

Abstract
Gliomas are highly aggressive and accompanied by numerous microglia/macrophages (MG/MP) in and about the tumor. Little is known about what MG/MP do in this setting, or whether modulating MG/MP activation might affect glioma progression. Here, we used a glioma-microglia in culture system to establish the effects the tumor and microglia have on each other. We assessed glioma progression in vivo after MG/MP ablation or in the setting of exaggerated MG/MP activation. We show that glioma cells activate microglia but inhibit their phagocytic activities. Local ablation of MG/MP in vivo decreased tumor size and improved survival curves. Conversely, pharmacological activation of MG/MP increased glioma size through stimulating tumor proliferation and inhibiting apoptosis. In agreement with recent reports, expression of the chemokine CCL21 is enhanced after MG/MP activation and correlates with tumor growth. Taken together, our findings demonstrate that inhibition of MG/MP activation may constitute a new and effective contribution towards suppressing glioma proliferation.
Keywords: Glioma, mice, microglia, ablation, tuftsin, MIF/TKP-tuftsin fragment 1–3
Introduction
Malignant gliomas are primary central nervous system (CNS) tumors arising from glial cells and are one of the deadliest cancers - median survival time is one year even with aggressive surgical resection combined with irradiation and chemotherapy. Although many therapeutic approaches have been explored, there has been no major improvement in survival over the last 30 years (DeAngelis, 2001; Legler et al., 1999).
Gliomas are infiltrated by MG/MP, and the extent of MG/MP infiltration correlates positively with malignancy (Morimura et al., 1990; Morris and Esiri, 1991; Roggendorf et al., 1996). Microglia are capable of antigen presentation to T cells patrolling the CNS (Kreutzberg, 1996). Upon injury, microglia undergo activation characterized by changes in morphology, gene expression, proliferation, phagocytic capacity, and migration towards the injury site (Kreutzberg, 1996; Streit et al., 1999).
The role of MG/MP in glioma progression remains controversial. Studies reported that the immune defensive functions of glioma-infiltrating MG/MP (GIMs) are compromised. Moreover, GIMs have been proposed to promote glioma growth by secreting growth factors, immune-suppressive cytokines and angiogenic factors (Alterman and Stanley, 1994; Demuth et al., 2007; Galasso et al., 2000; Lafuente et al., 1999; Wagner et al., 1999; Wesolowska et al., 2008), thus stimulating interest in therapies that modulate MG/MP activity/function. However, such approaches yielded conflicting results: injection of CpG-containing oligonucleotides, which stimulate MG/MP, induced glioma apoptosis and prolonged survival times of tumor-bearing animals in one report, whereas the same approach caused increased animal tumor size in others (El Andaloussi et al., 2006; Ginzkey et al., 2009).
Here we investigate the consequences of interaction of MG/MP and glioma cells in culture using MG/MP activation and glioma cell proliferation as functional endpoints. We examine glioma progression in a mouse model using pharmacogenetics to locally ablate MG/MP, and a pharmacological approach to exaggerate MG/MP activation. We show that manipulation of MG/MP activation state appears to be a potentially promising novel interventional approach for gliomas.
Materials and methods
Cell Lines
GL261, a glioma cell line derived from C57BL/6 mice, and CRL-2541, a mouse astrocyte cell line, were obtained from ATCC. The cells were cultured in DMEM with 10% heat-inactivated fetal bovine serum (FBS) and 1mM Na pyruvate (Fisher Mediatech). The cells were transfected with pEGFP-N1 using Lipofectamine (Invitrogen) and selected with 500μg/ml neomycin (Geneticin, Invitrogen) to generate the stable cell lines GL261-EGFP and CRL-2541-EGFP.
Animals
C57BL/6 mice (wild type, WT) were purchased from Jackson Laboratory. CD11b-HSVTK transgenic mice were described previously (Heppner et al., 2005). Female CD11b-HSVTK (+/−) mice were bred with male C57BL/6 mice and the offspring genotyped by PCR using primers 5′-GACTTCCGTGGCTTCTTGCTGC-3′ and 5′-GTGCTGGCATTACAGGCGTGAG-3′. All animal procedures were approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC). Mice were bred in-house under maximum isolation conditions on a 12:12 hour light: dark cycle with food ad libitum.
Primary microglia cultures
Mixed cortical cultures from newborn C57BL/6 mice (day 0–3) were made using standard protocols (Rogove and Tsirka, 1998). Ten days after plating, primary microglia were isolated by treatment with 15mM lidocaine (Sigma) and gentle rocking. Microglia were resuspended in an appropriate volume of DMEM with 1% FBS. After 24–48 hours of culture, microglia were used for experiments.
Microglia and glioma cell co-culture
GL261-EGFP cells were plated with rhodamine-labeled primary microglia. In brief, after isolation of primary microglia, the cells were resuspended at a density of 5×104 cells/ml in DMEM with 1% FBS and 20μg/ml mini-ruby (Invitrogen) (Ullrich et al., 2001). After 48 hours, the medium was removed and 2×104 GL261-EGFP cells seeded on top of the microglia in DMEM with 10% FBS and desired treatments, such as 150μg/ml tuftsin, MIF/TKP (Bachem), or 4μg/ml CCL21 neutralizing antibody (PeproTech). As controls, primary microglia were either switched to the same medium without the GL261-EGFP, or microglia were seeded with 2×104 CRL-2541-EGFP astrocytes. The microglia-glioma interactions were followed by confocal imaging over 5 days. To evaluate the growth rate, cell numbers were counted by hemacytometer for 5 days. The experiments were repeated 3 times with duplicate samples per group.
Segregated microglia-glioma co-cultures were set as follows: 5×104 microglia were seeded on 0.4μm inserts (Millicell) in DMEM with 1% FBS. After 48 hours, the inserts were moved to 24-well plates containing 5,000 GL261 cells/well in DMEM with 10% FBS. 150μg/ml tuftsin or MIF/TKP was added to the medium above the inserts. Empty inserts with the same medium were used as control. GL261 proliferation was measured every other day. The experiments were repeated 3 times with duplicate samples per group.
Cell proliferation assay
The CCK-8 kit (Dojindo Molecular Technologies) was used for cell proliferation. In brief, equal numbers of GL261 cells were seeded in 96-well or 24-well plates (co-culture) with respective treatments (duplicate samples for each treatment). At each time point, the medium was changed to include a 1:10 dilution of CCK-8. After 2 hours at 37°C, absorbance was read at 450nm. Cell proliferation was plotted by normalizing the absorbance values at each time point over the ones at day 0.
Western blotting of microglial markers
Primary microglia were seeded into 6-well plates at equal numbers in DMEM+1% FBS. After two days, the medium was replaced with DMEM+10% FBS or conditioned medium. Glioma-conditioned medium (GCM) was collected from an 80%-confluent GL261 plate and centrifuged at 10,000 rpm for 10 minutes to remove cell debris. Negative controls included GCM boiled for 10 minutes and astrocyte-conditioned medium (ACM) collected from a CRL-2541 plate. At different time points, cells were lysed and protein concentrations measured by the Bio-Rad Bradford detergent-compatible (DC) assay. 15% SDS-PAGE was used to separate protein samples (10μg), which were transferred to polyvinylidene difluoride (PVDF) membrane, probed with rabbit anti-mouse Iba1 antibody (1:1000, Wako) and mouse anti-mouse α-tubulin antibody (1:2000, Millipore) overnight at 4°C. Goat anti-rabbit and anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Immunoresearch) were used at 1:5000. HRP activity was detected with LumiGLO Chemiluminescent Substrate System (KPL). Expression levels were quantified using SCION Image software, and Iba1 expression level normalized to the α-tubulin.
In vivo mouse glioma model and local drug delivery
12–16 week old male CD11b-HSVTK (+/−) mice weighing 25–30 grams were used. Wild type (WT) littermates [CD11b-HSVTK(−/−) mice] were used as controls. For the tuftsin group, 12–16 week old C57BL/6 mice were used. Mice were anesthetized with atropine (0.6mg/kg, i.p.) and 2.5% Avertin (0.02mg/g, i.p.), and then a midline incision made on the scalp. At stereotaxic coordinates of bregma, −1mm anteroposterior and +2mm mediolateral, a small burr hole was drilled on the skull. 3×104 GL261-EGFP cells were delivered in 1μl PBS at a depth of 3mm over 2minutes (modified from El Andaloussi et al., 2006).
Reagents were administered locally using mini-osmotic pumps (Alzet®, DURECT) containing different concentrations of ganciclovir (GCV) (Calbiochem), 250μg/ml tuftsin, or 250μg/ml MIF/TKP. Each pump was connected to plastic tubing and a 4mm guide cannula (PlasticsOne). The pumps were placed subcutaneously on the mouse back with the cannulas inserted to the injection site. The drugs were infused at 0.25μl/hr over 14 or 28 days. Pumps with normal saline inserted right after tumor cell injection were used as negative control. Pumps were inserted either immediately after (D0 pump), or five days after (D5 pump) tumor injection. Animals accepting GCV pumps were administered 50μg/g GCV i.p. one day before tumor injection to ensure maximum ablation of MG/MP.
Tumor evaluation
Mice were deeply anesthetized with 2.5% Avertin and transcardially perfused with 50ml PBS followed by 50ml 4% paraformaldehyde (PFA) in PBS. Brains were removed and post-fixed in 4% PFA/PBS for 6 hours at 4°C, followed by 30% sucrose at 4°C until fully dehydrated. Brains were then frozen-embedded in optimal cutting temperature compound (Tissue-Tek) and cut using a Leica (Nussloch, Germany) cryostat. 10–15 series of 25μm coronal sections encompassing the entire tumor were generated for each animal. For general morphology, sections were stained with hematoxylin and eosin (H&E). GFP-fluorescent glioma cells were visualized using fluorescent microscopy (Nikon E600). To measure tumor size, randomly picked serial sections from each animal were used, and the GFP+ tumor area measured using NIS-Elements software. The tumor sizes were calculated as sum of (tumor area × section thickness) for each section with a tumor tissue. The sections were immunostained with Iba1 antibody or biotinylated tomato lectin (Sigma) to visualize MG/MP infiltration, Ki67 (Abcam) for proliferation, activated caspase-3 (Sigma) for apoptosis and CCL21 (PeproTech) to assess cytokine expression. For quantification, 3 randomly selected samples per group were used and at least 15 fields in different sections of the same sample captured by confocal microscopy.
Immunofluoresent staining
Primary microglia treated with GCM were fixed with 4% PFA/PBS for 15minutes at room temperature. Cells or brain sections were blocked in 5% goat serum (Sigma) in PBS-T for 1 hour at room temperature, and then incubated with Iba1 antibody (1:500), Ki67 (1:500), activated caspase-3 (1:500), CCL21 (1:500) and biotinylated tomato lectin (1:500) in PBS-T overnight at 4°C. The samples were incubated with corresponding secondary antibodies (Alexa Fluor-555 or 647, Invitrogen) for 1-hour at room temperature, and mounted with Fluoromount-G (Southern Biotech).
Statistical analysis
The statistical significance between two groups was determined by Student’s t-test. For comparison of more than two groups, one-way ANOVA followed by a Bonferroni-Dunn test was used. The Kaplan-Meier survival curve was calculated with MedCalc software. Data were expressed as mean ± standard error of the mean (SEM). The statistical significance is either described in figure legends, or indicated as asterisks (*). *:P <0.05; **: P<0.01; ***:P< 0.001.
Results
Glioma cells produce soluble factors that activate microglia
We exposed primary microglia to glioma-conditioned medium (GCM) to determine whether soluble factors secreted by glioma cells alter microglial activation. Microglial activation can be assessed by morphology changes. Resting microglia appear rod-shaped in culture, and upon activation, they transform into an ameboid shape with pseudopodia (Siao and Tsirka, 2002). At time 0, resting microglia had elongated cell bodies and thin processes; no change in the morphology was obvious 24 h later in the same medium (microglia, DMEM+1% FBS) or in GL261 control medium (DMEM+10% FBS) (Fig. 1A, B). When exposed to GCM for 16–24 hours, however, the microglia retracted their processes and became ameboid in shape. Iba1 (ionized calcium binding adaptor molecule 1) is a MG/MP marker that is upregulated upon activation (Ito et al., 1998). Iba1 expression levels increased 3–4 fold with GCM treatment (Fig. 1B, C), concomitant with morphological changes. However, no increase in Iba1 was observed when the microglia switched from 1% to 10% FBS, or were cultured with boiled GCM or with astrocyte-conditioned medium (ACM), indicating that the secreted factor is glioma-specific and most likely protein in nature (Fig 1C).
Figure 1. GCM activated primary microglia.

Microglia were treated with GCM or DMEM+1% FBS (microglial medium) or 10% FBS (glioma medium) as controls. Cells were observed under bright field (A), or immunofluorescently stained with Iba1 antibody and labeled with DAPI (B), or lysed to obtain protein extracts for western blotting (C). Iba1 expression was normalized to α-tubulin. The experiment was repeated 3 times. Arrows: amoeboid microglia.
Microglia in culture modestly affect glioma cell growth
Microglia play a major role in innate and adaptive immune responses in the CNS. As part of the innate immune response, they phagocytose pathogens and release proteases, cytokines and molecules that function to protect the host (Kreutzberg, 1996); this function may be compromised in a glioma-harboring environment (Hussain et al., 2006). To directly assess the effect of glioma cells on microglial function, we co-cultured rhodamine-labeled microglia and glioma GL261-EGFP cells. As control, the microglia were also plated alone or with the CRL-2541 astrocytes. In setting up this experiment, we first did a dose response by varying the microglial numbers. We used 4×103, 8×103, 2×104, 4×104, 8×104, 2×105, 4×105cells and found that 4–8×104 was the optimal microglia concentration. If the microglia concentration was lower than 4×104 cells, they could not spread evenly in the well (they distributed along the margin of culture plates) and could not form sufficient contact with GL261-EGFP cells. If the concentration was higher than 8×104 cells, the density of microglia was too high and led to microglia forming a barrier against GL261-EGFP cell attachment. Then we kept the microglia concentration constant (5×104 cells) and varied the GL261-EGFP concentrations (5×103, 104, 2×104, 4×104, 8×104 cells). We found that 2×104 GL261-EGFP cells worked best in the co-culture system showing sufficient interactions with microglia and adequate growth in a well of a 12-well plate.
Microglia plated alone remained rod-like throughout the 5-day experiment (Fig. 2A). Microglia co-cultured with CRL-2541-EGFP cells exhibited modest signs of morphological activation and phagocytic activity throughout the 5-day period (Fig. 2B, arrows and insets where the red–microglia- channel has been removed to display the green (astrocytic) fluorescence within microglial cells, indicative of phagocytic events). In contrast, within one day of being co-plated with GL261 cells, the microglia exhibited activated morphology and their pseudopodia were in contact with the GL261 cells (Fig. 2C). At days 2 and 3, a few phagocytic microglia containing GFP fluorescence (arrows in figure 2C–D) could be observed. However, by day 4, phagocytosis of GL261 cells was observed only rarely.
Figure 2. Microglia-glioma co-culture.

5×104 rhodamine-labeled microglia (M) were plated in DMEM+1% FBS (microglial medium) for 48 hours before 2×104 CRL-2541-EGFP (B) or GL261-EGFP (C, D) were plated in the same well. DMEM+10% FBS was used as negative control. Confocal images (A–D) were taken and cell numbers of each population (E) were counted over 5 days. (A–D) Red cells: primary microglia; green cells: CRL-2541-EGFP or GL261-EGFP as indicated. Arrows: phagocytic microglia with GFP protein in cell body. (E) *** indicates the statistical analysis of GL261 alone and GL261 cell numbers in co-culture. The experiment was repeated 3 times. The scale bar in panels A–C is 50μm, in D 20μm.
We next examined the growth rate of GL261 and microglia in the co-culture system by counting the cell numbers of each population every 24-hours (Fig. 2E). Switching primary microglia into DMEM+10% FBS (the GL261 medium) did not stimulate proliferation, nor was there an effect of adding GL261 cells. In contrast, when GL261 cells were cultured together with microglia, the GL261 growth rate was modestly higher than that observed for GL261cells cultured alone (15.3% more cells on day 5, slope of growth curve without microglia, 4.2; with microglia, 4.9). These results suggest that the innate phagocytic response of microglia in culture is inhibited by GL261 cells, while microglia promote glioma growth.
Establishment of an in vivo mouse glioma model with local MG/MPablation
To better understand the in vivo microglial response to a developing glioma, we employed a model of local microglial ablation. The CD11b-HSVTK transgenic mice express the herpes simplex thymidine kinase (HSVTK) protein in monocytic cells such as MG/MP (Heppner et al., 2005). When these animals are exposed to ganciclovir (GCV), the cells that express HSVTK are eliminated. Systemic administration (100μg/g GCV i.p. every 2days) resulted in death of all proliferating monocytic cells, which led to severe anemia and death around 10days after the GCV injection (Heppner et al., 2005). We modified this protocol by performing local infusion of GCV (Mirrione et al., 2010) at the tumor injection site, which ablated MG/MP in and around the tumor.
To establish the optimal dose of GCV, we examined whether GCV affected GL261-EGFP growth in culture and in vivo. GL261-EGFP cells treated with 2.5μg/ml and 10μg/ml of GCV (every other day for 8days) exhibited no difference in proliferation in comparison to the vehicle-treated control group, whereas cell growth was significantly inhibited at 25μg/ml (data not shown). In the in vivo setting, concentrations up to 1.25mg/ml had no significant effect on tumor size at day 14 after tumor cell injection (Fig. 3A). Higher concentrations appeared to inhibit the rate of tumor growth, however (data not shown). These results suggested that sufficiently high concentrations of GCV affect GL261-EGFP growth, since all cells have intrinsic thymidine kinases; however the HSV thymidine kinases are ~1000-fold more sensitive to GCV. For subsequent in vivo experiments we thus used 1mg/ml GCV.
Figure 3. MG/MP ablation decreased glioma size and slowed down glioma progression.

(A) WT mice were treated with GCV 14 days after GL261 injection (n= 5). (B) Tumor sizes in WT and HSVTK mice were measured 14 days after GL261-EGFP injection and infusion of 1mg/ml GCV or saline (WT saline and GCV group: n=5; HSVTK saline and D0 GCV group: n=6; HSVTK D5 GCV group: n=4). (C) Tumor sizes were measured at different time points after GL261-EGFP injection and infusion of GCV or saline (WT saline group: day10 n=6, day14 n=5, day20 n=8; HSVTK D0 GCV group: day14 n=6, day28 n=3, day42 n=2). (D) Kaplan-Meier survival curves of tumor-bearing mice. WT saline group: n=10, HSVTK saline and D0 GCV groups: n=5. Saline group of WT and HSVTK mice: no significant difference; HSVTK GCV group vs. saline groups: P<0.01. (E–I) Tumor morphology and MG/MP infiltration was evaluated at each time point. Representative images of (E) H&E staining (scale bar=1000μm; the tumor is demarcated by the dashed line); (F–G) GFP fluorescence (F: scale bar=1000μm; G: scale bar=100μm); (H–I) Iba1 immunofluorescence (H: scale bar=100μm; I: scale bar=50μm).
To investigate the role of MG/MP in the temporal progression of glioma, we treated CD11b-HSVTK mice with GCV: one group received GCV immediately after tumor injection (D0 GCV group), and the other was treated with GCV five days after tumor implantation (D5 GCV group) to mimic an early stage glioma. Control animals were either WT treated with saline or with GCV or CD11b-HSVTK mice treated with saline.
MG/MPablation inhibits glioma progression
Administration of clodronate for depletion of microglia was reported to attenuate glioma invasion in organotypic brain slices (Markovic et al., 2005), potentially by depriving the glioma cells of microglia-produced MT1-MMP (Markovic et al., 2009). We next performed a detailed evaluation of tumor growth in the presence and absence of microglia. Using the experimental and control groups described above, we found that the tumor size in control mice (WT receiving saline or GCV, or CD11b-HSVTK receiving saline) was similar at 14 days after GL261 injection, whereas the tumors were dramatically reduced in MG/MP-depleted mice (P<0.001; Figure 3B). At day 14 after tumor implantation, the average tumor sizes in the D0 GCV group and D5 GCV group of CD11b-HSVTK mice were respectively 87.5% and 76.3% less than that of CD11b-HSVTK mice receiving only saline (1.6mm3 and 2.9mm3 versus 12.38mm3). Long-term effects of MG/MP depletion on glioma progression were also followed. Twenty days after tumor inoculation, WT mice were morbid with an average tumor size of 55.6mm3 (Figure 3C), and exhibited extensive brain swelling, hemorrhage, and weight loss. In contrast, tumors in HSVTK mice treated with D0 GCV were very small (~0.15mm3) 28 days after inoculation, and undetectable by day 42.
Survival curves demonstrated no difference between WT and CD11b-HSVTK mice treated with saline (median survival time 24 days and 22 days respectively, Fig. 3D). In contrast, all of the CD11b-HSVTK mice receiving GCV concomitant with tumor inoculation (HSVTK D0 GCV) survived. These results indicate that glioma growth is dependent on the presence of MG/MP in this model system.
Tumor morphology was evaluated using a combination of H&E-staining (Figure 3E), GFP-fluorescence to visualize tumor cells (Figures 3F–I), and anti-Iba1immunostaining to identify MG/MP (Figures 3H–I). Both saline- and GCV-treated WT mice as well as saline-treated CD11b-HSVTK (labeled as HSVTK) mice displayed intact glioma tissue with numerous MG/MP in and around the tumor (Figure 3H, I). GCV treatment of CD11b-HSVTK mice significantly decreased MG/MP, especially in the animals that received D0 pumps (although there was a significant decrease in those with the D5 pump as well). The decrease of MG/MP was accompanied by a reduction of tumor size as indicated by the limited amount of residual GFP signal. At late time points, day 20 and day 42, the tumor was almost undetectable.
The number of MG/MP, after an initial decrease at earlier time-points, increased later (day 28–42), and the microglial morphology indicated that MG/MP were activated, as they presumably were recruited to the injury site to clean up tissue debris (Figure 3H, I).
Exaggerated activation of MG/MPand glioma growth
Tuftsin (threonine-lysine-proline-arginine, TKPR), which is derived from the proteolytic degradation of immunoglobulin G (Nishioka et al., 1973), is a potent stimulator of MG/MP, enhancing the phagocytic activity, migration and antigen presentation of monocytic cells (Siemion and Kluczyk, 1999). Tuftsin has been widely used as an anti-tumor agent in animal cancer models (Banks et al., 1985; Noyes et al., 1981; Wleklik et al., 1986). GL261-EGFP cells cultured with tuftsin did not exhibit alterations in cell proliferation (data not shown). We thus examined whether tuftsin stimulation of microglia affected growth of GL261-EGFP cells, using a tissue-culture insert system to restrict the effects to soluble factors released by microglia. Microglia growing in inserts were placed in wells containing GL261-EGFP cells and control or tuftsin-supplemented medium, and the glioma cells counted every second day. When compared to GL261-EGFP alone, GL261-EGFP growth was modestly slower in the presence of microglia (Figure 4A); but after day 6 of co-culture, glioma proliferation was accelerated (P<0.01). Using a co-culture system with physical interaction between microglia and glioma, growth was increased at all timepoints (Figure 4B). Similarly incubation with tuftsin resulted in increased glioma cell numbers at the same rate as in the presence of microglia alone, therefore in this culture system tuftsin does not provide additional stimulation to microglial activation.
Figure 4. Different effects of tuftsin and MIF/TKP on GL261 growth in the presence of microglia.
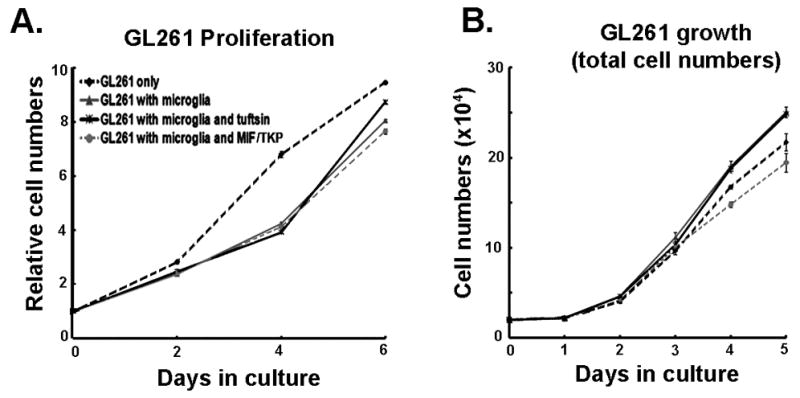
(A) Primary microglia on inserts were treated with 150μg/ml tuftsin or MIF/TKP. The inserts were placed in wells containing GL261 cells. GL261 proliferation was measured every 2 days. (B) GL261-EGFP cells were cultured with primary microglia in the presence of 150μg/ml tuftsin or MIF/TKP, and GL261-EGFP cell number was counted every day for 5 days. Experiments were repeated 3 times.
To assess effects on glioma growth in vivo, tuftsin was delivered either at day 0 or 5 after tumor injection to the implantation site. Tumor size was analyzed 14 days after tumor inoculation. Tuftsin at day 0 (immediately after tumor implantation) significantly increased the tumor size (mean=21.0mm3) compared to the average tumor size of the control group (mean=11.7mm3) (P<0.05), while tuftsin infusion starting 5 days after tumor injection had no significant effect (Table 1, Fig. 5A). Immunofluorescence for Iba1 revealed that tuftsin activated MG/MP around the infusion site in the absence of glioma cell implantation (supplementary figure 1). MG/MP numbers and Iba1 intensity increased in and around the tumor by tuftsin administration immediately after tumor cell injection (Figure 5B, C). When the proliferation (Ki67) and apoptosis (activated caspase-3) in the saline and D0 tuftsin group were quantified, tuftsin infusion increased the proliferation in tumor tissue volume and cell numbers compared to the saline control group (Table 2), and decreased the apoptosis of both tumor cells and GIMs (Table 2).
Table 1.
Tumor size after tuftsin and MIF/TKP treatment
| Treatment | Tumor size at Day 14 (mm3) |
|---|---|
| Saline | 11.16±2.01 |
| Tuftsin Day0 pump | 20.99±3.91*** |
| Tuftsin Day5 pump | 9.86±2.57 |
| MIF/TKP Day0 pump | 17.97±17.06 |
| MIF/TKP Day5 pump | 5.46±0.69** |
Tumor sizes were measured 14 days after GL261-EGFP injection and infusion of 250μg/ml tuftsin, MIF/TKP or saline.
Data are presented as mean±SEM. and
indicate the statistical analysis between the experimental group and saline control group.
Figure 5. Tuftsin and MIF/TKP treatment affect glioma progression in vivo.

(A) Tumor sizes and MG/MP infiltration (Iba1+ cells) were evaluated 14 days after GL261-EGFP injection and infusion of 250μg/ml tuftsin (TF), MIF/TKP or saline (scale bar 500μm). The proliferation and apoptosis of tumor cells and GIM in saline group, D0 tuftsin and D5 MIF/TKP groups were quantified with (B) Ki67 (scale bar 50μm) and (C) activated caspase-3 (scale bar 100μm) immunofluorescent staining (n = 3).
Table 2.
Quantification of Glioma Infiltrating Microglia/Macrophages (GIMs) density, proliferation and apoptosis
| Tumor treatment |
Saline | tuftsin | MIF/TKP |
|---|---|---|---|
| Measurement | |||
| GIM density (lectin+ cells/mm2) | 687±23.9 | 1014±176.8* | 495±51.7** |
| Total proliferation (Ki67+ cells/mm2) | 2354.7±225.0 | 2839.5±164.0* | 1386.0±219.7** |
| Tumor cell proliferation (Ki67+GFP+ cells/mm2) | 1848.1±130.3 | 2207.6±78.5** | 1322.7±267.9* |
| Total apoptosis (activated caspase 3+/mm2) | 48.6±4.6 | 28.0±0.9** | 60.4±5.2* |
| Tumor cell apoptosis (activated caspase 3+GFP+ clls/mm2) | 20.9±3.8 | 13.4±0.8* | 15.2±2.1 |
| GIM apoptosis (activated caspase 3+lectin+ cells/mm2) | 4.2±0.5 | 2.8±0.5* | 17.3±4.1** |
GIM density was measured by counting MG/MP number per field inside tumor area. The proliferation and apoptosis of tumor tissue, tumor cells and GIM in saline group, D0 tuftsin group and D5 MIF/TKP group were quantified by Ki67 and activated caspase-3 immunofluorescent staining (n = 3)
Data are presented as mean±SEM. and
indicate the statistical analysis between the experimental group and saline control group.
MIF/TKP decreases the growth of glioma cells in the presence of microglia
Based on our results and previous findings (Ginzkey et al., 2009), the size of gliomas in vivo appears to moderately increase upon administration of stimulators of MG/MP. We used therefore an inhibitor of MG/MP, MIF/TKP, to evaluate whether it would reduce glioma growth. MIF/TKP (tuftsin fragment 1–3) is a tripeptide, that blocks the activity of monocytic cells in vitro and in vivo (Thanos et al., 1993; Rogove and Tsirka, 1998; Bhasin et al., 2007). The infusion of MIF/TKP to the site of excitotoxic injury inhibits microglia activation, and MIF/TKP treatment of primary microglia inhibits lipopolysaccharide (LPS)-mediated TNF-α release (Rogove and Tsirka, 1998). As shown above, an indirect (segregated) and direct (physical interaction) co-culture system was utilized to assess the effects of MIF/TKP on glioma growth. In the indirect system, in the presence of MIF/TKP and microglia the growth of GL261-EGFP cells slightly slowed down 6 days after co-culture compared to that of GL261-EGFP cells with microglia only (P<0.05) (Figure 4A). In the direct co-culture in the presence of microglia, MIF/TKP treatment had a very modest effect on GL261-EGFP growth 2 days after co-culture compared to that of GL261-EGFP cells with microglia only (P<0.01) (Figure 4B). MIF/TKP-treated microglia resulted in lower total GL261-EGFP cell numbers than GL261 alone at day 4 (P<0.01). Since MIF/TKP had no effect on GL261-EGFP growth in the absence of microglia (not shown), the MIF/TKP-mediated inhibitory effect on glioma cell proliferation in microglial presence was probably due to attenuated microglial activation. These results were then explored in vivo: delivery of MIF/TKP 5 days after tumor injection inhibited tumor growth (Figure 5A) and MG/MP infiltration. The quantification of Ki67 (cell proliferation) and activated caspase-3 (cell death) in saline control mice and D5 MIF/TKP-treated mice revealed that MIF/TKP treatment inhibited glioma cell proliferation, but had little effect on glioma cell apoptosis (Figures 5B, C). However MIF/TKP induced apoptosis of GIMs, which may contribute to the lower level of MG/MP infiltration (Table 2).
Tuftsin and MIF/TKP treatment had different effects on survival time of tumor-bearing mice
Since D0 tuftsin and D5 MIF/TKP treatment can affect glioma progression, we examined the survival time of glioma-bearing mice with D0 tuftsin and D5 MIF/TKP pumps, which continuously delivered the drugs for 28 days. The Kaplan-Meier survival curve (Figure 6) revealed that the D0 tuftsin pump group survived significantly shorter time than the control group animals, while the D5 MIF/TKP group survived significantly longer (P<0.001). These results suggest that exaggerated MG/MP activation had a positive effect on glioma progression, while the inhibition of MG/MP was beneficial to the survival of mice with gliomas.
Figure 6. Kaplan-Meier survival curves of tumor-bearing mice with tuftsin or MIF/TKP (tuftsin fragment 1–3) treatment.
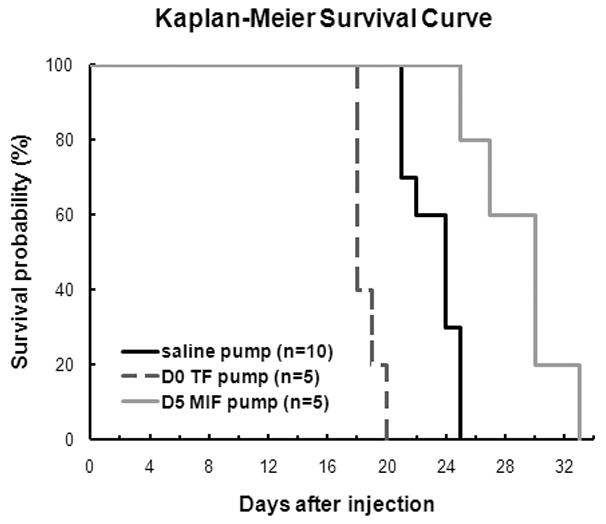
The survival time of tumor-bearing mice infused with D0 tuftsin pumps (n=5) and D5 MIF/TKP pumps (n=5) was compared to the D0 saline pumps (n=10). (P < 0.001)
Tuftsin and MIF/TKP treatment had different effects on CCL21 expression in glioma tissue
The chemokine CCL21 is expressed by many cancers, including breast cancer and melanoma, which was recently shown to function as an immune suppressive chemokine, facilitating the immune escape of tumors (Shields et al., 2007; 2010). Here we examined whether the tuftsin or MIF/TKP treatment could modulate CCL21 expression in glioma tissue. The immunofluorescent staining of CCL21 revealed that primarily stromal and some glioma cells expressed CCL21 (supplementary figure 2), while the adjacent normal brain tissue didn’t show any expression (Figure 7A). The D0 tuftsin group had more CCL21+ cells in glioma tissue compared to saline control group. In contrast, the D5 MIF/TKP group revealed a significant decrease in the number of CCL21+ cells (Figures 7A, B).
Figure 7. Tuftsin and MIF/TKP treatment had different effects on CCL21 expression in glioma tissue.

14 days after GL261-EGFP injection and infusion of 250μg/ml tuftsin (TF), MIF/TKP or saline, CCL21 expression was quantified by counting CCL21+ cells within the tumor tissue (B) (n=3). Representative images from tumor border and center shown in (A). Dashed lines indicate the tumor borders. (C) The tumor-promoting function of CCL21 was confirmed in microglia-glioma co-culture. (n = 4) ** and *** indicates the statistical significance between the cell numbers of GL261 in co-culture with and without 4μg/ml CCL21 antibody (ab). There is no significant difference among the cell numbers of GL261 alone, or cultured with 4μg/ml CCL21 antibody, or in co-culture with microglia and CCL21 antibody.
To further confirm the immunosuppressive role of CCL21 in glioma progression, we performed a microglia-glioma co-culture experiment in the presence of 4μg/ml CCL21 neutralizing antibody (CCL21 ab) and counted the GL261-EGFP cell numbers over 5 days (Figure 7C). When GL261-EGFP cells were cultured alone, the inhibition of CCL21 had no effect on GL261-EGFP growth. However, the presence of CCL21 ab in microglia-glioma co-culture abolished the glioma-promoting effect of microglia, indicating that CCL21 is a key regulator of the interactions between microglia and glioma cells.
Taken together, these results suggest that modulating microglial activation, potentially using MIF/TKP, may be beneficial for the treatment of glioma by attenuating the immune microenvironment surrounding the gliomas.
Discussion
The components of tumor mass are tumor and stromal cells, which interact with each other and ultimately affect tumor growth. Among the stromal cells, macrophages have been shown to have dual roles, which are both tumor-rejecting and tumor-promoting (Allavena et al., 2008; Solinas et al., 2009). Macrophages secrete anti-tumor cytokines and interact with T cells to destroy tumor cells (Galani et al., 2009). On the other hand they can be recruited by tumor-released chemokines and switch to tumor-supporting behavior in the tumor microenvironment (Demuth and Berens, 2004; Pollard, 2004).
Glioma cells release factors such as colony stimulating factor-1, granulocyte-macrophage stimulating factor, monocyte chemoattractant protein-1, and hepatocyte growth factor/scatter factor, which recruit and promote the growth of tumor-infiltrating MG/MP (Alterman and Stanley, 1994; Badie et al., 1999; Kielian et al., 2002; Leung et al., 1997; Nitta et al., 1992). Moreover, tumor-infiltrating MG/MP are commonly activated and proliferate in gliomas (Klein and Roggendorf, 2001). In our study, we show that glioma conditioned medium, but not astrocyte-derived medium, activated microglia, as previously reported (Klein and Roggendorf, 2001). In the GL261 mouse glioma model, as in human glioma specimens, activated MG/MP infiltrated within and around the tumor, and the density of MG/MP in the tumor area was higher than the MG/MP density that surrounded relatively normal brain tissue.
MG/MP are crucial to CNS innate and adaptive immune responses. Patients with gliomas often have deficiency in immunologic responses against the tumor (Morford et al., 1997). Although up to one third of all cells in glioma specimens were MG/MP, T cells were rarely seen in gliomas (Morimura et al., 1990; Roggendorf et al., 1996). Independent studies showed that the microglial antigen-presenting function was compromised in gliomas, since the major histocompatibility complex (MHC) class II molecules and co-stimulatory molecules exhibited decreased expression levels (Badie et al., 2002; Flugel et al., 1999). The studies on GIM innate immune responses demonstrated that GIMs expressed Toll-like receptors (TLR) and mediated phagocytosis, but lacked IL-1β and TNF-α production, which are important cytokines for tumor rejection (Frei et al., 1994; Hussain et al., 2006). Moreover in a CD11b-HSVTK glioma model, Markovic et al. (2009) reported that activation of MMPs in glioma is promoted via activation of TLRs and MT1-MMP in microglia. Our data shed light on the MG/MP innate immune response against gliomas. When microglia encountered glioma cells in culture, they first became activated, but at later timepoints phagocytosis was rarely observed and the growth of glioma cells was unaffected by microglia, suggesting the presence of immunosuppressive environment within gliomas. The components contributing to this suppression could be anti-inflammatory cytokines released by glioma cells, such as interleukin (IL)-6, IL-11, leukemia inhibitory factor, oncostatin-M, and TGF-β, which can inhibit cytotoxic T-cell activation and MG/MP activation (Constam et al., 1992; Goswami et al., 1998; Halfter et al., 1998; Hao et al., 2002; Murphy et al., 1995).
On the other hand, glioma infiltrating microglia/macrophages (GIMs) release many factors, such as cytokines, growth factors and proteases, which directly or indirectly influence tumor progression (Graeber et al., 2002). It has been shown that GL261 cell migration occurred earlier and faster in the presence of primary microglia or microglia-conditioned medium, which indicated that microglia-released soluble factors can promote glioma migration (Bettinger et al., 2002). GIMs are the major source of IL-10, an immunosuppressive cytokine that promotes glioma proliferation and migration (Huettner et al., 1997; Wagner et al., 1999). Moreover, the glioma- and GIM-derived TGF-β has multiple effects on glioma progression: it can inhibit microglia proliferation and release of cytokines in vitro (Suzumura et al., 1993), can also induce metalloproteinase (MMPs) expression and promote the invasion of glioma (Markovic et al., 2009; Wesolowska et al., 2008; Wick et al., 2001). Both GIMs and glioma cells secrete vascular endothelial growth factor (Lafuente et al., 1999; Tsai et al., 1995) that mediates angiogenesis. These findings suggested that gliomas recruit MG/MP, inhibit the immune response and induce the MG/MP production of tumor survival factors, which in turn facilitates glioma growth and malignancy.
Our experiments further supported this hypothesis and provide new evidence of the tumor-promoting function of GIMs. In vivo, we demonstrated that GIM depletion led to significant decrease in glioma size and prolonged survival of tumor-bearing mice. The results presented here agree with recent data (Markovic et al., 2009), but provide more detailed characterization of the model. Since GIMs are a significant source of tumor survival factors, GIM loss may result in decrease of immunosuppressive cytokines, growth factors and proteases, which are essential for glioma growth.
It was shown that macrophage depletion in CD11b-TKmt-30 mice via systemic GCV injection resulted in glioma growth increase (Galarneau et al., 2007). These results agree with the early-time points of our data, but the final outcome is different. The difference may lie in the experimental system (administration route of GCV, reconstitution with bone-marrow derived wild-type cells).
The recent treatments for glioma are–among others - manipulations of the activity of tumor-associated macrophages, including inhibition of their recruitment to and survival in tumor tissue and restoration of their anti-tumor immunity (Allavena et al., 2005; Sessa et al., 2005; Wu et al., 2009). However, the development of therapeutic anti-glioma options by modulating the activity of GIM has not been thoroughly investigated. Glioma cells used in animal studies are of different immunogenicity and thus probably a reason for controversial results in the literature. For example, the injection of CpG ODN to GL261 mouse glioma cells resulted in contradictory data in a 9L rat glioma model (El Andaloussi et al., 2006; Ginzkey et al., 2009), which suggests that MG/MP stimulators may have different effects on MG/MP activities depending on the tumor microenvironment. Our results suggest that tuftsin will not be an effective treatment option for glioma patients. Although tuftsin has been widely used in anticancer studies (Banks et al., 1985; Noyes et al., 1981; Wleklik et al., 1986), its infusion into the glioma model presented here exaggerated tumor growth and shortened the survival time of tumor-bearing mice. Tuftsin activates monocytes and macrophages leading to IL-1, TNF-α and nitric oxide release (Cillari et al., 1994; Robey et al., 1987; Wleklik et al., 1987), which possibly contributed to the initial slowing down of glioma growth. However, due to the immunosuppressive environment within gliomas, the activation effects of tuftsin on GIMs may switch them towards a tumor-promoting phenotype at later stages. Similar results were obtained in a mouse model of multiple sclerosis, where tuftsin infusion altered the host immune response towards favoring the expression of immunosuppressive Th2 genes (Bhasin et al., 2007).
In contrast, the presence of MIF/TKP (tuftsin fragment 1–3) and microglia slowed down glioma cell growth. Delivery of MIF/TKP to the tumor inhibited tumor proliferation and induced GIM apoptosis, in agreement with the current trend of tumor therapy (Allavena et al., 2005; Meng et al., 2010; Miselis et al., 2008). Similarly, it was shown that MIF/TKP delivery to the injury site after intracerebral hemorrhage (ICH) inhibited MG/MP infiltration and activation, and afforded neuroprotection (Wang and Tsirka, 2005).
One possible mechanism contributing to the above results could be the change of CCL21 expression level in glioma tissue. A recent report (Shields et al., 2010) showed that melanoma cells could shift their microenvironment to an immune tolerant one by expressing CCL21. This led to the formation of lymphoid-like stromal structures, increased the levels of TGF-β1 and the number of myeloid-derived suppressor cells. Melanoma cells upregulated CCL21 in stromal cells, and this induced expression was a major contributor to an immunotolerant microenvironment, which resulted in promotion of melanoma progression. Consistently, we found that addition of CCL21 neutralizing antibody to microglia-glioma co-culture inhibited the glioma-promoting effects of microglia, suggesting that CCL21 is an important mediator of glioma growth. Previously it has been shown that CCL21 is up-regulated in neurons after injury, which activates microglia through the chemokine receptor CXCR3 and induces microglia migration (Biber et al., 2001; de Jong et al., 2005; Rappert et al., 2002). We found here that glioma and surrounding stromal cells expressed CCL21 two weeks after glioma inoculation, which suggested that the glioma microenvironment is immune suppressive. Moreover, tuftsin treatment increased CCL21 levels in the glioma tissue, whereas MIF/TKP treatment had the opposite effect. In GCV-treated CD11b-HSVTK mice, the expression of CCL21 was not detectable (data not shown), indicating that the modulation of GIM activities can possibly change the tumor microenvironment and modulate tumor progression.
Taken together, our data demonstrate that gliomas activate microglia and suppress immune responses (phagocytosis) by microglia, even when microglia are activated. On the other hand we find that microglia only modestly stimulate glioma growth in culture, even when activated (or inhibited); nonetheless, glioma growth in vivo is profoundly affected by microglia activation. We think that this effect is due to the environment, as CCL21 is upregulated to immunoprotect tumors, while in absence of microglia (or microglial activation), no immune protection is afforded to the tumor. GIMs promote glioma growth and invasion, and, GIMs depletion or pharmacological inhibition is in the longer-term beneficial to glioma-bearing animals. Thus, inhibiting GIM activity by local delivery of factors such as MIF/TKP may be a potential novel interventional, adjuvant regimen to tackle gliomas.
Supplementary Material
Acknowledgments
We would like to thank Drs Michael Frohman and Howard Crawford for critical reading of the manuscript. We thank Diandra Ayala and Dorothy Konomos for setting the glioma cell lines and injection parameters. This work was supported by National Institutes of Health, RO1NS42168 to SET, R01NS046006 and DFG SFB TRR43 and Exc 25 to FLH.
References
- Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65(7):2964–71. doi: 10.1158/0008-5472.CAN-04-4037. [DOI] [PubMed] [Google Scholar]
- Alterman RL, Stanley ER. Colony stimulating factor-1 expression in human glioma. Mol Chem Neuropathol. 1994;21(2–3):177–88. doi: 10.1007/BF02815350. [DOI] [PubMed] [Google Scholar]
- Badie B, Bartley B, Schartner J. Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J Neuroimmunol. 2002;133(1–2):39–45. doi: 10.1016/s0165-5728(02)00350-8. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner J, Klaver J, Vorpahl J. In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery. 1999;44(5):1077–82. doi: 10.1097/00006123-199905000-00075. discussion 1082–3. [DOI] [PubMed] [Google Scholar]
- Banks RA, Nishioka K, Kazazi F, Babcock GF. Effect of tuftsin on in vivo development of 3-methylcholanthrene-induced primary fibrosarcoma and Lewis lung carcinoma in mice. J Natl Cancer Inst. 1985;74(5):1079–83. [PubMed] [Google Scholar]
- Bettinger I, Thanos S, Paulus W. Microglia promote glioma migration. Acta Neuropathol. 2002;103(4):351–5. doi: 10.1007/s00401-001-0472-x. [DOI] [PubMed] [Google Scholar]
- Bhasin M, Wu M, Tsirka SE. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunol. 2007;8:10. doi: 10.1186/1471-2172-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HW. Ischemia-induced neuronal expression of the microglia attracting chemokine Secondary Lymphoid-tissue Chemokine (SLC) Glia. 2001;34(2):121–33. doi: 10.1002/glia.1047. [DOI] [PubMed] [Google Scholar]
- Cillari E, Arcoleo F, Dieli M, D’Agostino R, Gromo G, Leoni F, Milano S. The macrophage-activating tetrapeptide tuftsin induces nitric oxide synthesis and stimulates murine macrophages to kill Leishmania parasites in vitro. Infect Immun. 1994;62(6):2649–52. doi: 10.1128/iai.62.6.2649-2652.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148(5):1404–10. [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25(33):7548–57. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis LM. Brain tumors. N Engl J Med. 2001;344(2):114–23. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70(2):217–28. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- Demuth T, Reavie LB, Rennert JL, Nakada M, Nakada S, Hoelzinger DB, Beaudry CE, Henrichs AN, Anderson EM, Berens ME. MAP-ing glioma invasion: mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol Cancer Ther. 2007;6(4):1212–22. doi: 10.1158/1535-7163.MCT-06-0711. [DOI] [PubMed] [Google Scholar]
- El Andaloussi A, Sonabend AM, Han Y, Lesniak MS. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia. 2006;54(6):526–35. doi: 10.1002/glia.20401. [DOI] [PubMed] [Google Scholar]
- Flugel A, Labeur MS, Grasbon-Frodl EM, Kreutzberg GW, Graeber MB. Microglia only weakly present glioma antigen to cytotoxic T cells. Int J Dev Neurosci. 1999;17(5–6):547–56. doi: 10.1016/s0736-5748(99)00020-9. [DOI] [PubMed] [Google Scholar]
- Frei K, Malipiero U, Piani D, Fontana A. Microglia and tumor rejection. Neuropathol Appl Neurobiol. 1994;20(2):206–8. [PubMed] [Google Scholar]
- Galani IE, Wendel M, Stojanovic A, Jesiak M, Muller MM, Schellack C, Suri-Payer E, Cerwenka A. Regulatory T cells control macrophage accumulation and activation in lymphoma. Int J Cancer. 2009 doi: 10.1002/ijc.25132. [DOI] [PubMed] [Google Scholar]
- Galarneau H, Villeneuve J, Gowing G, Julien JP, Vallieres L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007;67(18):8874–81. doi: 10.1158/0008-5472.CAN-07-0177. [DOI] [PubMed] [Google Scholar]
- Galasso JM, Stegman LD, Blaivas M, Harrison JK, Ross BD, Silverstein FS. Experimental gliosarcoma induces chemokine receptor expression in rat brain. Exp Neurol. 2000;161(1):85–95. doi: 10.1006/exnr.1999.7249. [DOI] [PubMed] [Google Scholar]
- Ginzkey C, Eicker SO, Marget M, Krause J, Brecht S, Westphal M, Hugo HH, Mehdorn HM, Steinmann J, Hamel W. Increase in tumor size following intratumoral injection of immunostimulatory CpG-containing oligonucleotides in a rat glioma model. Cancer Immunol Immunother. 2009 doi: 10.1007/s00262-009-0771-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Gupta A, Sharma SK. Interleukin-6-mediated autocrine growth promotion in human glioblastoma multiforme cell line U87MG. J Neurochem. 1998;71(5):1837–45. doi: 10.1046/j.1471-4159.1998.71051837.x. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40(2):252–9. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- Halfter H, Kremerskothen J, Weber J, Hacker-Klom U, Barnekow A, Ringelstein EB, Stogbauer F. Growth inhibition of newly established human glioma cell lines by leukemia inhibitory factor. J Neurooncol. 1998;39(1):1–18. doi: 10.1023/a:1005901423332. [DOI] [PubMed] [Google Scholar]
- Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol. 2002;103(2):171–8. doi: 10.1007/s004010100448. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11(2):146–52. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997;17(5A):3217–24. [PubMed] [Google Scholar]
- Hussain SF, Yang D, Suki D, Grimm E, Heimberger AB. Innate immune functions of microglia isolated from human glioma patients. J Transl Med. 2006;4:15. doi: 10.1186/1479-5876-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57(1):1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Kielian T, van Rooijen N, Hickey WF. MCP-1 expression in CNS-1 astrocytoma cells: implications for macrophage infiltration into tumors in vivo. J Neurooncol. 2002;56(1):1–12. doi: 10.1023/a:1014495613455. [DOI] [PubMed] [Google Scholar]
- Klein R, Roggendorf W. Increased microglia proliferation separates pilocytic astrocytomas from diffuse astrocytomas: a double labeling study. Acta Neuropathol. 2001;101(3):245–8. doi: 10.1007/s004010000286. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lafuente JV, Adan B, Alkiza K, Garibi JM, Rossi M, Cruz-Sanchez FF. Expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor-beta (PDGFR-beta) in human gliomas. J Mol Neurosci. 1999;13(1–2):177–85. doi: 10.1385/JMN:13:1-2:177. [DOI] [PubMed] [Google Scholar]
- Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, Linet MS. Cancer surveillance series [corrected]: brain and other central nervous system cancers: recent trends in incidence and mortality. J Natl Cancer Inst. 1999;91(16):1382–90. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol. 1997;93(5):518–27. doi: 10.1007/s004010050647. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol. 2005;64(9):754–62. doi: 10.1097/01.jnen.0000178445.33972.a9. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kalin R, van Rooijen N, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106(30):12530–5. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, Weichselbaum RR. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70(4):1534–43. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrione MM, Konomos DK, Gravanis I, Dewey SL, Aguzzi A, Heppner FL, Tsirka SE. Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiol Dis. 2010;39(1):85–97. doi: 10.1016/j.nbd.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 2008;7(4):788–99. doi: 10.1158/1535-7163.MCT-07-0579. [DOI] [PubMed] [Google Scholar]
- Morford LA, Elliott LH, Carlson SL, Brooks WH, Roszman TL. T cell receptor-mediated signaling is defective in T cells obtained from patients with primary intracranial tumors. J Immunol. 1997;159(9):4415–25. [PubMed] [Google Scholar]
- Morimura T, Neuchrist C, Kitz K, Budka H, Scheiner O, Kraft D, Lassmann H. Monocyte subpopulations in human gliomas: expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990;80(3):287–94. doi: 10.1007/BF00294647. [DOI] [PubMed] [Google Scholar]
- Morris CS, Esiri MM. Immunocytochemical study of macrophages and microglial cells and extracellular matrix components in human CNS disease. 1. Gliomas. J Neurol Sci. 1991;101(1):47–58. doi: 10.1016/0022-510x(91)90017-2. [DOI] [PubMed] [Google Scholar]
- Murphy GM, Jr, Bitting L, Majewska A, Schmidt K, Song Y, Wood CR. Expression of interleukin-11 and its encoding mRNA by glioblastoma cells. Neurosci Lett. 1995;196(3):153–6. doi: 10.1016/0304-3940(95)11862-q. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Sato PS, Constantopoulos A, Najjar VA. The chemical synthesis of the phagocytosis-stimulating tetrapeptide tuftsin (Thr-Lys-Pro-Arg) and its biological properties. Biochim Biophys Acta. 1973;310(1):230–7. doi: 10.1016/0005-2795(73)90025-1. [DOI] [PubMed] [Google Scholar]
- Nitta T, Sato K, Allegretta M, Brocke S, Lim M, Mitchell DJ, Steinman L. Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Res. 1992;571(1):19–25. doi: 10.1016/0006-8993(92)90505-4. [DOI] [PubMed] [Google Scholar]
- Noyes RD, Babcock GF, Nishioka K. Antitumor activity of tuftsin on murine melanoma in vivo. Cancer TreatRep. 1981;65(7–8):673–5. [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H. Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol. 2002;168(7):3221–6. doi: 10.4049/jimmunol.168.7.3221. [DOI] [PubMed] [Google Scholar]
- Robey FA, Ohura K, Futaki S, Fujii N, Yajima H, Goldman N, Jones KD, Wahl S. Proteolysis of human C-reactive protein produces peptides with potent immunomodulating activity. J Biol Chem. 1987;262(15):7053–7. [PubMed] [Google Scholar]
- Roggendorf W, Strupp S, Paulus W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996;92(3):288–93. doi: 10.1007/s004010050520. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8(1):19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Sessa C, De Braud F, Perotti A, Bauer J, Curigliano G, Noberasco C, Zanaboni F, Gianni L, Marsoni S, Jimeno J, et al. Trabectedin for women with ovarian carcinoma after treatment with platinum and taxanes fails. J Clin Oncol. 2005;23(9):1867–74. doi: 10.1200/JCO.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11(6):526–38. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749–52. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- Siao CJ, Tsirka SE. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci. 2002;22(9):3352–8. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemion IZ, Kluczyk A. Tuftsin: on the 30-year anniversary of Victor Najjar’s discovery. Peptides. 1999;20(5):645–74. doi: 10.1016/s0196-9781(99)00019-4. [DOI] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57(6):563–81. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Sawada M, Yamamoto H, Marunouchi T. Transforming growth factor-beta suppresses activation and proliferation of microglia in vitro. J Immunol. 1993;151(4):2150–8. [PubMed] [Google Scholar]
- Thanos S, Mey J, Wild M. Treatment of the adult retina with microglia-suppressing factors retards axotomy-induced neuronal degradation and enhances axonal regeneration in vivo and in vitro. J Neurosci. 1993;13(2):455–66. doi: 10.1523/JNEUROSCI.13-02-00455.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JC, Goldman CK, Gillespie GY. Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB, and bFGF. J Neurosurg. 1995;82(5):864–73. doi: 10.3171/jns.1995.82.5.0864. [DOI] [PubMed] [Google Scholar]
- Ullrich O, Diestel A, Eyupoglu IY, Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3(12):1035–42. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- Wagner S, Czub S, Greif M, Vince GH, Suss N, Kerkau S, Rieckmann P, Roggendorf W, Roosen K, Tonn JC. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int J Cancer. 1999;82(1):12–6. doi: 10.1002/(sici)1097-0215(19990702)82:1<12::aid-ijc3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005;36(3):613–8. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion--an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27(7):918–30. doi: 10.1038/sj.onc.1210683. [DOI] [PubMed] [Google Scholar]
- Wick W, Platten M, Weller M. Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J Neurooncol. 2001;53(2):177–85. doi: 10.1023/a:1012209518843. [DOI] [PubMed] [Google Scholar]
- Wleklik M, Levy SB, Luczak M, Najjar VA. Suppression of Friend virus-induced leukaemia in mice by tuftsin. J Gen Virol. 1986;67 (Pt 9):2001–4. doi: 10.1099/0022-1317-67-9-2001. [DOI] [PubMed] [Google Scholar]
- Wleklik MS, Luczak M, Najjar VA. Tuftsin induced tumor necrosis activity. Mol Cell Biochem. 1987;75(2):169–74. doi: 10.1007/BF00229905. [DOI] [PubMed] [Google Scholar]
- Wu QL, Buhtoiarov IN, Sondel PM, Rakhmilevich AL, Ranheim EA. Tumoricidal effects of activated macrophages in a mouse model of chronic lymphocytic leukemia. J Immunol. 2009;182(11):6771–8. doi: 10.4049/jimmunol.0801847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.