

Abstract
The ability to develop counter-regulatory mechanisms to maintain energy balance in response to environmental and physiologic insults is essential for survival, but the mechanisms underlying these compensatory regulations are poorly understood. Agouti-related peptide (AGRP) and Neuropeptide Y are potent orexigens and are coexpressed in neurons in the arcuate nucleus of the hypothalamus. Acute ablation of these neurons leads to severe anorexia and weight loss, whereas progressive degeneration of these neurons has minimal impact on food intake and body weight, suggesting that compensatory mechanisms are developed to maintain orexigenic drive. In this study, we show that cell proliferation is increased in the hypothalamus of adult mutant animals in which AgRP neurons undergo progressive neurodegeneration due to deletion of mitochondrial transcription factor A, and that a subset of these newly generated cells differentiate into AgRP neurons along with other resident neuronal subtypes. Furthermore, some of the newly generated cells are capable of responding to leptin, and a central blockade of cell proliferation in adult animals results in decreases in food intake and body adiposity in mutant but not in control animals. Our study indicates that neurons important for energy homeostasis can be regenerated in adult feeding centers under neurodegenerative conditions. It further suggests that de novo neurogenesis might serve as a compensatory mechanism contributing to the plastic control of energy balance in response to environmental and physiologic insults.
Introduction
The ability to maintain energy balance in an ever-changing environment or in response to physiologic insults is essential for survival. Agouti-related peptide (AGRP) and Neuropeptide Y (NPY) are potent orexigens agents and are coexpressed in neurons within the arcuate nucleus (ARC) of the hypothalamus. Located adjacent to the median eminence, a circumventricular organ, AgRP and Npy neurons are in constant communication with metabolic signals such as leptin, insulin, ghrelin, and glucocorticoids. AgRP neurons project to the proopiomelanocortin (Pomc) neurons in the ARC, and the release of NPY and GABA from AgRP neuronal terminals potently inhibits Pomc neuronal activities. AgRP neurons also project to neurons expressing melanocortin-4 receptor (MC4R), where AGRP acts as an antagonist and an inverse agonist (for review, see Morton et al., 2006; Williams et al., 2009).
It is known that central administration of AGRP or NPY causes hyperphagia and that overexpression of AgRP in transgenic mice causes severe obesity (Ollmann et al., 1997). However, targeted deletion of the AgRP gene, or AgRP and Npy combined, has minimal impact on feeding (Qian et al., 2002; Wortley et al., 2005). Previously, we generated mice in which the mitochondrial transcription factor A (Tfam) gene was specifically deleted from the AgRP neurons (designated AgRP-Tfam mutant mice). As a result, the AgRP neurons undergo progressive neurodegeneration due to depletion of mitochondria over a period of several months (Sorensen et al., 2001; Xu et al., 2005b). Despite the loss of ∼85% of AgRP neurons, mutant animals exhibit no difference in food intake and body weight (Xu et al., 2005b). In stark contrast, mice exhibit severe anorexia and weight loss within days when AgRP neurons are acutely ablated using diphtheria toxin-mediated strategies (Gropp et al., 2005; Luquet et al., 2005), despite the use of the same Tg.AgRP-Cre line in both the acute and progressive ablation studies (Gropp et al., 2005; Xu et al., 2005b). These results demonstrate that the AgRP neurons are essential for feeding and energy homeostasis. They further suggest that compensatory mechanisms may have developed during the progressive degeneration of the AgRP neurons, allowing the animals to escape the life-threatening effects of severe anorexia.
Although compensatory regulation of energy balance is frequently observed, the underlying mechanisms are largely unexplored. In this study, we show that cell proliferation increases in the AgRP-Tfam mutant hypothalamus, and a subset of newly generated cells take on AgRP neuronal identity. In addition, newly proliferated cells are capable of responding to leptin by activating signal transducer and activator of transcription 3 (Stat3) signaling. Finally, we show that inhibiting cell proliferation in the adult brain significantly decreases food intake and body adiposity in the mutants but not in the controls, suggesting that de novo cell proliferation is functionally required for maintenance of energy balance in the mutant mice. Our study indicates that neurons important for energy balance regulation can be regenerated in adult feeding centers, suggesting that de novo neurogenesis is a novel mechanism contributing to the plastic control of energy balance.
Materials and Methods
Animals.
Mice used this study were generated by using two different breeding strategies, and male mice were used all experiments. For the LacZ colocalization experiment, Tg.AgRP-Cre transgenic mice that were heterozygous for the floxed Tfam allele (Xu et al., 2005b) and homozygous for the R26R-LacZ Cre reporter (B6.129S4-Gt(ROSA)26Sortm1Sor/J, The Jackson Laboratory) were bred with mice that were heterozygous for the floxed Tfam allele. Resultant offspring produced control mice that were wild type or heterozygous for floxed Tfam, heterozygous for Tg.AgRP-Cre, and heterozygous for the R26R-LacZ gene. Resultant mutant offspring were homozygous for floxed Tfam, heterozygous for Tg.AgRP-Cre, and heterozygous for the R26R-LacZ gene.
For other experiments, female mice that were homozygous for the floxed Tfam allele were bred with male mice that were homozygous for the floxed Tfam allele and heterozygous for the AgRP-Cre transgene. The conditional allele for Tfam has been characterized in multiple studies with different Cre lines (Silva et al., 2000; Sorensen et al., 2001; Wang et al., 2001; Hansson et al., 2004), and the specificity of the Tg.AgRP-Cre transgenic mice has been characterized and validated by multiple studies (Gropp et al., 2005; Xu et al., 2005a,b; Kitamura et al., 2006; Konner et al., 2007; van de Wall et al., 2008; Zhang et al., 2008). The effect of the loss of Tfam in neurons, including AgRP-expressing neurons, has been previous described in detail (Sorensen et al., 2001; Xu et al., 2005b).
All mice were housed in the mouse barrier facility of the University of California, San Francisco in a room with a 7 A.M.–7 P.M. light/dark cycle. Mice were fed standard mouse chow and given access to water ad libitum. All experiments were carried out under a protocol approved by the University of California, San Francisco Institutional Animal Care and Use Committee.
Intracerebroventricular infusion.
In a single surgical procedure, 12-week-old mice were anesthetized with ketamine and xylazine (45 and 5 mg/kg, respectively) supplemented with isofluorane inhalation. Mice were mounted on a stereotaxic apparatus (model 1900; David Kopf Instruments) and implanted with an Alzet steel guide cannula (Durect) into the right lateral cerebroventricle (anteroposterior, −0.3 mm to bregma; lateral, +1.0 mm to bregma; and dorsoventral, −2.5 mm below skull). The cannula was connected to an osmotic minipump (flow rate of 0.15 μl/h for 42 d; Alzet model 2006; Durect) via a 50-mm-long vinyl tubing (inner diameter 1.22 mm, Durect). Each minipump was filled with either vehicle solution alone or vehicle solution with Ara-C (10.9 μg/μl; Sigma). The vehicle solution was artificial CSF containing 3.2 μg/μl Ig-free bovine serum albumin (BSA; Jackson ImmunoResearch Laboratories) and 3.2 μg/μl 5-bromodeoxyuridine (BrdU; Sigma). Pumps connected to the intracerebroventricular cannulae were primed overnight at 37°C in 0.9% saline. Mice were housed singly and monitored for body weight, body composition, and food intake. At the end of the study, 10 μl of trypan blue dye was injected through the tube attached to the cannula to validate ventricular placement of the cannulae, and residual volumes in the pumps were checked to ensure that proper infusion took place. In addition, correct placement of the cannulae was verified by histological examination.
Body weight, food intake, and body composition analysis.
Animals were weighed regularly before intracranial surgery. Following surgery, all mice were weighted again and underwent dual energy x-ray absorptiometry (DEXA; Lunar PixiMus II; GE Healthcare) scans to determine body composition. Body weight and food intake were determined daily for 1 week following surgery and weekly thereafter. Body composition was determined every 2 weeks following surgery. Food intake was determined by measuring the amount initially in the food hopper and food remaining in the food hopper at the end of week. Cages were monitored for evidence of grinding and food spillage, and any mice that showed excess food grinding were removed from the study.
Immunohistochemical analysis.
Ad libitum fed mutant and control mice were injected with either PBS or leptin (5 mg/kg, i.p.). Forty-five minutes later, mice were anesthetized and perfused transcardially with normal PBS followed by 4% paraformaldehyde in normal buffered PBS (Electron Microscopy Supply). Brains were postfixed in perfusion solution at 4°C overnight, infiltrated with 30% sucrose in PBS at 4°C overnight, and sectioned at 10 μm in the coronal plane using a cryostat.
For immunohistochemical and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) staining, the procedures used were similar to those previously described (Xu et al., 2005a,b; Xu et al., 2007). Briefly, sections were washed in Tris-buffered saline (TBS) with 0.2% Tween 20. X-gal staining was carried out overnight at 37°C in PBS that contained 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mm MgCl2, and 1 mg/ml X-gal. After X-gal staining, sections were washed with 0.25% Triton X-100 in PBS (three times, 10 min each time), incubated in 0.3% hydrogen peroxide and 0.3% normal goat serum in PBS for 5 min at room temperature, and then washed in 0.2% Tween 20 in TBS (three times, 10 min each time). Antigen unmasking was then done by boiling the sections in sodium citrate and holding at a sub-boil for 10 min. After cooling for 30 min at room temperature, sections were washed (three times for 10 min each time) and then incubated in 50% normal goat serum and 1% BSA in PBS for 1 h at room temperature. Sections were subsequently immunostained with a rabbit polyclonal Ki-67 antibody (Novocastra Laboratories) at a 1:1000 dilution in blocking buffer or a mouse monoclonal proliferating cell nuclear antigen (PCNA) antibody (BD Pharmingen) at a 1:4000 dilution overnight at 4°C. Sections were washed with 0.2% Tween 20 in TBS (three times for 10 min each time) and then incubated with the appropriate biotinylated goat anti-rabbit or biotinylated goat anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) at a 1:500 dilution in blocking buffer for 1 h at room temperature. After washing with 0.2% Tween 20 in TBS (three times for 10 min each time), biotin was detected using a horseradish peroxidase detection kit (Vector Laboratories). Sections were mounted and coverslipped using Vectashield mounting media. Bright-field images were captured using a Zeiss Axioscope imaging system.
Double immunofluorescent experiments were carried out using procedures similar to those described previously (Xu et al., 2007). To visualize cells expressing AGRP neuropeptides, mutant and control mice were injected with 1 μl of colchicine (20 μg/μl) into the third ventricle as described previously (Xu et al., 2005a). Forty-eight hours after colchicine injection, mice were transcardially perfused and sections were prepared as described above. Sections were washed and the antigen retrieval with sodium citrate was accomplished as described above. Following blocking, sections were incubated with a rabbit polyclonal AGRP antibody (Xu et al., 2005a,b) and a mouse monoclonal anti-human PCNA antibody described above (1:4000 dilution; BD Pharmingen).
For all other colocalization studies, mice did not receive colchicine injection. The staining procedure was similar to that described above, but a polyclonal anti-rat ACTH (from Dr. A. F. Parlow, National Hormone and Peptide Program, Torrance, CA) or a polyclonal anti-phosphorylated (Tyr-705) STAT3 (pSTAT3; Cell Signaling Technology) was used at a dilution of 1:500 or 1:200, respectively (Hentges et al., 2004; Piper et al., 2008). For BrdU immunofluorescence staining, antigen retrieval was performed by washing the sections in glycine for 10 min followed by SDS for 10 min at room temperature. After antigen retrieval, DNA was denaturated in 1N HCl for 6 min at 60°C. Immunofluorescence analysis was carried out using a rat anti-BrdU primary antibody (1:200 dilution; Serotec). For antibody detection, the sections were incubated in the appropriate Alexa Fluor 488 or 555 secondary antibodies (Invitrogen) at a 1:200 dilution in 1% BSA with PBS. Fluorescent images were captured using a Zeiss Axioscope 2. Cell counts were quantified by visually counting positive cells at the microscope and by examining photomicrographs of each section. The observer was blind to the identity of each section at the time of counting.
Cell counting.
Photographs of immunohistochemical sections of the control and mutant animals were taken using the same exposures and visualized in Photoshop Creative Suite 2 (Adobe Systems). Nuclear localization of Ki-67, PCNA, and BrdU signals was verified by colocalization with 4′,6-diamidino-2-phenylindole (DAPI). Regions of the hypothalamus, such as the ARC, ventromedial hypothalamus (VMH), dorsomedial hypothalamus (DMH), and lateral hypothalamic area (LHA), were defined using Paxinos and Franklin (2004). The observer was blinded to the identity of the sections at the time of counting. Positive signals were gauged by comparing the intensity of the Ki-67, PCNA, or BrdU signals in dentate gyrus, an adult neurogenic region, with signals from non-neurogenic regions such as cortex and thalamus. Positive cells in the hypothalamus of intensity similar to that of cells in the dentate gyrus and having a nuclear staining pattern were considered positive. For BrdU analysis, sections of 10 μm thickness were sampled from the rostral, medial, and caudal thirds of the ARC between bregma −1.82 to −2.30. Each glass slide contained two adjacent sections, but only one of them was used for cell counting (randomly selected or based on tissue integrity). An independent cohort of mice was used for Ki-67 and PCNA analysis. Sections of approximately equal spacing were sampled between bregma −2.06 to −2.30. Double counting was avoided by analyzing nonadjacent sections. To avoid overestimation of cell numbers due to cell fractionation in thin sections, cell counts were corrected using the Abercrombie method (Abercrombie, 1946). Nuclear diameters of 25–35 cells for each proliferation marker were measured using NIH ImageJ. Four transects, at 45° angles to one another, were placed across each nucleus. The average transect length was used as the diameter for the nucleus being measured. The mean diameter of Ki-67, PCNA, and BrdU-positive nuclei was ∼8 μm in length. The crude cell count was corrected following the correction formula P = A × M/(L + M), where P is the corrected cell count, A is the crude cell count, M is the section thickness (in micrometers), and L is the mean cell diameter (in millimeters).
Statistical analysis.
All statistical analyses were done using the SPSS 14.0 program. T tests were used to analyze differences between genotypes in immunofluorescent experiments. A two-way ANOVA (treatment × genotype) was used to test for differences among BrdU cell counts and for food intake in vehicle and Ara-C-infused mutants and controls. Significant main effects were evaluated by a posteriori multiple pairwise comparisons using the Holm–Sidak method. Two-way repeated measures ANOVA was used to analyze the changes in body weight and body composition. A significant interaction term (genotype × treatment) was found in terms of body fat content over time; therefore, we evaluated the within-subjects effects of the different treatments. Because there was a significant main effect, we performed a posteriori multiple pairwise comparisons using the Holm–Sidak method. Statistical differences were accepted at p < 0.05.
Results
Cell proliferation increases in the hypothalamus of adult mutant mice in response to AgRP neurodegeneration
Deletion of the Tfam gene from cells leads to progressive depletion of mitochondria and apoptosis (Wang et al., 2001). Cells positive for active caspase 3, an apoptosis marker, can be detected in the ARC of the AgRP-Tfam mice as early as 2.5 month of age. Thus, if neurodegeneration induces a microenvironment permissive for cell proliferation, such inductive signals would be present following the onset of apoptosis. In this study, we first sought to determine whether cell proliferation increased in the hypothalamus of male mutants at 2.5–4 months of age. AgRP-Tfam mutant mice were generated by crossing the Tfam floxed allele with Tg.AgRP-Cre mice that were used in the previous study (Xu et al., 2005b). The specificity of the Tg.AgRP-Cre animals has been previously characterized and validated by a number of independent research groups (Gropp et al., 2005; Xu et al., 2005a,b; Kitamura et al., 2006; Konner et al., 2007; van de Wall et al., 2008; Zhang et al., 2008).
Immunohistochemical analysis was carried out using Ki-67 and PCNA, two commonly used proliferation markers that mark cells in the active cell cycle. AgRP-Tfam mutant mice exhibited an over threefold increase in the number of Ki-67-positive cells in the ARC of the hypothalamus (Fig. 1A,B,G; Table 1). Similarly, a significant increase in the number of cells expressing PCNA, which has a longer half-life than that of Ki-67, was detected in the ARC of the AgRP-Tfam mutant animals (Fig. 1C,D,G; Table 1). To further confirm the increase in cell proliferation, we infused BrdU, which is incorporated into newly replicating DNA, intracerebroventricularly into 3-month-old controls and mutants for 6 weeks via osmotic minipumps. A significantly higher number of BrdU-positive cells were detected in the ARC of the AgRP-Tfam mutant mice (Fig. 1E–G, Table 1), consistent with the increase in the endogenous cell proliferation markers Ki-67 and PCNA. Many of the Ki-67- and PCNA- and some of the BrdU-positive cells were present in doublets, suggesting that these cells had just undergone cell divisions and still remained in close proximity.
Figure 1.
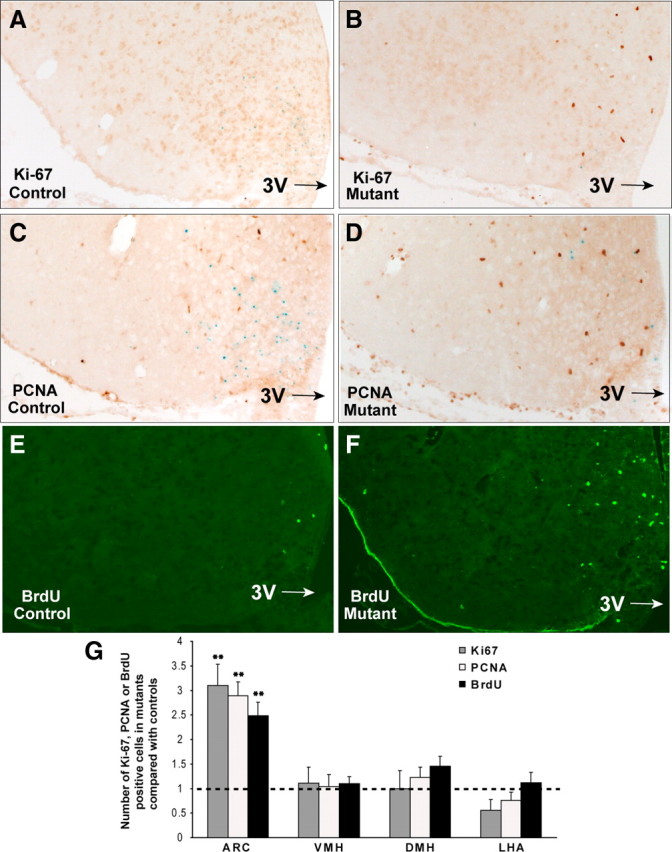
Cell proliferation is increased in the hypothalamus of AgRP-Tfam mutant mice. A–D, Immunohistochemical analyses for endogenous cell proliferation markers Ki-67 (A, B) and PCNA (C, D) were carried out in 2.5- to 4-month-old male control and AgRP-Tfam mutant mice. E, F, A separate cohort of control (E) and mutant (F) mice was infused intracerebroventricularly with BrdU (3.2 μg/μl) via osmotic minipumps (flow rate of 0.15 μl/h for 6 weeks; Alzet model 2006; Durect). Immunofluorescence analysis of BrdU in the hypothalamus of controls and mutants is shown. G, Proportional increase of Ki-67-, PCNA-, or BrdU-positive cells in the mutants was compared with that of the controls. Detailed quantification of positive cells is shown in Table 1. Error bars represent SEM; **p ≤ 0.01. 3V, Third ventricle.
Table 1.
Quantification of cell proliferation in the hypothalamus of adult AgRP-Tfam mutant mice
| ARC |
VMH |
DMH |
LHA |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Count | Mean ± SEM | n, N | Count | Mean ± SEM | n, N | Count | Mean ± SEM | n, N | Count | Mean ± SEM | n, N | |
| Ki-67 | ||||||||||||
| Control | 28 | 1.8 ± 0.4 | 16, 5 | 14 | 1.0 ± 0.3 | 13, 5 | 9 | 0.7 ± 0.2 | 13, 5 | 14 | 1.0 ± 0.2 | 13, 5 |
| Mutant | 153 | 5.5 ± 0.7** | 28, 6 | 28 | 1.2 ± 0.3 | 23, 6 | 16 | 0.7 ± 0.2 | 23, 6 | 15 | 0.6 ± 0.2 | 23, 6 |
| PCNA | ||||||||||||
| Control | 44 | 4.0 ± 1.2 | 11, 5 | 49 | 5.5 ± 2.4 | 9, 4 | 26 | 2.9 ± 0.7 | 9, 4 | 31 | 3.4 ± 1.1 | 9, 4 |
| Mutant | 195 | 11.5 ± 1.1** | 17, 6 | 85 | 5.7 ± 1.3 | 15, 6 | 57 | 3.6 ± 0.6 | 16, 6 | 41 | 2.6 ± 0.6 | 16, 6 |
| BrdU | ||||||||||||
| Control | 114 | 3.9 ± 0.4 | 29, 4 | 62 | 4.5 ± 0.8 | 14, 4 | 36 | 2.6 ± 0.6 | 14, 4 | 24 | 1.9 ± 0.4 | 13, 4 |
| Mutant | 332 | 9.8 ± 1.0** | 34, 4 | 79 | 4.9 ± 0.6 | 16, 4 | 60 | 3.8 ± 0.5 | 16, 4 | 33 | 2.1 ± 0.4 | 16, 4 |
Number of cells positive for Ki-67, PCNA, or BrdU was quantified in different regions of the hypothalamus. Cell counts were corrected using the Abercrombie correction method (Abercrombie, 1946). The Count columns contain the total number of positive cells corrected using the Abercrombie method. Mean is the average number of positive cells per section. All sections were 10 μm. n, Number of sections evaluated; N, number of mice from which the sections were evaluated. **p ≤ 0.01, the difference between mutant and control mice.
To determine whether increased cell proliferation was occurring in specific hypothalamic nuclei, Ki-67-, PCNA-, and BrdU-positive cells were quantified in the ARC, VMH, DMH, and LHA. Ki-67, PCNA, and BrdU cell counts were all significantly higher in the ARC of mutants relative to controls. However, Ki-67, PCNA, and BrdU did not differ significantly in the VMH, DMH, or LHA of mutants and controls (Fig. 1G, Table 1). Taken together, our results suggest that degeneration of AgRP neurons in the ARC leads to increased cell proliferation in this region of the adult mutant animals.
A subset of the newly generated cells take on AgRP neuronal identity
Because AgRP neurons are specifically degenerated in the AgRP-Tfam mutant animals, we wished to determine whether AgRP neurons could be regenerated in the mutant hypothalamus. By double immunofluorescence analysis, some PCNA-positive cells were found to express AGRP in the mutant hypothalamus, suggesting that AgRP neurons were regenerated (Fig. 2A–D). Using an independent approach, we generated control and mutant mice that carry the Tg.AgRP-Cre and Cre-activatable Rosa26-LacZ reporter so that AgRP neurons can be identified by LacZ expression in control and mutant animals (Xu et al., 2005a,b; Xu et al., 2007). Zero of 1055 AgRP neurons (LacZ positive) were positive for Ki-67 in the controls, whereas 39 of 2498 AgRP neurons coexpressed Ki-67 in the mutants. Conversely, we found that 0 of 48 Ki-67-positive and 1 of 83 PCNA-positive cells expressed LacZ in the controls, whereas 33 of 278 Ki-67-positive and 27 of 373 PCNA-positive cells expressed LacZ in the mutants (Fig. 2E,F). Taken together, these results suggest that a small population (7.2–11.8%) of dividing cells in the hypothalamus differentiates into AgRP neurons at a given time.
Figure 2.
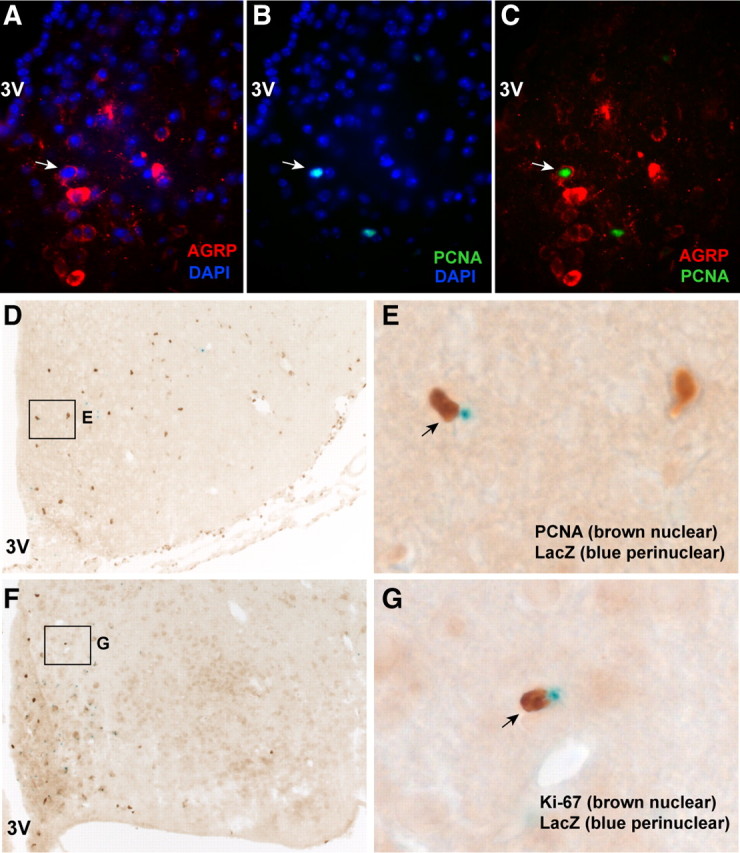
Newborn hypothalamic cells take on AgRP neuronal identity. A–C, Representative field showing the colocalization of AGRP immunoreactivity (cytoplasmic signals) and PCNA nuclear signals from a colchicine-injected AgRP-Tfam mutant mouse. The arrows show the colocalized cell. DAPI staining revealed all nuclei in the field. The specificity of the AGRP antibody was previously characterized (Xu et al., 2005a,b). D–G, X-gal staining combined with immunohistochemical analysis of PCNA or Ki-67 in hypothalamic sections from the AgRP-Tfam mutant animals. Control and mutant animals carried the Tg.AgRP-Cre transgene and the Cre-activatable R26R-LacZ reporter so that Agrp neurons were identified by LacZ expression (Xu et al., 2005a,b). In this preparation, LacZ expression is characteristically seen as perinuclear blue dots, whereas brown nuclear staining represents the PCNA or Ki-67 signal as indicated. Arrows show a LacZ/PCNA coexpressing neuron and a LacZ/Ki-67 coexpressing neuron. E, Magnified view of the boxed area in D; G, magnified view of the boxed area in F. The specificity of the Tg.AgRP-Cre transgenic mice has been characterized and validated by multiple studies (Gropp et al., 2005; Xu et al., 2005a,b; Kitamura et al., 2006; Konner et al., 2007; van de Wall et al., 2008; Zhang et al., 2008). 3V, Third ventricle.
A subset of the newly generated cells take on Pomc neuronal cell fate
Because neurodegeneration was specific to the AgRP neurons in the AgRP-Tfam mutant mice (Xu et al., 2005b), we wished to evaluate whether de novo neurogenesis is confined to neurons that would restore orexigenic drive. Pomc neurons are situated in the vicinity of AgRP neurons in the ARC, and Pomc and AgRP neurons have opposite functions in energy balance regulation. Thus, we investigated whether Pomc neurons were regenerated in the hypothalamus of the same AgRP-Tfam mutant mice. Double immunofluorescence analysis was carried out to detect ACTH- (the product of Pomc neurons) and PCNA-coexpressing cells. The ACTH antibody was used previously and was highly specific (Hentges et al., 2004; Piper et al., 2008). Although there was no significant difference in the number of Pomc neurons between the controls and mutants (Xu et al., 2005b), zero Pomc neurons were seen to coexpress PCNA in the controls, whereas 9.3% of PCNA-positive cells were identified as Pomc-expressing neurons (14 Pomc cells out of 150 PCNA-positive cells from six different mutant mice) (Fig. 3). These data suggest that anorexigenic Pomc neurons are generated in the AgRP-Tfam mutant mice along with orexigenic AgRP neurons. In addition, the proliferation of non-AgRP neurons argues against the notion that the incorporation of BrdU and the increased expression of PCNA and Ki-67 in the AgRP-Tfam mutant mice are the result of DNA repairs in degenerating AgRP neurons.
Figure 3.

Newborn hypothalamic cells take on Pomc neuronal identity. A, Representative field of the ARC showing expression of PCNA (nuclear signal) and ACTH (product of the Pomc neurons; cytoplasmic signal) in an AgRP-Tfam mutant animal. DAPI marked all nuclei in the field. B–D, Magnified views of the boxed area in A, showing a pair of PCNA-positive cells that expressed ACTH. Arrows indicate the two colocalized cells. Fourteen Pomc cells were identified out of 150 PCNA-positive cells from six different mutant mice. The ACTH antibody was previously used and it was highly specific (Piper et al., 2008). 3V, Third ventricle.
A subset of the newly generated cells are responsive to leptin
Both AgRP and Pomc neurons express the leptin receptor long form. Leptin administration robustly stimulates pSTAT3 in Pomc, AgRP, and other leptin target neurons in the hypothalamus. To determine whether some of the newly proliferated neurons in AgRP-Tfam mutant mice acquired leptin responsiveness, control and mutant mice were injected intraperitoneally with saline or leptin (5 mg/kg) and the mice were killed 45 min later. Leptin but not saline treatment induced strong pSTAT3 immunoreactivity in the hypothalamus (Fig. 4A,B). Double immunofluorescence analysis revealed that 22.6% of PCNA-positive cells expressed pSTAT3 in the ARC of leptin-treated mutant animals (Fig. 4C–F). These results suggest that some of the newly proliferated cells in the AgRP-Tfam mutant animals are leptin responsive. Similar proportion of newborn cells was shown to be leptin responsive in the hypothalamus of CNTF-treated mice (Kokoeva et al., 2005).
Figure 4.
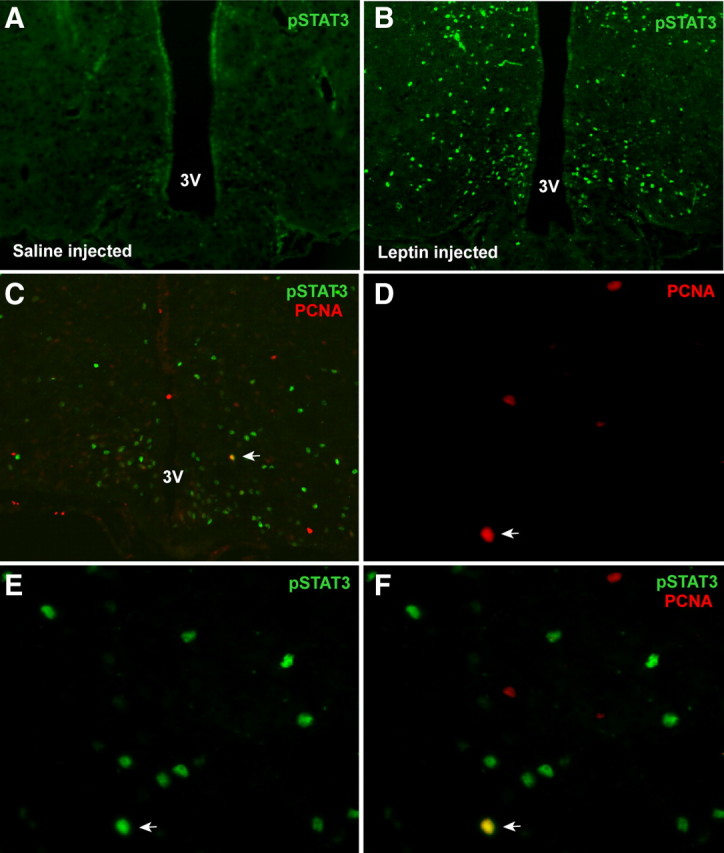
Newly generated hypothalamic cells are responsive to leptin. Four-and-a-half-month-old AgRP-Tfam mutant mice were treated with either vehicle or leptin (5 mg/kg, i.p.), and mice were killed 45 min later. Immunofluorescence analysis was carried out to identify cells that express pSTAT3 and PCNA. A, B, Leptin but not saline treatment activated pSTAT3 in the ARC of the hypothalamus. C, A field in the ARC of the hypothalamus showing expression of PCNA and leptin-induced pSTAT3 expression. Arrow indicates a colocalized cell. D–F, Magnified views of the colocalized cell indicated by the arrow in C showing PCNA, pSTAT3 immunoreactivity, and merged images. Arrow indicates a colocalized cell. Twenty-four of 106 PCNA cells (22.6%) were found to be positive for pSTAT3. Fourteen sections from six mice were analyzed. 3V, Third ventricle.
Central blockade of cell proliferation results in decrease in food intake and body adiposity in the AgRP-Tfam mutants but not in controls
Acute ablation of AgRP neurons results in severe anorexia and weight loss, whereas progressive ablation of AgRP neurons in the AgRP-Tfam mutants causes little change in feeding and body weight (Gropp et al., 2005; Luquet et al., 2005; Xu et al., 2005b). Thus, we sought to determine whether the newly proliferated cells in the mutant hypothalamus functionally compensated for the loss of AgRP neurons and helped the AgRP-Tfam mutant mice escape starvation. To this end, we inhibited cell proliferation in the brains of 3-month-old male AgRP-Tfam mutants and controls by intracerebroventricular infusion of the antimitotic compound arabinosylcytosine (AraC; 10.9 μg/μl at a flow rate of 0.15 μl/h for 4 weeks via osmotic minipumps). This dose of AraC effectively blocked cell proliferation in the hypothalamus (Fig. 5A,B) without causing overt adverse effects on health (Kokoeva et al., 2005). Although AraC-treated control mice did not show significant change in food intake compared with vehicle-treated controls, AgRP-Tfam mutant animals decreased their feeding significantly during the 4 week AraC treatment compared with vehicle-treated mutants (Fig. 5C). In addition, body composition was measured by DEXA before and after AraC or vehicle treatments. Whereas 4 weeks of AraC treatment did not affect lean body mass in either controls or mutants, it significantly reduced body fat mass in the mutants but not in the controls (Fig. 5E,F). Body weight followed the same trend, although it did not reach statistical significance (Fig. 5D). This experiment demonstrates that newly generated cells in the mutant hypothalamus are functionally required, at least in part, for the maintenance of feeding and body adiposity.
Figure 5.

Blockade of cell proliferation in the adult brain leads to decreased food intake and body adiposity in AgRP-Tfam mutants but not in control mice. Vehicle or mitotic blocker AraC (10.9 μg/μl) was infused into the lateral ventricle of 3-month-old control and AgRP-Tfam mutant mice via osmotic minipumps (flow rate of 0.15 μl/h). A, B, AraC effectively blocked cell proliferation in the hypothalamus of mutant mice as evident by reduced number of BrdU-positive cells upon AraC treatment. C, Average weekly food intake during a 4 week AraC treatment in control and mutant mice. D, Body weight of mice after 4 weeks of AraC or vehicle treatment was compared with their own pretreatment body weight. E, F, Body composition was measured by DEXA before and after 4 weeks of AraC or vehicle treatment. Four weeks of AraC treatment did not change body lean mass in either controls or mutants, whereas it significantly decreased body fat mass in the mutants but not in the controls. n = 5–6 mice for each group; *p ≤ 0.05; **p ≤ 0.01. 3V, Third ventricle.
Discussion
AgRP and Npy neurons are key first-order neurons in the ARC of the hypothalamus that sense and integrate diverse metabolic signals. Paradoxically, chronic and acute disruptions of AgRP gene or AgRP neurons have produced very different outcomes. Although acute reduction of AgRP gene expression by 50% using RNA interference results in significant weight loss days after small interfering RNA administration (Makimura et al., 2002), germline mutation of AgRP gene has minimal consequence on body weight (Qian et al., 2002; Wortley et al., 2005). Similarly, acute ablation of AgRP neurons leads to severe anorexia and weight loss, while progressive degeneration of AgRP neurons in the AgRP-Tfam mutants has minimal impact on feeding and body weight although the same Tg.AgRP-Cre line was used in both studies (Gropp et al., 2005; Xu et al., 2005b). These results clearly demonstrate that compensatory mechanisms develop during the gradual loss of AgRP neuronal function, which allow the mice to escape the deleterious effects of weight loss. In this study, we provide evidence that de novo cell proliferation increases in the hypothalamus of adult AgRP-Tfam mutant mice in response to degeneration of the AgRP neurons, and that some of the newly generated cells take on AgRP neuronal cell fate. We further show that a subset of the newly proliferative cells are capable of responding to leptin and that central blockade of cell proliferation leads to decreased feeding and body adiposity in the mutants but not in the controls. Thus, our study suggests that de novo neurogenesis provides a new level of neural plasticity in reshaping the hypothalamic feeding circuits.
It should be noted that there are methodological limitations in the detection of neurogenesis in the adult brain (Emsley et al., 2005), and the one most relevant to this study is the potential incorporation of BrdU into AgRP neurons that are undergoing DNA repair. However, evidence of mitotic divisions indicated by closely situated cell doublets, the generation of non-AgRP neurons such as Pomc neurons, and alteration of energy balance upon inhibition of CNS cell proliferation strongly argue that cell proliferation is indeed increased in the hypothalamus of AgRP-Tfam mutant mice. The nuclear localization of BrdU signals also excludes mitochondrial labeling. The basal levels of BrdU-positive cells in our control hypothalamus appear to be lower than those previously reported by Kokoeva et al. (2005, 2007). There are several possible reasons for this discrepancy. First, there are differences in experimental procedures. For example, Kokoeva and colleagues prepared hypothalamic sections at 25 μm and we prepared ours at 10 μm, so our sections were thinner, which may contribute to the lower cell counts. Also, different types of osmotic minipumps were used. Second, genetic background may influence the rate of adult neurogenesis. Whereas Kokoeva and colleagues used C57BL/6 mice, our AgRP-Tfam mice were on a mixed background with contributions from 129/SvJ, C57BL/6, and FVB mice. It has been shown that C57BL/6 mice exhibit a higher degree of adult neurogenesis in the hippocampus region compared with other mouse strains, including 129/SvJ (Kempermann et al., 1997; Kempermann and Gage, 2002). Thus, it is possible that genetic background variation may contribute to the discrepant cell counts in these studies.
AGRP and NPY are two of the most potent orexigens identified to date. Disruption of AgRP/Npy neuronal function due to mutations, injuries, or neurodegeneration can lead to severe anorexia and weight loss, which is life threatening. Thus, evolution may have favored the development of multiple mechanisms to ensure that the orexigenic drive is not lost. The fact that AgRP and Npy are coexpressed in the same neurons in the ARC suggests that one such safeguarding mechanism is by coexpression of genes that perform similar functions. Another level of compensation could be provided by the redundant wiring of AgRP/Npy neurons on their targets. AgRP/Npy neurons are known to project to MC4R neurons and also to their upstream Pomc neurons that express the ligand for MC4R (Bagnol et al., 1999; Cowley et al., 1999; Cowley et al., 2001; Roseberry et al., 2004). Thus, AgRP/Npy neurons exert their orexigenic functions by inhibiting two different anorexigenic neurons, the Pomc and MC4R neurons, via regulation of neuronal activities and receptor signaling. The redundant wiring and differential regulation of Pomc and MC4R neurons provide another safeguarding mechanism to ensure sufficient inhibition of anorexigenic drive. The current study provides an additional compensatory mechanism in that de novo cell proliferation functions to replenish the diminishing AgRP neuronal cell pool. Previous studies indicate that neurons can survive and remain functional for a few months following Tfam deletion (Sorensen et al., 2001). Thus, the ability to regenerate neurons in the AgRP-Tfam mutants would allow other compensatory mechanisms to develop, which would act in concert to provide full functional compensation. Some of these potential compensatory mechanisms include modulation of synaptic densities on MC4R, Pomc, or other downstream neurons, change in MC4R and NPY receptor sensitivity, and alterations of gene expression in downstream neurons.
Adult neurogenesis is readily observed in the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus in the hippocampus but is limited in the adult hypothalamus under normal conditions (Ming and Song, 2005). However, resident neural stem cells have been described in the adult hypothalamus, where de novo neurogenesis occurs at a low rate (Kokoeva et al., 2007). A number of growth factors, and neurotrophic factors such as FGF2, BDNF, CNTF, VEGF (vascular endothelial growth factor), and TGFα, have been shown to regulate neural stem cells and neural progenitor proliferation in the adult rodent brain (Hagg, 2005; Lledo et al., 2006). Infusion of BDNF into the lateral ventricle of the adult rat leads to generation of new neurons in the hypothalamus (Pencea et al., 2001). In addition, central administration of CNTF to mice stimulates neurogenesis in the adult hypothalamus (Kokoeva et al., 2005). Together, these results suggest that neurogenesis in the adult hypothalamus can be stimulated under certain conditions. It is likely that neurodegeneration in the AgRP-Tfam mutant mice activates this latent pool of neural stem cells, and they differentiate into AgRP neurons along with other resident cell types, such as Pomc neurons and glial cells. Because AgRP neurons, but not Pomc or other neurons, are specifically degenerated in the AgRP-Tfam mutant animals, regeneration of AgRP neurons would be expected to have greater impact on the AgRP neuronal cell pool, whereas the addition of new Pomc neurons may not affect the overall Pomc population. It is difficult to directly compare the relative abundance of newborn AgRP neurons with newborn Pomc neurons due to differences in antibody sensitivity. However, it is conceivable that the increase in cell proliferation in the mutant hypothalamus would partially restore orexigenic drive while having a smaller impact on anorexigenic drive.
Neurogenesis in the adult brain, although at low rates, could be a critical mechanism to replenish degenerated neurons due to aging, gene mutations, or environmental toxins, and impairment in this process could lead to disease onset or aggravate disease progression. Impaired adult neurogenesis has been implicated in the etiology and progression of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, or amyotrophic lateral sclerosis, which are characterized by loss of neurons in particular brain regions (for review, see Abdipranoto et al., 2008). Stimulation of neurogenesis has been found to associate with functional recovery of memory and learning in models of Alzheimer's disease. In addition, it has been shown that neurogenesis increases following ischemia or stroke in both humans and rodents, suggesting that stroke-induced neurogenesis serves as a self-repair mechanism (Okano et al., 2007; Wiltrout et al., 2007). Interestingly, recent studies show that consumption of a high-fat diet in rodents causes hypothalamic endoplasmic reticulum stress (Zhang et al., 2008; Ozcan et al., 2009), and a high-fat diet induces apoptosis of hypothalamic neurons (Moraes et al., 2009). Thus, these findings raise the possibility that diet-induced neurodegeneration may play a role in the etiology of obesity. Our current study provides new evidence that AgRP neurons, critical regulators of energy balance, can be regenerated in the adult hypothalamus, suggesting that adult neurogenesis is one adaptive mechanism to reshape the feeding circuits in response to hypothalamic neurodegeneration. Understanding the mechanisms underlying neural stem cell activation would pave the way for future therapeutic intervention to treat feeding disorders that are associated with neurodegeneration.
Footnotes
This work was supported by National Institutes of Health Grant 5R21DK80509 (A.W.X.) and an American Heart Association Postdoctoral Fellowship (A.A.P.).
References
- Abdipranoto A, Wu S, Stayte S, Vissel B. The role of neurogenesis in neurodegenerative diseases and its implications for therapeutic development. CNS Neurol Disord Drug Targets. 2008;7:187–210. doi: 10.2174/187152708784083858. [DOI] [PubMed] [Google Scholar]
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Bagnol D, Lu XY, Kaelin CB, Day HE, Ollmann M, Gantz I, Akil H, Barsh GS, Watson SJ. Anatomy of an endogenous antagonist: relationship between Agouti-related protein and proopiomelanocortin in brain. J Neurosci. 1999;19:RC26. doi: 10.1523/JNEUROSCI.19-18-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol. 2005;75:321–341. doi: 10.1016/j.pneurobio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Bruning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Hagg T. Molecular regulation of adult CNS neurogenesis: an integrated view. Trends Neurosci. 2005;28:589–595. doi: 10.1016/j.tins.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci U S A. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci. 2004;24:1578–1583. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gage FH. Genetic determinants of adult hippocampal neurogenesis correlate with acquisition, but not probe trial performance, in the water maze task. Eur J Neurosci. 2002;16:129–136. doi: 10.1046/j.1460-9568.2002.02042.x. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci U S A. 1997;94:10409–10414. doi: 10.1073/pnas.94.19.10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310:679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J Comp Neurol. 2007;505:209–220. doi: 10.1002/cne.21492. [DOI] [PubMed] [Google Scholar]
- Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Bruning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Makimura H, Mizuno TM, Mastaitis JW, Agami R, Mobbs CV. Reducing hypothalamic AGRP by RNA interference increases metabolic rate and decreases body weight without influencing food intake. BMC Neurosci. 2002;3:18. doi: 10.1186/1471-2202-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, Romanatto T, Carvalheira JB, Oliveira AL, Saad MJ, Velloso LA. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009;4:e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Okano H, Sakaguchi M, Ohki K, Suzuki N, Sawamoto K. Regeneration of the central nervous system using endogenous repair mechanisms. J Neurochem. 2007;102:1459–1465. doi: 10.1111/j.1471-4159.2007.04674.x. [DOI] [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in sterotaxic coordinates. Ed 2. London: Elsevier; 2004. [Google Scholar]
- Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci. 2001;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper ML, Unger EK, Myers MG, Jr, Xu AW. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol. 2008;22:751–759. doi: 10.1210/me.2007-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, Van der Ploeg LH, Marsh DJ. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol. 2002;22:5027–5035. doi: 10.1128/MCB.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM. Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron. 2004;41:711–722. doi: 10.1016/s0896-6273(04)00074-1. [DOI] [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci. 2001;21:8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, Elmquist J, Lowell BB, Barsh GS, de Luca C, Myers MG, Jr, Schwartz GJ, Chua SC., Jr Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A. 2001;98:4038–4043. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Scott MM, Elmquist JK. From observation to experimentation: leptin action in the mediobasal hypothalamus. Am J Clin Nutr. 2009;89:985S–990S. doi: 10.3945/ajcn.2008.26788D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltrout C, Lang B, Yan Y, Dempsey RJ, Vemuganti R. Repairing brain after stroke: a review on post-ischemic neurogenesis. Neurochem Int. 2007;50:1028–1041. doi: 10.1016/j.neuint.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Wortley KE, Anderson KD, Yasenchak J, Murphy A, Valenzuela D, Diano S, Yancopoulos GD, Wiegand SJ, Sleeman MW. Agouti-related protein-deficient mice display an age-related lean phenotype. Cell Metab. 2005;2:421–427. doi: 10.1016/j.cmet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005a;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS. Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol. 2005b;3:e415. doi: 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology. 2007;148:72–80. doi: 10.1210/en.2006-1119. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]