOI] [PMC free article] [PubMed]
- Boughton, B. A., Dobson, R. C. J., Gerrard, J. A. & Hutton, C. A. (2008). Bioorg. Med. Chem. Lett. 18, 460–463. [DOI] [PubMed]
- Burgess, B. R., Dobson, R. C. J., Bailey, M. F., Atkinson, S. C., Griffin, M. D. W., Jameson, G. B., Parker, M. W., Gerrard, J. A. & Perugini, M. A. (2008). J. Biol. Chem. 283, 27598–27603. [DOI] [PubMed]
- Burgess, B. R., Dobson, R. C. J., Dogovski, C., Jameson, G. B., Parker, M. W. & Perugini, M. A. (2008). Acta Cryst. F64, 659–661. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dobson, R. C. J., Atkinson, S. C., Gorman, M. A., Newman, J. M., Parker, M. W. & Perugini, M. A. (2008). Acta Cryst. F64, 206–208. [DOI] [PMC free article] [PubMed]
- Dobson, R. C. J., Griffin, M. D. W., Jameson, G. B. & Gerrard, J. A. (2005). Acta Cryst. D61, 1116–1124. [DOI] [PubMed]
- Dobson, R. C. J., Valegård, K. & Gerrard, J. A. (2004). J. Mol. Biol. 338, 329–339. [DOI] [PubMed]
- Domigan, L. J., Scally, S. W., Fogg, M. J., Hutton, C. A., Perugini, M. A., Dobson, R. C., Muscroft-Taylor, A. C., Gerrard, J. A. & Devenish, S. R. (2009). Biochim. Biophys. Acta, 1794, 1510–1516. [DOI] [PubMed]
- Fan, C., Clay, M. D., Dehyholos, M. K. & Vederas, J. C. (2010). Bioorg. Med. Chem. 18, 2141–2151. [DOI] [PubMed]
- Griffin, M. D., Dobson, R. C. J., Pearce, F. G., Antonio, L., Whitten, A. E., Liew, C. K., Mackay, J. P., Trewhella, J., Jameson, G. B., Perugini, M. A. & Gerrard, J. A. (2008). J. Mol. Biol. 380, 691–703. [DOI] [PubMed]
- Hudson, A. O., Gilvarg, C. & Leustek, T. (2008). J. Bacteriol. 109, 3256–3263. [DOI] [PMC free article] [PubMed]
- Hudson, A. O., Singh, B. K., Leustek, T. & Gilvarg, C. (2006). Plant Physiol. 140, 292–301. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 26
- Liu, Y., White, R. H. & Whitman, W. B. (2010). J. Biol. Chem. 192, 3304–3310.
- McCoy, A. J., Adams, N. E., Hudson, A. O., Gilvarg, C., Leustek, T. & Maurelli, A. T. (2006). Proc. Natl Acad. Sci. USA, 103, 17909–17914. [DOI] [PMC free article] [PubMed]
- Newman, J., Egan, D., Walter, T. S., Meged, R., Berry, I., Ben Jelloul, M., Sussman, J. L., Stuart, D. I. & Perrakis, A. (2005). Acta Cryst. D61, 1426–1431. [DOI] [PubMed]
- Newman, J., Pham, T. M. & Peat, T. S. (2008). Acta Cryst. F64, 991–996. [DOI] [PMC free article] [PubMed]
- Nishida, H., Nishiyama, M., Kobashi, N., Kosuge, T., Hoshino, T. & Yamane, H. (1999). Genome Res. 9, 1175–1183. [DOI] [PubMed]
- Velasco, A. M., Leguina, J. I. & Lazcano, A. (2002). J. Mol. Evol. 55, 449–455. [DOI] [PubMed]
- Watanabe, N., Cherney, M. M., van Belkum, M. J., Marcus, S. L., Flegel, M. D., Clay, M. D., Deyholos, M. K., Vederas, J. C. & James, M. N. (2007). J. Mol. Biol. 317, 685–702. [DOI] [PubMed]
- Watanabe, N., Clay, M. D., van Belkum, M. J., Cherney, M. M., Vederas, J. C. & James, M. N. G. (2008). J. Mol. Biol. 134, 1314–1329. [DOI] [PubMed]