

Abstract
It has been hypothesized that ionizing radiation-induced disruptions in mitochondrial O2 metabolism lead to persistent heritable increases in steady-state levels of intracellular superoxide (O2•−) and hydrogen peroxide (H2O2) that contribute to the biological effects of radiation. Hamster fibroblasts (B9 cells) expressing a mutation in the gene coding for the mitochondrial electron transport chain protein succinate dehydrogenase subunit C (SDHC) demonstrate increases in steady-state levels of O2•− and H2O2. When B9 cells were exposed to low-dose/low-LET radiation (5–50 cGy), they displayed significantly increased clonogenic cell killing compared with parental cells. Clones derived from B9 cells overexpressing a wild-type human SDHC (T4, T8) demonstrated significantly increased surviving fractions after exposure to 5–50 cGy relative to B9 vector controls. In addition, pretreatment with polyethylene glycol-conjugated CuZn superoxide dismutase and catalase as well as adenoviral-mediated overexpression of MnSOD and/or mitochondria-targeted catalase resulted in significantly increased survival of B9 cells exposed to 10 cGy ionizing radiation relative to vector controls. Adenoviral-mediated overexpression of either MnSOD or mitochondria-targeted catalase alone was equally as effective as when both were combined. These results show that mammalian cells over expressing mutations in SDHC demonstrate low-dose/low-LET radiation sensitization that is mediated by increased levels of O2•− and H2O2. These results also support the hypothesis that mitochondrial O2•− and H2O2 originating from SDH are capable of playing a role in low-dose ionizing radiation-induced biological responses.
Introduction
Mammalian cells obtain energy required for metabolism through the biochemical oxidation of substrates, such as carbohydrates, fats and amino acids. The electrons that are extracted during this process drive oxidative phosphorylation via mitochondrial electron transport chains (ETC) to produce ATP, with O2 acting as the terminal electron acceptor (1, 2). Mutations in genes encoding mitochondrial ETC proteins have been hypothesized to lead to oxidative stress and thereby to result in genomic instability, increased mutation rates, and age-related diseases (3–8). Mitochondrial ETC complex II, known as succinate dehydrogenase (SDH), plays major biological roles in both the Krebs cycle and oxidative phosphorylation. During normal metabolism complex II is thought to produce less than 1% of the ROS resulting from mitochondrial metabolism (9–11). However, recent studies have suggested that defects in complex II can cause increased univalent reduction of O2 to O2•−, leading to oxidative stress, which contributes to genomic instability, aging and cancer (7, 12, 13). In addition, mutations in genes coding for SDHB, C and D have been associated with increased susceptibility to induction of paragangliomas and pheochromocytomas in humans (14, 15).
Analogous to the mitochondrial production of reactive oxygen species during aberrant oxidative phosphorylation, exposure of cells to ionizing radiation also causes immediate formation of free radicals [i.e. hydroxyl radical (OH•), superoxide (O2•−), and organic radicals] that damage critical biomolecules (16–22). These radical species lead to the generation of other reactive oxygen species such as hydrogen peroxide (H2O2) and organic hydroperoxides (ROOH) in the presence of O2 (17, 18, 21) and are believed to be the primary source of ionizing radiation-induced damage to biomolecules such as DNA, lipids and proteins along with causing perturbations in intracellular metabolic oxidation/reduction processes (7). In addition, previous studies have shown that antioxidant enzymes and thiols involved with the metabolic detoxification of free radials (as well as O2•− and H2O2) are capable of mediating radioprotection when administered both before and after irradiation, suggesting that these reactive species contribute significantly to radiation injury both at the time of and after ionizing radiation exposure (21, 23).
While evidence in favor of the hypothesis that metabolic sources of reactive oxygen species (ROS) contribute to radiation response after exposure is becoming fairly robust (24–26), the exact nature of the intracellular sources of these species and the radiation dose–response relationships are less well characterized. Previous studies have clearly indicated the general importance of mitochondrial ROS in the biological effects of radiation (27–33), and mitochondrial electron transport chain (ETC) complex II (a.k.a. SDH) dysfunction has been causally linked to persistent genomic instability induced in hamster fibroblasts exposed to high-dose (10 Gy) low-LET radiation (34). To determine what role O2•− and H2O2 from SDH might play in the clonogenic survival response after exposure to low-dose/low-LET radiation, B9 hamster fibroblasts expressing a mutation in succinate dehydrogenase subunit C (SDHC) resulting in a 33-amino acid truncation of the protein were irradiated and compared to the B1 parental cells as well as the T4 and T8 clones isolated from B9 cells stably overexpressing wild-type human SDHC (hSDHC).
Materials and Methods
Cell and Culture Conditions
The immortalized Chinese hamster lung fibroblast cell lines B1 (wild-type) and B9 (mutant containing truncated form of SDHC protein) were a gift from Dr. Immo Scheffler (University of California San Diego). B9 cells were derived from B1 cells after exposure to the mutagen ethyl methane sulfonate (EMS) for 24 h and were allowed to grow for at least eight generations. The mutation and selection process of B9 cells is described in detail by Ditta et al. (35). Stable transfection of human SDHC into B9 cells and clonal selection and characterization of T4 and T8 clones as well as V8 vector control cells were performed as described (7). All cells were grown in DMEM containing 4.5 g/ml of glucose and l-glutamine, 10% fetal bovine serum (FBS), 2 ml gentamycin, and 5 ml nonessential amino acids. Cells were maintained in 95% air/5% CO2 and humidified in a 37°C incubator and were assayed between passages 10 and 25.
Irradiation
Cells were plated in 60-mm dishes with 5 ml of complete medium and allowed to attach and incubate at 37°C for 24 h to obtain exponentially growing cultures at 50% confluence. All cells, experimental and control, were transported to the radiation facility. Cells were irradiated at doses ranging from 5 cGy to 1 Gy in the Radiation and Free Radical Research Core laboratory at the University of Iowa using a 137Cs source at a dose rate of 0.87 Gy min−1 (J. L. Shepherd & Associates, San Fernando, CA). After irradiation all cells were incubated at 37°C for 1 h before being plated for the survival assay.
Clonogenic Survival Assay
After the cells were trypsinized and counted, they were plated for clonogenic survival as described previously (36–38). Proper plating dilutions were determined by experimentation and by the anticipated survival at each time or dose of ionizing radiation. After 8 days (B1 and hSDHC-transfected T4 and T8) or 13 days (B9 and empty vector-transfected V8), colonies were fixed with 70% ethyl alcohol, stained with Coomassie blue, and counted. Plating efficiency was determined by dividing the number of cells plated by the number of colonies formed. The surviving fraction was determined by dividing the plating efficiency of experimentally manipulated cells by the plating efficiency of sham-treated control cells.
Thenoyltrifuoroacetone (TTFA) Treatment
The cells were treated with 20 μM TTFA (or 0.01% DMSO as the vehicle control) 30 min before irradiation in complete medium. The drug was also present during the 1-h incubation at 37°C after irradiation before the cells were trypsinized and plated for clonogenic assay. TTFA or DMSO (20 μM) was also added and left in the dishes during 8–13 days of incubation. The treatment with 20 μM TTFA slightly decreased the plating efficiency of both B1 (from 42% to 38%) and B9 (from 25% to 19%) cells; therefore, the data were normalized to the respective treatment group of unirradiated B1 and B9 cells.
Measurement of Intracellular Superoxide Levels
Steady-state levels of superoxide were estimated 1 h after exposure to 10 cGy using oxidation of the fluorescent dye dihydroethidium (DHE) (Molecular Probes, Eugene, OR), as described previously (7). After labeling with 10 μM dihyrdoethidium (DHE) at 37°C for 40 min, the samples were analyzed using a FACScan flow cytometer (Becton Dickinson, excitation 488 nm, emission 585 nm band-pass filter). The mean fluorescence intensity (MFI) of 10,000 cells was analyzed in each sample and corrected for autofluorescence from unlabeled cells.
Detection of Apoptosis and DNA Damage
Apoptosis after exposure to ionizing radiation was determined via flow cytometry using the BD-Pharmingen (San Jose, CA) apoptosis detection kit. Briefly, B1 and B9 cells were exposed to 10 cGy ionizing radiation and incubated at 37°C for 24 h. The cells were then washed twice with cold PBS and resuspended in binding buffer. The cells were then incubated with Annexin V-FITC and propidium iodide according to the manufacturer's directions at 25°C for 15 min in the dark in a 100-μl volume. The volume of the cells was raised to 500 μl by addition of binding buffer and immediately analyzed by flow cytometry. The percentage of cells residing in the “Annexin V-FITC” high and “propidium iodide” low quadrant were calculated as the percentages of apoptotic cells.
Oxidative DNA damage was assessed using a kit based on an FITC-conjugated 8OHdG antibody (OxyDNA, Calbiochem, EMD Biosciences, San Diego, CA). The cells were plated on chamber slides the day before irradiation. B1 and B9 cells were irradiated with 10 cGy and fixed on the slides with 4% paraformaldehyde on ice at 0, 15 and 60 min after irradiation. The cells were then dehydrated in 70 and 95% ethanol and permeated in 99% ethanol on ice for 30 min. After the cells were rehydrated through 95 and 70% ethanol and washed in Tris-buffered saline-Tween 20 (TBST), they were blocked for nonspecific biding sites at 37°C for 1 h. The slides were washed in TBST and incubated with FITC-8OHdG Ab at 4°C overnight. The cells were then washed in TBST and distilled water. The 8OHdG-positive cells were counted using a fluorescence microscope equipped with FITC filters and expressed as a percentage of 200–300 total cells counted per group. Treatment with 1 mM H2O2 for 30 min was used as a positive control.
Adenoviral Transduction of Antioxidant Enzymes
The replication-incompetent adenovirus vectors AdBgl II, AdCMV MnSOD (AdMnSOD), and AdMitochondrialCAT (AdMitoCAT) were purchased from Viraquest (North Liberty, IA) (39). B1 and B9 cells were seeded in 60-mm tissue culture plates at 1.0 × 105 cells/plate and 3.0 × 105 cells/plate, respectively, and incubated for 24 h. The adenovirus at 50 MOI individually and 25 MOI in combination was then added to cells in 1.8 ml of complete medium and incubated at 37°C for 24 h. The adenovirus-containing medium was then replaced with 4 ml of medium and cells were incubated for an additional 24 h to allow expression of the protein before irradiation. After irradiation, cells were incubated at 37°C for 1 h and plated for clonogenic survival.
Polyethylene (PEG)-Conjugated Antioxidant Enzyme Treatment
PEG-conjugated CuZnSOD (PEG-SOD) and PEG-conjugated catalase (PEG-CAT) (Sigma-Aldrich, St. Louis, MO) were used to pretreat both B1 and B9 cells. B1 cells were seeded at 1.0 × 105 cells/60-mm dish and B9 cells were seeded at 3.0 × 105 cells/60-mm dish. After a 48-h delay to allow recovery from trypsinization, cells were pretreated with PEG alone, PEG-SOD (200 U/ml), PEG-CAT (200 U/ml), or PEG-SOD + PEG-CAT (100 U/ml each) for 4 h, then irradiated and plated for survival. PEG alone prepared at the same concentration (18 μM) was used as the control.
Western Blotting
Both B1 and B9 cells were grown to 80% confluence on 60-mm dishes. The cells were scraped in cold PBS and centrifuged at 4°C at 1200 rpm for 5 min. The resulting pellet was frozen overnight at −20°C and resuspended in 50 mM PBS (pH 7.8) containing 1.34 mM DETAPAC. Protein concentrations, which were used to normalize the biochemical analyses, were determined by the Lowry assay (40). Samples were electrophoresed on a 15% polyacrylamide gel. Proteins were then transferred to nitrocellulose paper, and the blots were blocked for 1 h in 5% milk containing Tris-buffered saline (TBS)-Tween at room temperature. Blots were incubated with primary antibody (1:1000; MnSOD from Millipore, Billerica, MA, and catalase from Athens Inc., Athens, GA) at 4°C overnight in 5% milk containing TBS-Tween, washed and then incubated in HRP-conjugated secondary antibody (1:10,000) at room temperature for 1 h in TBS-Tween. HRP-conjugated antibody signal was detected using the ECL Plus detection system (Amersham Pharmacia Biotech) and visualized by exposure to Kodak X-Omat LS film (Kodak, New York, NY).
Measurement of Antioxidant Enzyme Activities
Both B1 and B9 cells were grown to 80% confluence on 60-mm dishes. The cells were scraped in cold 50 mM phosphate-buffered saline (PBS) buffer on ice and centrifuged at 4°C at 1200 rpm for 5 min. The resulting pellet was frozen overnight at −20°C and resuspended in 50 mM PBS (pH 7.8) containing 1.34 mM DETAPAC. Protein concentrations used to normalize the biochemical analyses were determined by the Lowry assay (40). The total SOD activity of whole cell homogenates prepared on ice in 50 mM PBS was determined by the indirect competitive inhibition assay originally developed by Spitz and Oberley (41). In this assay, O2•− is generated from xanthine by xanthine oxidase and detected by recording nitroblue tetrazolium (NBT) reduction. SOD can scavenge O2•− and competitively inhibit the reduction of NBT, and one unit of SOD activity is defined as the amount of protein required to inhibit 50% of the maximal NBT reduction. SOD activity was also measured in the presence of sodium cyanide, which inhibits the activity of CuZnSOD, to assess the level of MnSOD. This value was then subtracted from total SOD activity to provide a measure CuZnSOD activity (41). Catalase activity was determined following the method of Beers and Sizer, with analysis according to Aebi (42, 43). The decomposition of 10 mM H2O2 in 50 mM K2HPO4 (pH 7.0) containing 10–250 μg of total cellular protein was followed by ultraviolet spectroscopy at 240 nm (42, 43).
Statistics
Results are expressed as means ± 1 SEM unless otherwise specified. Comparisons within individual cell lines were made using a one-way analysis of variance (ANOVA). Statistical significance was accepted if the level of probability was less than 0.05. ANOVA was used to compare mean cell survival across the five treatment groups (Fig. 2 control, PEG alone, PEG-SOD, PEG-CAT and PEG-SOD+PEG-CAT, and Figs. 3 and 4 control, AdBglII, AdMitoCAT, AdMnSOD and AdMnSOD + AdMitoCAT) and two radiation doses (0 and 10 cGy). Multiple pairwise comparisons of the experimental conditions were performed with the Tukey's HSD test to control for the overall Type I error rate.
FIG. 2.
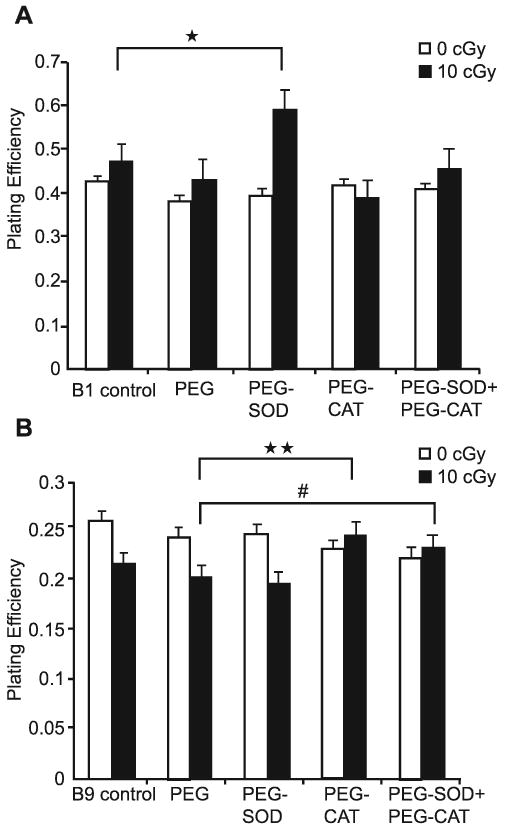
Treatment with PEG-CAT alone or combination with PEG-SOD inhibits sensitivity of B9 cells to low-dose ionizing radiation. Cells were pretreated with PEG alone, PEG-SOD and/or PEG-CAT (200 U/ml individually or 100 U/ml each in combination) for 4 h prior to irradiation. Panel A: B1 cells. Panel B: B9 cells. Bars represent means of three separate experiments. Error bars represent ±1 SEM of 12–18 dishes from three different experiments. *Significantly different from 10-cGy irradiated B1 cells, P < 0.001; **significantly different from 10-cGy irradiated, “PEG alone” treated B9 cells, P < 0.001; #significantly different from 10-cGy irradiated, “PEG alone” treated B9 cells, P < 0.05.
FIG. 3.
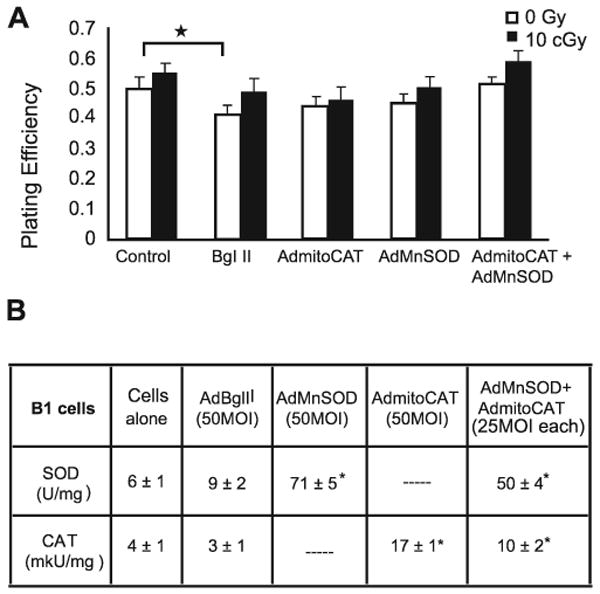
Overexpression of mitochondria-targeted antioxidant enzymes MnSOD and CAT delivered via adenoviral vectors did not alter the response of B1 cells to low-dose ionizing radiation. Panel A: The plating efficiency of B1 cells transfected with 50 MOI AdBglII is significantly decreased in unirradiated cells compared to cells that were not transduced with adenovirus (*P < 0.001). However, no survival advantage was observed in any irradiated groups of B1 cells transfected with AdMnSOD and/or AdMitoCAT. Columns represent means of three separate experiments. Error bars represent ±1 SEM of 12–18 dishes from three different experiments. Panel B: B1 cells transduced with AdMnSOD and/or AdMitoCAT showed elevated enzyme activity relative to untransduced cells (P < 0.001). Errors represent ±1 SD. Data are averages of three independent experiments.
FIG. 4.
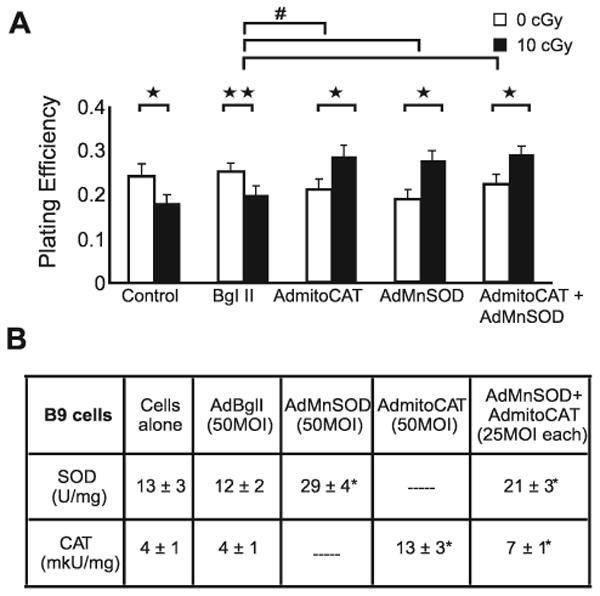
Overexpression of mitochondria-targeted antioxidant enzymes MnSOD and CAT reduces cytotoxicity of low-dose radiation in B9 cells. Panel A: Plating efficiency. Error bars represent ±1 SEM of 12–18 cloning dishes from three different experiments. *Significantly different from unirradiated matched B9 cells, P < 0.001; **significantly different from unirradiated matched B9 cells, P < 0.05; #significantly different from AdBglII-transduced unirradiated B9 cells, P < 0.001. Panel B: Enzyme activity. Errors represent ±1 SD. Data are averages of three independent experiments. *Significantly different from cells alone, P < 0.001, N = 3.
Results
B9 Cells Expressing the SDHC Mutation Display Increased Sensitivity to Low-Dose Radiation
Previously characterized B9 cells expressing a mutant SDHC and known to demonstrate increased steady-state levels of O2•− and H2O2 were exposed to low doses of radiation (0–1 Gy), and clonogenic survival was compared to that of the parental B1 cells expressing wild-type SDHC (Fig. 1A). B9 cells displayed a significant decrease in clonogenic survival at doses below 1 Gy compared with B1 cells, with the largest difference in survival (relative to B1) occurring at 10 cGy. These data support the hypothesis that a mutation in SDHC that led to elevated steady-state levels of O2•− and H2O2 derived from mitochondrial metabolism (7) might contribute to increased radiosensitivity to low-dose/low-LET radiation.
FIG. 1.
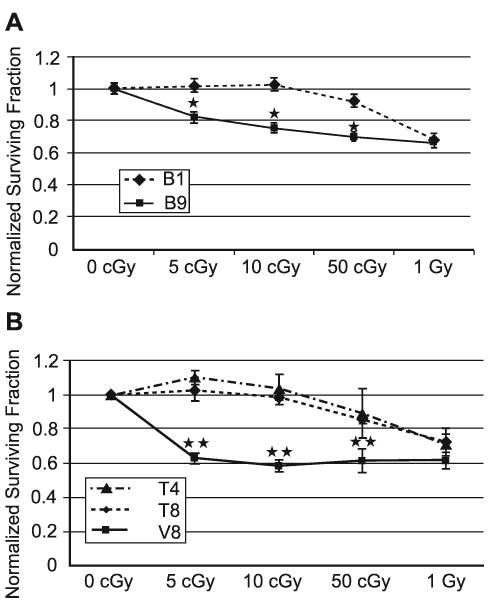
SDHC mutant cells display increased radiosensitivity and decreased survival after exposure to low-dose ionizing radiation compared to wild-type cells. Panel A: SDHC mutant B9 and wild-type cells. Panel B: B9 cells overexpressing wild-type hSDHC (T4 and T8) exhibited decreased radiosensitivity to low-dose ionizing radiation. Points represent means of three separate experiments. Error bars represent ±1 SEM of 12–18 dishes from three different experiments. *Significantly different from 10-cGy irradiated B1 cells, P < 0.001; **Significantly different from 10-cGy irradiated T4 or T8 cells, P < 0.001.
Since B9 cells were generated via exposure to an alkylating agent (EMS), which might have induced other mutations (44–46) that contributed to increased sensitivity to low-dose ionizing radiation, B9 cells were stably transfected with human wild-type SDHC (hSDHC) to determine whether the biological effects on radiation survival could be directly attributed to the SDHC mutation. Previously published results had shown that the transfection of B9 cells harboring the SDHC mutation with an expression vector encoding wild-type human SDHC (hSDHC) was sufficient to reduce the steady-state levels of ROS (i.e. O2•− and H2O2) relative to B1 and vector control cells (7), indicating that the mutation in SDHC was directly responsible for increased steady-state levels of ROS. When low-dose radiation sensitivity was determined in T4 and T8 clones (derived from B9 cells by overexpressing hSDHC) and compared to the V8 vector control, the T4 and T8 cells displayed significant increases in clonogenic survival after exposure to 5–50 cGy (Fig. 1B). The largest differences in survival in T4 and T8 cells occurred at 5 and 10 cGy. These results show that stable overexpression of hSDHC in B9 cells reverses low-dose radiosensitivity, strongly supporting the conclusion that the expression of the SDHC mutation in B9 cells is directly responsible for increased low-dose radiation sensitivity.
Treatment with O2•− and H2O2 Scavenging Enzymes Increases Clonogenic Survival in Low-Dose Irradiated B9 Cells
Polyethylene glycol (PEG) conjugation is used to help deliver antioxidant enzymes into cells to determine the involvement of ROS in biological responses to oxidative stress (47). To determine whether antioxidant enzymes with O2•− and H2O2 scavenging ability were capable of protecting against low-dose ionizing radiation-induced cell killing in B9 cells, cells were treated with PEG-SOD and PEG-CAT 4 h before irradiation at 0 and 10 cGy (Fig. 2A and B). Incubation of cells with PEG alone had no significant effect on plating efficiency at either 0 or 10 cGy. In contrast, pretreatment of PEG-SOD and PEG-CAT had significant effects in B1 and B9 cells exposed to 10 cGy (Fig. 2A and B). The presence of 200 U/ml PEG-SOD significantly increased plating efficiency after 10 cGy ionizing radiation (Fig. 2A). In addition, pretreatment with either PEG-CAT alone or PEG-CAT+PEG-SOD significantly protected B9 cells from low-dose ionizing radiation-induced cytotoxicity. Interestingly, when B9 cells were pretreated with PEG-SOD alone, they showed no protection after 10 cGy ionizing radiation, suggesting that differences in endogenous levels of O2•− and H2O2 might have differential outcomes on the cells' response to low-dose ionizing radiation (Fig. 2B). These results support the hypothesis that metabolic oxidative stress produced by persistently elevated levels of mitochondrial ROS enhances low-dose radiosensitivity.
AdMnSOD and AdMitoCAT-Transduced B9 Cells Demonstrate Increased Clonogenic Survival after Exposure to Low-Dose/Low-LET Radiation
After determining the potential protective role of PEG-SOD and PEG-CAT against low-dose radiation-induced cell killing in B9 cells, we shifted our focus to determining whether mitochondria-targeted antioxidant enzymes MnSOD and MitoCAT could protect cells from low-dose radiation. B1 and B9 cells were transduced with AdMnSOD and AdMitoCAT prior to exposure to 10 cGy, and the resulting surviving fractions were compared (Figs. 3 and 4). The overexpression of MnSOD and CAT activity in transduced cells was determined using standard activity assays (41, 42) (Figs. 3B and 4B), and similar increases in immunoreactive protein were confirmed using Western blotting techniques (data not shown).
Figure 3A shows that there was a reduction in plating efficiency in B1 cells transduced with 50 MOI AdBglII, indicating that the replication-incompetent adenoviral vector did cause modest biological effects independent of the target gene of interest. B1 cells also showed slightly higher plating efficiencies when irradiated with 10 cGy (similar to the results presented in Figs. 1 and 2) regardless of whether they had been transduced with 50 MOI AdBglII, 50 MOI AdMnSOD, 50 MOI AdMitoCAT, or 25 MOI AdMnSOD + 25 MOI AdMitoCAT. In contrast to the results with B1 cells, B9 cells transfected with 50 MOI AdMnSOD or 50 MOI AdMitoCAT alone or 25 MOI AdMnSOD + 25 MOI AdMitoCAT displayed significantly increased cell survival compared to empty vector-transfected B9 cells when exposed to 10 cGy ionizing radiation (Fig. 4). Transduction with the combination of AdMnSOD and AdMitoCAT prior to 10 cGy did not provide additional protection compared to either one alone. These data demonstrate that overexpression of mitochondria-targeted MnSOD and catalase protect B9 cells from cell killing induced by low-dose/low-LET radiation and continue to support the conclusion that elevated levels of O2•− and H2O2 may play a significant role in the radiosensitivity demonstrated by B9 cells harboring a mutation in SDHC.
Low-Dose/Low-LET Ionizing Radiation did not Cause an Increase in the Intracellular Superoxide Levels in B1 or B9 Cells 1 h after Exposure
When cells were exposed to 10 cGy ionizing radiation and incubated in 37°C for 1 h after radiation and then labeled using the superoxide-sensitive dye DHE, the results confirmed previous findings (7) demonstrating that B9 cells have two- to threefold baseline increases in DHE oxidation relative to B1 cells (data not shown). Furthermore, there were no additional increases in DHE oxidation in either B1 or B9 cells 1 h after 10 cGy low-LET ionizing radiation exposure (data not shown). These results support the hypothesis that the sensitivity to low-dose ionizing radiation in B9 cells is due to the differences in endogenous steady-state levels of superoxide between B9 and B1 cells (caused by a mutation in the SDHC gene), which can be reversed by overexpression of MnSOD and mitoCAT (Fig. 4).
Low-Dose/Low-LET Ionizing Radiation did not Induce Apoptosis or DNA Damage in B1 or B9 Cells
To determine the percentage of apoptosis after low-dose/low-LET ionizing radiation, B1 and B9 cells exposed to 10 cGy were stained with Annexin V-FITC/propidium iodide labeling 24 h after irradiation (48, 49). B9 cells had a fivefold higher baseline percentage of apoptotic cells compared to wild-type B1 cells, but there were no significant differences in the percentage of cells with Annexin V-positive/PI-negative labeling between the 10-cGy irradiated and nonirradiated groups (data not shown). These results imply that differential sensitivity of B9 cells to low-dose ionizing radiation is probably due to mitosis-linked death and not to apoptosis.
When oxidative DNA damage was evaluated in B1 and B9 cells immediately, 15 min or 60 min after 10 cGy ionizing radiation using immunofluorescence, the percentages of 8-OHdG-positive cells were not significantly different in either B1 or B9 cells at any time after irradiation. In contrast, the positive control cells (1 mM H2O2 for 30 min) demonstrated two- to fourfold increases in the percentage of 8-OHdG-positive cells in both B1 and B9 cells (data not shown). These results support the conclusion that bulk oxidative DNA damage does not affect the differential sensitivity of B9 cells to low-dose/low-LET ionizing radiation.
Inhibition of Complex II Protects against Low-Dose/Low-LET Ionizing Radiation
To test whether inhibition of complex II protected B9 cells against the low-dose ionizing radiation-induced cytotoxicity, a known complex II blocker, thenoyltrifuoroacetone (TTFA), which was reported to inhibit complex II by binding to iron-sulfur clusters (50, 51), was used. The treatment of B9 cells with 20 μM TTFA before and after irradiation resulted in a significant protection against the increased radiation sensitivity of B9 cells (Table 1), supporting the hypothesis that the mutation in complex II is significantly contributing to the low-dose radiation sensitivity seen in these cells.
TABLE 1. Normalized Surviving Fractions of B1 and B9 Cells Treated with 20 μM TTFA (complex II blocker) 1 h after Irradiation.
| Dose | Complex II blocker | B1 cells | B9 cells |
|---|---|---|---|
| 0 Gy | 0 μM TTFA | 1.00 ± 0.04 | 1.00 ± 0.02 |
| 10 cGy | 0 μM TTFA | 1.08 ± 0.07 | 0.68 ± 0.06* |
| 0 Gy | 20 μM TTFA | 1.00 ± 0.03 | 1.00 ± 0.03 |
| 10 cGy | 20 μM TTFA | 1.00 ± 0.03 | 0.89 ± 0.03** |
Notes. Plating efficiency is expressed as the surviving fraction normalized to that of unirradiated cells. Errors represent ±1 SD of six dishes counted from each treatment.
Significantly different from unirradiated B9 cells, P < 0.05;
significantly different from B9 cells irradiated with 10 cGy, P < 0.01.
Discussion
The current study demonstrates that B9 cells harboring a mutation in an ETC protein (SDHC), which was previously shown to result in increased steady-state levels of O2•− and H2O2 (7), also displayed elevated sensitivity to low-dose/low-LET ionizing radiation. Furthermore, when B9 cells were stably transfected with wild-type hSDHC, which was previously shown to reverse metabolic oxidative stress and genomic instability in these cells (7), the increased radiosensitivity was abrogated. The results presented in the current report provide the first direct evidence that expressing a mutation in a gene coding for a mitochondrial ETC protein causes radiosensitization to low-dose/low-LET ionizing radiation.
During respiration in mitochondria from normal tissues, the SDH complex (aka ETC complex II) produces less than 1% of the ROS resulting from ETC activity (9, 10). However, previous work with hamster fibroblasts showed that a single point mutation leading to a premature stop codon in SDHC was sufficient to produce significantly elevated steady-state levels of O2•− and H2O2 in addition to increased glucose metabolism, increased sensitivity to glucose deprivation-induced cytotoxicity, increases in steady-state levels of GSH/GSSG, and nuclear aneuploidy (13, 52–54). It has also been shown that exposure of hamster fibroblasts to higher-dose (10 Gy) low-LET radiation results in persistent heritable mitochondrial dysfunction in chromosomally unstable surviving cells that is characterized by alterations in O2 consumption, alterations in mitochondrial membrane potential, and increased ROS production that contributes to increased point mutation rates as well as genomic instability (55). In more recent studies, 10-Gy irradiated chromosomally unstable surviving cells have also been shown to demonstrate specific alterations in SDH complex structure and function that appear to significantly contribute to the genomic instability phenotype as well as elevated ROS levels (34).
Given these findings, it is tempting to speculate that at higher doses radiation-induced mutations in genes coding for complex II (or other ETC) proteins may contribute to persistent heritable phenotypes resulting from ROS-mediated genomic instability. If this speculation were confirmed, it is possible that any ionizing radiation-induced damage to genes coding for proteins necessary for the proper functioning and assembly of mitochondrial electron transport chains could result in increases in residence times or accessibility of electrons at sites where one-electron reductions of O2 to form ROS (i.e. O2•− and H2O2) could occur (34, 55, 56). The resulting increased mitochondrial flux of O2•− and H2O2 might then lead to a persistent heritable condition of metabolic oxidative stress that could continue to contribute to the biological effects for many cell generations after exposure to higher doses of radiation.
In contrast to higher doses of radiation, far less is known about the involvement of mitochondrial dysfunction leading to ROS production in low-dose irradiated cells. Some of the earlier work conducted by Marples and Joiner demonstrated a decrease in low-dose hypersensitivity in Chinese hamster V79-379A cells after pretreatment with X rays as well as low doses of H2O2, suggesting that alterations in redox status might have significant effects in response to low-dose ionizing radiation (57). A more recent report showed that in normal human fibroblasts, protein import into mitochondria isolated from low-dose irradiated cells (10 cGy) is enhanced, suggesting that alterations in mitochondrial assembly may play a crucial role in low-dose-induced biological effects (58). Another recent report showed that in mouse skin cells low-dose/low-LET ionizing radiation was capable of increasing the activity of a mitochondrial enzyme that scavenges superoxide (MnSOD) via the activation of NFκB (59). In that report, the authors showed that induction of MnSOD was at least partially required to induce an adaptive response to subsequent high-dose radiation. Another report presented data showing that MnSOD overexpression suppressed micronucleus formation in low-dose irradiated cells, again suggesting the involvement of mitochondrial ROS in damage caused by low-dose radiation exposure (60). There are also reports demonstrating the induction of antioxidant enzymes (i.e. total SOD and glutathione peroxidase) in vivo by low-dose ionizing radiation (61–63). These publications suggest that mitochondrial ROS production can contribute to the biological effects of low-dose radiation, but the involvement of specific components of mitochondrial electron transport chains in this process has not been demonstrated.
The data in the current report clearly demonstrate that overexpression of antioxidant enzymes (SOD and catalase) that scavenge O2•− and H2O2 in the B9 cells expressing the SDHC mutation inhibited clonogenic cell killing caused by low-dose/low-LET ionizing radiation. This represents the first clear evidence showing that O2•− and H2O2 derived from a mutation at a specific site in mitochondrial electron transport chains (SDHC) can contribute significantly to the biological effects of low-dose/low-LET ionizing radiation. It is also of interest to note that when using polyethylene-conjugated CuZnSOD and catalase (which are most likely to have a cytosolic localization), inhibition of low-dose ionizing radiation-induced cell killing in B9 cells was achieved with catalase alone but not SOD alone (Fig. 2), emphasizing the importance of H2O2. In contrast, when mitochondria-targeted MnSOD and catalase were used, both enzymes were equally effective at inhibiting low-dose radiation-induced cell killing in B9 cells, pointing to an important role for both mitochondrial O2•− and H2O2 in this biological effect (Figs. 3 and 4). While the exact mechanism for this difference is not currently known, these results clearly support the conclusion that intracellular localization and mitochondrial targeting are important considerations when using these antioxidants to mitigate the effects of low-dose radiation.
The cell cycle distribution of B9 cells has been reported to be different from that of B1 cells (B1, 36% G1, 28% S, 36% G2/M; B9, 46%G1, 30% S, 24% G2/M) (7). Furthermore, hyper-sensitivity to low-dose ionizing radiation is associated with a higher fraction of G2/M cells (64), suggesting that subtle differences in cell cycle distribution might explain the differential effects seen with low-dose ionizing radiation. However, since asynchronously growing B9 cells have a lower percentage of G2/M cells, B1 cells, the increased sensitivity of B9 cells after exposure to low-dose ionizing radiation relative to B1 cells is not likely to be caused by differences in the fraction of cells in G2/M at the time of irradiation.
In summary, this study demonstrates that increased steady-state levels of mitochondrial O2•− and H2O2 contribute significantly to radiosensitivity and decreased cell survival after exposure to low-dose/low-LET ionizing radiation. We have shown that the radiosensitivity observed in SDHC mutant B9 cells can be eliminated by the stable transfection of wild-type hSDHC, which has been shown to reduce the steady-state levels of O2•− and H2O2, as well as by blocking electron transport chain Complex II using TTFA. In addition, our results show that overexpression of the mitochondrial antioxidant enzymes MnSOD and catalase prior to exposure of B9 cells to low-dose/low-LET ionizing radiation produces a radioprotective effect seen as increased cell survival. This work suggests a direct causal relationship between elevated steady-state levels of mitochondrial O2•− and H2O2 and sensitivity to low-dose/low-LET ionizing radiation.
Acknowledgments
The authors would like to thank Amanda Kalen from the Radiation and Free Radical Research Core in the Holden Comprehensive Cancer Center for assistance with all irradiations and Gareth Smith for his editorial assistance. This work was supported by Department of Energy grants DE-FG02-05ER64050 and DE-SC0000830 as well as NIH P30-CA086862, R01-CA115438 and T32 CA078586.
References
- 1.Lehninger AL. Principles of Biochemistry. Worth, New York: 2000. Oxidative phosphorylation and photophosphorylation; pp. 690–745. [Google Scholar]
- 2.Mitchell P. Keilin's respiratory chain concept and its chemiosmotic consequences. Science. 1979;206:1148–1159. doi: 10.1126/science.388618. [DOI] [PubMed] [Google Scholar]
- 3.Arthur CR, Morton SL, Dunham LD, Keeney PM, Bennett JP., Jr Parkinson's disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener. 2009;4:37. doi: 10.1186/1750-1326-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10:131–138. doi: 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 6.Nooteboom M, Johnson R, Taylor RW, Wright NA, Lightowlers RN, Kirkwood TB, Mathers JC, Turnbull DM, Greaves LC. Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts. Aging Cell. 2010;9:96–99. doi: 10.1111/j.1474-9726.2009.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slane BG, Aykin-Burns N, Smith BJ, Kalen AL, Goswami PC, Domann FE, Spitz DR. Mutation of succinate dehydrogenase subunit C results in increased O2•−, oxidative stress, and genomic instability. Cancer Res. 2006;66:7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 8.Zuin A, Gabrielli N, Calvo IA, Garcia-Santamarina S, Hoe KL, Kim DU, Park HO, Hayles J, Ayte J, Hidalgo E. Mitochondrial dysfunction increases oxidative stress and decreases chronological life span in fission yeast. PLoS One. 2008;3:e2842. doi: 10.1371/journal.pone.0002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boveris A, Cadenas E. In: Superoxide Dismutase. Oberley LW, editor. CRC Press; Boca Raton, FL: 1982. pp. 15–30. [Google Scholar]
- 10.Boveris A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol. 1977;78:67–82. doi: 10.1007/978-1-4615-9035-4_5. [DOI] [PubMed] [Google Scholar]
- 11.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 12.Ishii N, Ishii T, Hartman PS. The role of the electron transport SDHC gene on lifespan and cancer. Mitochondrion. 2007;7:24–28. doi: 10.1016/j.mito.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65:203–209. [PubMed] [Google Scholar]
- 14.Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003;14:453–459. doi: 10.1016/j.tem.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 15.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 16.Hall E. Radiation and Life. Pergamon; Oxford: 1984. Of cells, mice and men; pp. 21–61. [Google Scholar]
- 17.Hall E. Radiobiology for the Radiologist. 5th. Lippincott Williams and Wilkins; Philadelphia: 2000. Clinical response of normal tissues; pp. 327–348. [Google Scholar]
- 18.Biaglow JE, Mitchell JB, Held K. The importance of peroxide and superoxide in the X-ray response. Int J Radiat Oncol Biol Phys. 1992;22:665–669. doi: 10.1016/0360-3016(92)90499-8. [DOI] [PubMed] [Google Scholar]
- 19.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karbownik M, Reiter RJ. Antioxidative effects of melatonin in protection against cellular damage caused by ionizing radiation. Proc Soc Exp Biol Med. 2000;225:9–22. doi: 10.1177/153537020022500102. [DOI] [PubMed] [Google Scholar]
- 21.Oberley LW, Lindgren LA, Baker SA, Stevens RH. Superoxide ion as the cause of the oxygen effect. Radiat Res. 1976;68:320–328. [PubMed] [Google Scholar]
- 22.Sener G, Kabasakal L, Atasoy BM, Erzik C, Velioglu-Ogunc A, Cetinel S, Contuk G, Gedik N, Yegen BC. Propylthiouracil-induced hypothyroidism protects ionizing radiation-induced multiple organ damage in rats. J Endocrinol. 2006;189:257–269. doi: 10.1677/joe.1.06574. [DOI] [PubMed] [Google Scholar]
- 23.Petkau A, Chelack WS, Pleskach SD. Protection of post-irradiated mice by superoxide dismutase. Int J Radiat Biol Relat Stud Phys Chem Med. 1976;29:297–299. doi: 10.1080/09553007614550341. [DOI] [PubMed] [Google Scholar]
- 24.Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis. 1996;17:1633–1639. doi: 10.1093/carcin/17.8.1633. [DOI] [PubMed] [Google Scholar]
- 25.St Clair DK, Wan XS, Oberley TD, Muse KE, St Clair WH. Suppression of radiation-induced neoplastic transformation by overexpression of mitochondrial superoxide dismutase. Mol Carcinog. 1992;6:238–242. doi: 10.1002/mc.2940060404. [DOI] [PubMed] [Google Scholar]
- 26.Wu LJ, Randers-Pehrson G, Xu A, Waldren CA, Geard CR, Yu Z, Hei TK. Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells. Proc Natl Acad Sci USA. 1999;96:4959–4964. doi: 10.1073/pnas.96.9.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epperly MW, Epstein CJ, Travis EL, Greenberger JS. Decreased pulmonary radiation resistance of manganese superoxide dismutase (MnSOD)-deficient mice is corrected by human manganese superoxide dismutase-plasmid/liposome (SOD2-PL) intratracheal gene therapy. Radiat Res. 2000;154:365–374. doi: 10.1667/0033-7587(2000)154[0365:dprrom]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 28.Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, Ogi J, Khaletskiy A, Li Z, Weydert C, Li JJ. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol Cell Biol. 2003;23:2362–2378. doi: 10.1128/MCB.23.7.2362-2378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 2006;21:361–367. doi: 10.1093/mutage/gel048. [DOI] [PubMed] [Google Scholar]
- 30.Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 2006;66:10377–10383. doi: 10.1158/0008-5472.CAN-05-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61:3894–3901. [PubMed] [Google Scholar]
- 33.Limoli CL, Giedzinski E, Morgan WF, Swarts SG, Jones GD, Hyun W. Persistent oxidative stress in chromosomally unstable cells. Cancer Res. 2003;63:3107–3111. [PubMed] [Google Scholar]
- 34.Dayal D, Martin SM, Owens KM, Aykin-Burns N, Zhu Y, Boominathan A, Pain D, Limoli CL, Goswami PC, Spitz DR. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat Res. 2009;172:737–745. doi: 10.1667/RR1617.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ditta G, Soderberg K, Landy F, Scheffler IE. The selection of Chinese hamster cells deficient in oxidative energy metabolism. Somat Cell Genet. 1976;2:331–344. doi: 10.1007/BF01538838. [DOI] [PubMed] [Google Scholar]
- 36.Blackburn RV, Spitz DR, Liu X, Galoforo SS, Sim JE, Ridnour LA, Chen JC, Davis BH, Corry PM, Lee YJ. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic Biol Med. 1999;26:419–430. doi: 10.1016/s0891-5849(98)00217-2. [DOI] [PubMed] [Google Scholar]
- 37.Puck TT, Marcus PI. A rapid method for viable cell titration and clone production with HeLa cells in tissue culture: the use of x-irradiated cells to supply conditioning factors. Proc Natl Acad Sci USA. 1955;41:432–437. doi: 10.1073/pnas.41.7.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puck TT, Morkovin D, Marcus PI, Cieciura SJ. Action of x-rays on mammalian cells. II. Survival curves of cells from normal human tissues. J Exp Med. 1957;106:485–500. doi: 10.1084/jem.106.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 41.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 42.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 43.Beers RF, Jr, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 44.Chasin LA, Urlaub G. Mutant alleles for hypoxanthine phosphoriboxyltransferase: codominant expression, complementation, and segregation in hybrid Chinese hamster cells. Somat Cell Genet. 1976;2:453–467. doi: 10.1007/BF01542725. [DOI] [PubMed] [Google Scholar]
- 45.Okagaki RJ, Neuffer MG, Wessler SR. A deletion common to two independently derived waxy mutations of maize. Genetics. 1991;128:425–431. doi: 10.1093/genetics/128.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soderberg K, Mascarello JT, Breen GA, Scheffler IE. Respiration-deficient Chinese hamster cell mutants: genetic characterization. Somat Cell Genet. 1979;5:225–240. doi: 10.1007/BF01539163. [DOI] [PubMed] [Google Scholar]
- 47.Liu TH, Beckman JS, Freeman BA, Hogan EL, Hsu CY. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am J Physiol. 1989;256:H589–H593. doi: 10.1152/ajpheart.1989.256.2.H589. [DOI] [PubMed] [Google Scholar]
- 48.Boustany NN, Tsai YC, Pfister B, Joiner WM, Oyler GA, Thakor NV. BCL-xL-dependent light scattering by apoptotic cells. Biophys J. 2004;87:4163–4171. doi: 10.1529/biophysj.104.048736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krueger SA, Joiner MC, Weinfeld M, Piasentin E, Marples B. Role of apoptosis in low-dose hyper-radiosensitivity. Radiat Res. 2007;167:260–267. doi: 10.1667/RR0776.1. [DOI] [PubMed] [Google Scholar]
- 50.Nohl H, Jordan W, Hegner D. Mitochondrial formation of OH radicals by an ubisemiquinone-dependent reaction an alternative pathway to the iron-catalysed Haber-Weiss cycle. Hoppe Seylers Z Physiol Chem. 1982;363:599–607. doi: 10.1515/bchm2.1982.363.1.599. [DOI] [PubMed] [Google Scholar]
- 51.Tappel AL. Inhibition of electron transport by antimycin A, alkyl hydroxy naphthoquinones and metal coordination compounds. Biochem Pharmacol. 1960;3:289–296. doi: 10.1016/0006-2952(60)90094-0. [DOI] [PubMed] [Google Scholar]
- 52.Ackrell BA. Cytopathies involving mitochondrial complex II. Mol Aspects Med. 2002;23:369–384. doi: 10.1016/s0098-2997(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 53.Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 54.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 55.Dayal D, Martin SM, Limoli CL, Spitz DR. Hydrogen peroxide mediates the radiation-induced mutator phenotype in mammalian cells. Biochem J. 2008;413:185–191. doi: 10.1042/BJ20071643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 57.Marples B, Joiner MC. The elimination of low-dose hypersensitivity in Chinese hamster V79-379A cells by pretreatment with X rays or hydrogen peroxide. Radiat Res. 1995;141:160–169. [PubMed] [Google Scholar]
- 58.Pandey BN, Gordon DM, De Toledo SM, Pain D, Azzam EI. Normal human fibroblasts exposed to high- or low-dose ionizing radiation: differential effects on mitochondrial protein import and membrane potential. Antioxid Redox Signal. 2006;8:1253–1261. doi: 10.1089/ars.2006.8.1253. [DOI] [PubMed] [Google Scholar]
- 59.Fan M, Ahmed KM, Coleman MC, Spitz DR, Li JJ. Nuclear factor-kappaB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 2007;67:3220–3228. doi: 10.1158/0008-5472.CAN-06-2728. [DOI] [PubMed] [Google Scholar]
- 60.de Toledo SM, Asaad N, Venkatachalam P, Li L, Howell RW, Spitz DR, Azzam EI. Adaptive responses to low-dose/low-dose-rate gamma rays in normal human fibroblasts: the role of growth architecture and oxidative metabolism. Radiat Res. 2006;166:849–857. doi: 10.1667/RR0640.1. [DOI] [PubMed] [Google Scholar]
- 61.Yamaoka K, Edamatsu R, Itoh T, Mori A. Effects of low-dose X-ray irradiation on biomembrane in brain cortex of aged rats. Free Radic Biol Med. 1994;16:529–534. doi: 10.1016/0891-5849(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 62.Yamaoka K, Edamatsu R, Mori A. Increased SOD activities and decreased lipid peroxide levels induced by low dose X irradiation in rat organs. Free Radic Biol Med. 1991;11:299–306. doi: 10.1016/0891-5849(91)90127-o. [DOI] [PubMed] [Google Scholar]
- 63.Yamaoka K, Kojima S, Takahashi M, Nomura T, Iriyama K. Change of glutathione peroxidase synthesis along with that of superoxide dismutase synthesis in mice spleens after low-dose X-ray irradiation. Biochim Biophys Acta. 1998;1381:265–270. doi: 10.1016/s0304-4165(98)00021-x. [DOI] [PubMed] [Google Scholar]
- 64.Krueger SA, Wilson GD, Piasentin E, Joiner MC, Marples B. The effects of G2-phase enrichment and checkpoint abrogation on low-dose hyper-radiosensitivity. Int J Radiat Oncol Biol Phys. 2010;77:1509–1517. doi: 10.1016/j.ijrobp.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]