

Overview
The etiology of multiple sclerosis (MS) remains elusive. However, clues to its pathogenesis have traditionally been derived from the basic pathologic characterization of central nervous system (CNS) tissues of MS patients. While ongoing debate lingers over the autoimmune nature of MS, it is well established that the immune system directly participates in the destruction of myelin and nervous cells. Understanding the mechanisms of immune-mediated destruction of CNS components in MS promises to not only promote effective design of MS therapeutics, but also provides a broader understanding of immune-mediated diseases affecting the CNS.
This review will explore the principle features of the immuno-pathology of MS, in particular of relapsing-remitting MS (RR-MS). Herein, emerging concepts in the pathogenesis of MS in the context of known features of pathology have been highlighted. These include the characterization of cytokine networks promoting inflammatory damage of the CNS, B cell involvement, and inflammatory damage of axons and neurons. An effort has been made to preferentially focus on MS rather than animal models of the disease, such as experimental autoimmune encephalomyelitis (EAE). For a more comprehensive examination of data derived from studies on animal models, readers are referred to other outstanding reviews 52,53. From human studies alone, it is clear that the past 5–10 years have been highly productive in advancing the understanding of the pathogenesis of MS. For both the general neurologist and MS specialist, fundamental appreciation of the immunopathology of MS should lead to a broader understanding of the disease course and the emerging MS therapeutics that are now more than ever tailored to the intricacies of the immune system.
Pathologic characterization of MS lesions
Plaques of inflammatory demyelination within the CNS are the pathologic hallmark of MS 29,32,47. Myelin destruction is an essential element of the plaque. Yet the MS plaque is not simply a static entity of myelin loss in isolation; rather, lesions are comprised of a wide variation of immunologic and pathologic features. These features have been categorized in an effort to understand the neural-immune mechanisms underlying MS. Constructing a framework around which myelin loss and neuronal/axonal injury occur entails a close examination of the cellular and molecular constituents, timing of damage, and repair processes. Thus, the various features of plaques provide a platform on which hypotheses regarding pathogenic mechanisms underlying MS have been formulated for over a century.
Traditionally, MS plaque classification has been based on temporal progression, or stages, of inflammatory destruction. Accordingly, acute, chronic active, and chronic silent lesions are thought to occur along a continuous timeline, eventually producing the scarred and hardened areas within the CNS that can be appreciated grossly (FIGURE 1). The acute MS plaque represents the earliest stage of lesion formation. It is typified by robust inflammatory infiltration combined with demyelination distributed throughout the lesion 47,54,98 (FIGURE 1). Typical features of the acute plaque include ill-defined margins of myelin loss, infiltration of immune cells and parenchymal edema 47. The constituents of immune cell influx centered around vessels (termed perivascular cuffing) include lymphocytes (predominantly T cells), monocytes and macrophages. A portion of major histocompatibility class II (MHCII)-expressing cells, distributed evenly throughout the lesion, are loaded with lipids (foamy macrophages; FIGURE 1) and participate in active stripping of myelin from axons. While oligodendrocyte apoptosis has been observed 61, the degree of oligodendrocyte loss within active lesions can be variable 54. In spite of the relative degree of axonal sparing, axonal injury can be extensive in acute lesions (see section below; 129). Glial reactivity throughout the lesion is noted, particularly hypertrophic astrocytes. However, dense glial scarring is not typical of the acute plaque.
Figure 1.

Gross and histologic features of MS plaques. (A) Gross examination of the brain at autopsy of a 79 year old with RR-MS. A dorsal view of the corpus callosum after separation of the cerebral hemispheres reveals a hardened, discolored area within the body of the corpus callosum (arrow). Photo courtesy of Dr. Robert Schmidt. (B) Acute MS plaque stained with Hematoxylin and Eosin revealing hypercellularity due to the perivascular and parenchymal infiltration of leukocytes. (C) Luxol fast blue/periodic acid–Schiff (LFB-PAS) stained section of a plaque margin reveals a blurred but discrete edge (arrows). (D) Inactive plaque demonstrating borders that are distinct and devoid of inflammation (E) CD3 + lymphocytes clustering in a perivascular cuff within an active lesion area (F) Foamy macrophages characterized by fragments of myelin (arrow) engulfed by macrophages are identified using LFB-PAS at a plaque margin. Images in B-F used with permission; 98 (copyright Elsevier, 2010).
The chronic plaque is characterized by a region of hypocellularity with loss of myelin and glial scarring. On gross examination of post-mortem tissue, the hardened and discolored appearance of chronic plaques is often appreciable in frequently targeted areas of the CNS (e.g. the corpus callosum; FIGURE 1). Histologically, the lesion borders of chronic plaques are more distinct than those of acute plaques (FIGURE 1). A division into two forms of chronic plaque is made to signify temporal evolution from active destruction at the edge of the lesion (chronic active plaque) to an entirely “burned out” lesion devoid of active inflammatory destruction (chronic silent plaque). In chronic active lesions, inflammation continues along the outer border with the histologic appearance comparable to acute lesions 54. Thus, borders of the chronic active plaque are populated with activated microglia and macrophages, vessels demonstrating perivascular cuffing, and reactive astrocytes. The presence of antibody and complement are more prominent in chronic active lesions. Areas of re-myelination are often observed on the edge of lesions, but can encompass the entire of lesion 77. The core of the chronic plaque is typically hypocellular, though, and often contains thickened vessels with enlarged perivascular spaces. Chronic silent lesions are characterized by loss of the inflammatory traits along the border of chronic active lesions. Remyelination and the presence of oligodendrocyte progenitors are uncommon 136. Essentially, the chronic silent plaque is “burned out”, containing minimal inflammation and devoid of the active inflammatory border. This is accompanied by a complete loss of oligodendrocytes and a variable but demonstrable reduction in axonal density (FIGURE 1). Overall, the gross and histologic features of the MS plaque imply a complex progression of inflammatory damage culminating in a scarred region of demyelination.
Extensive variability across MS plaques exists, including different degrees of inflammation, demyelination, re-myelination and axonal injury. Controversy has emerged in the “taxonomy” by which lesions are grouped, possibly in part as a result of studying a combination of autopsy and biopsy tissues at different stages of the disease. A relatively recent attempt was made to re-define the process of categorizing MS lesions by proposing a system for classification based on suspected pathologic mechanisms. Alongside a temporally-based system, Lucchinetti et al described various types of lesions based on the pattern of leukocyte markers, myelin proteins, immunoglobulin and complement within lesions 79. From 83 pathologic specimens obtained at autopsy or from biopsy, four distinct patterns of immunologic and pathologic features of actively demyelinating lesions were discerned, patterns II and III being the most common. Pattern I is characterized by the predominance of T cell and macrophage inflammatory content; pattern II is characterized by T cell and macrophage infiltration, along with accentuated immunoglobulin deposition and myelin degradation products within macrophages; pattern III is distinguished by pronounced oligodendrocyte loss at the active edge of the lesion and preferential loss of myelin-associated glycoprotein (MAG); and pattern IV is characterized by oligodendrocyte dystrophy and the absence of remyelination or shadow plaques. Importantly, these distinct patterns were consistently observed within samples from the same patient, but varied between patients. The authors, based on the results from one open-label clinical trial in patients who had brain biopsies, hypothesized that deficit recovery after plasma exchange was indicative of the individual pattern of MS pathology. Plasma exchange was effective in patients with biopsy-proven pattern II (with prominent antibody and complement involvement) but not for patients with pattern I or III lesions 66. Overall, categorization of MS lesions based on immuno-pathologic mechanisms has added to the complexity of paradigms for lesion classification.
While the thought that distinct pathologic processes underlie a singular clinical entity referred to as MS is attractive, this concept has recently been disputed. In particular, challenges have emerged to the idea that there is a clear delineation between the different patterns of pathology 106. Prineas and colleagues have reported acute lesions from the same patient consisting of features from several of the four pathologic patterns 12. Furthermore, in a recent study, 131 tissue specimens from 39 MS patients all had lesions with little inter-individual variation in pathologic features. Indeed, lesions from this study exhibited traits consistent with inclusion into pattern II (i.e. deposition of complement and antibodies in proximity to macrophages and the absence of oligodendrocyte apoptosis) 22. Therefore, various pathologic patterns of disease may more precisely refer to the stage of lesion; the relevance of the classification system based on four individual subtypes of plaques remains to be confirmed.
The timing of lesion evaluation may be one critical factor in reconciling the conflicting data described above. One hypothesis proposed regarding initial events during plaque genesis is that early intrinsic oligodendrocyte injury leads to the inflammatory damage historically associated with the pathologic features of MS plaques. Thus, toxic insults directly affecting the oligodendrocyte could serve as a trigger for a common immuno-pathologic pathway associated with evolution into later stages of the plaque. That 30 out of 39 cases examined by Breij et al 22 were from patients who were in the progressive phase of disease would suggest that early heterogeneity of lesions could have been missed. Further support of this hypothesis comes from a study in which 26 active lesions were examined from 15 patients at autopsy, 11 of whom were early in the course of their disease 61. In this study, tissue immediately adjacent to lesion borders showed microscopic evidence of cellular injury without the presence of immune infiltration. Prineas and colleagues speculate that there are possibly toxic factors diffusible to the edge of the lesion resulting in oligodendrocyte fragility, and refer to these areas of initial injury as “pre-phagocytic” 61. Therefore, a critical question remaining in the field of neuropathology is whether differences exist between borders of acute versus chronic active lesions. Further work is required in order to more clearly identify white matter within the CNS that is undergoing early changes or at risk for doing so and how this relates to features described in more established MS plaques.
Normal appearing white matter
There has been a long-standing interest in early white matter changes in MS, with some of the earliest studies on myelin examining tissue outside of plaque regions that appears grossly unaffected 7,123. More recently, investigation into normal appearing white matter (NAWM) defined on conventional MRI sequences has been explored extensively using a variety of novel neuro-imaging techniques that show abnormalities in these areas suggestive of decreased myelin integrity and diminished axonal density within non-lesional regions 44,141 27.
Histo-pathologic examination of NAWM in patients with MS also support the concept that areas outside of plaques have immuno-pathologic changes. Microglial activation, T cell infiltration and perivascular cuffing have been reported in NAWM 1. 72. Not surprisingly, these features were found more diffusely in cases of progressive MS. As a transition zone, dirty appearing white matter (DAWM) - defined by MRI as having intermediate signal intensity between NAWM and T2 lesions - showed diffuse loss of myelin histologically 88. Thus, greater involvement of NAWM in MS, associated with the progression of disease, may be related to the extent to which inflammation extends beyond focal lesions 72. Overall, the extent of white matter abnormalities and the immuno-pathologic features of NAWM have yet to be precisely defined.
Gray matter plaques
MS plaques are not confined to the white matter. Gray matter lesions are detected by MRI and by examination of pathologic specimens 50. Almost all of the gray matter nuclei within the CNS can be affected, as observed in a cohort of mostly progressive MS patients 51, but out of several regions of the CNS, including motor cortex, the spinal cord and cerebellum are particularly vulnerable, resulting in demyelination in up to 28.8% of the gray matter on average. As might be expected, inflammatory lesions within the gray matter are associated with neuronal loss and transected axons, which are more common in active lesions 99. While gray matter lesions seem to be more common in patients with progressive forms of MS 72, they can develop early in the disease process and possibly account for some of the disability seen in MS patients that cannot be explained by white matter lesions. For example, lesions located within the gray matter correlate better with cognitive disability than white matter lesions 3.
There are several unique features of pathology associated with gray matter lesions. Histologically, gray matter lesions are less inflammatory with fewer infiltrating T-lymphocytes and microglia/macrophages 99. Purely cortical gray matter lesions have also been described as lacking complement deposition 23 and blood-brain barrier breakdown 132. Whether gray matter plaques arise from distinct immunologic mechanisms is unclear at this time. In an attempt to more carefully evaluate the processes involved, cortical lesions have been separated into several categories based on the depth of penetration from the surface into the brain (FIGURE 2). Type I lesions include discernable injury to both white and gray matter; Type II lesions have perivascular areas of demyelination isolated to the cortex; and Type III lesions demonstrate cortical demyelination below the pial surface that often cover several gyri and stop at cortical layers three or four 99. Others have proposed a fourth category, Type IV, to describe lesions that affect all cortical layers without extending into the white matter 18 19 (FIGURE 2). Type III and IV lesions are the most extensive and difficult to visualize. Although scarce inflammatory cells are found within these lesions, the meninges overlying them contain inflammatory cells that collect in structures resembling ectopic B-cell follicles 72,80. In support of a role for lymphoid neo-genesis in the pathogenesis of these lesions is that patients with ectopic B-cell follicles (41% of secondary progressive MS patients) had a more rapid disease progression 80. More solid determination of the role for ectopic B cell follicle formation in MS will depend upon the reliable detection of cortical lesions by MRI and identification of the immuno-pathologic mechanisms leading to lymphoid neogenesis within the CNS.
Figure 2.

Cortical lesions in MS revealed by sections immunohistochemically labeled for MBP. (A) Type 1 cortical lesions encompass both white and gray matter. Solid arrows indicate the cortex (CTX)/white matter (WM) border, while the lesion border is delineated by open arrows. (B) Type 2 lesions are contained entirely within the CTX and do not extend to the subcortical WM (solid arrow) or pial surface. The border of this type 2 lesion is indicated by the open arrows. (C) Type III cortical lesions represent cortical demyelination below the pial surface that often cover several gyri and stop at cortical layers three or four. Open arrows indicate the lesion border. (D) Type 4 lesions (border represented by filled arrow) span the entirety of the CTX without involvement of WM (solid arrow). The arrowhead indicates a small area likely undergoing remyelination. Figure from 19 used with permission (copyright Wolters Kluwer Health, 2003).
The role of T lymphocytes in multiple sclerosis
The lymphocytic presence within plaques and bordering areas suggests that inflammatory destruction in MS is driven by antigen-specific targeting of myelin and other CNS components. In particular, adaptive immune responses by T lymphocytes are thought to mediate injury to myelin and nerves within the CNS during MS. The determination that EAE can be mediated by CD4 T cells has promoted intense investigation into the potential CD4 T cell targets in MS. The relevance of antigenspecific CD4 T cell responses in MS was highlighted by the results of trials using an altered peptide ligand of MBP designed for therapeutic suppression of CD4 T cell responses, which resulted in disease exacerbations in multiple patients16. T cells from MS patients can recognize a variety of myelin protein targets, including MBP 100,131, PLP 55, MOG 139 and MOBP 36, among others. Non-myelin T cell antigens have also been described, including αB crystalline 6 and neuronal proteins such as contactin-2 38. Further, auto-reactive CD8 T cell are also observed 15. Although similar frequencies of auto-reactive T cells are found in MS patients and healthy subjects15,60, myelin-specific T cell avidity 17 and activation profiles 139 appear to be elevated in MS patients.
Newer technologies such as arrays of protein 58, lipid 64 and gene expression 78 have allowed fresh insight into targets that were heretofore unknown. For example, highly expressed genes in MS plaques compared to non-lesional CNS tissue include osteopontin 28; however, elevated gene expression does not necessarily imply targeting by the adaptive immune response in MS. Genes encoding myelin proteins actually display reduced expression 78. Thus, while several of these targets have been validated in EAE 28,75, their involvement in MS has yet to be fully demonstrated. Again, it is likely that multiple CNS antigens exist for T cells during the inflammatory targeting in MS; results from array experiments highlight how diverse and extensive these targets may be.
How these T cells become abnormally activated toward CNS antigens remains unclear. A popular concept invoked to explain how T cells become pathogenic in MS patients is that of molecular mimicry 89,119. Several infectious agents have been postulated to serve as activation triggers for auto-reactive T cells. The most consistently reported one is Epstein Barr virus (EBV) 113. Other infectious agents may also serve to trigger cross-reactivity with myelin components 124,126. One advantage of the adaptive immune response in MS lies in the ability to harness the specificity of T cell responses 122. As such, very discrete immunologic targets of therapy may be available for treating MS, including vaccines 9 and antigen presenting cell (APC)-mediated delivery of antigens 39 designed to induce antigenspecific tolerance. Clearly, antigen-specific therapeutics for MS must address the potentially wide array of T cell targets as well as HLA-specific presentation of antigens intrinsic to the genetic heterogeneity of individuals with MS.
The role of Th17 cells in MS
Since the identification of IL-17 as a novel cytokine in 1993, an intense inquiry into the role of IL-17 in EAE and MS has taken place. Two members of the IL-17 gene family, IL-17A and IL-17F, are expressed by CD4 T cells 87. In 2003, Cua and colleagues identified IL-23, and not IL-12, as a critical cytokine regulator of EAE 34. This quickly led to a new pathway for investigation into the immunologic basis for MS after the discovery that IL-23 regulates IL-17 production by CD4 T cells 34,74,82. Subsequently, IL-17-secreting CD4 T cells have emerged as a distinct lineage of T helper cells, termed Th17 cells. In EAE, Th17 cells participate in early infiltration of the CNS 109 and alone induce a unique neutrophil-predominant pathology 70. However, unlike IL-23, neutralization or genetic deletion of IL-17 in EAE does not result in a complete absence of disease 57. Hence, the direct contribution of Th17 cells during autoimmune demyelination remains unclear.
Human studies have emerged to bolster the relevance of IL-17 to the pathogenesis of MS. A greater proportion of IL-17-secreting cells, but not IFNγ+ CD4 T cells, is found in the CSF of MS patients compared to those with non-inflammatory neurologic diseases 24. The percentage of IL-17-producing memory CD4 T cells is elevated in peripheral blood from MS patients experiencing relapses 41, suggesting a prominent role for Th17 cells in MS. Further, IL-17 gene expression is up-regulated in lesions of MS patients 78, and Th17 cells are found in perivascular cuffs and borders of active lesions 130, indicating Th17 cells home to areas of inflammatory demyelination (FIGURE 3). These studies clearly highlight the association between Th17 cells and MS immune pathology.
Figure 3.
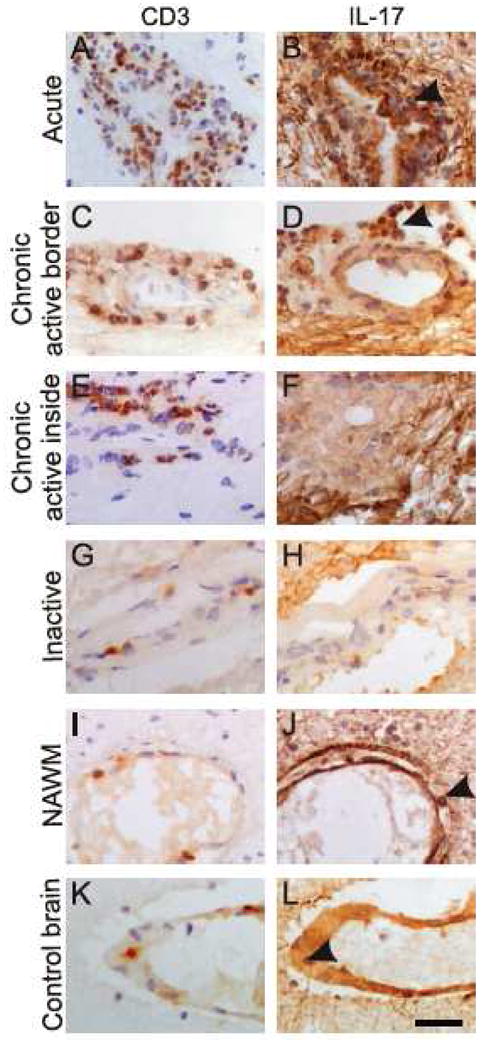
Involvement of IL-17 in MS lesions. Immunohistochemical identification of CD3 (left column) and IL-17 (right column) in consecutive sections. Abundant IL- 17 staining (arrowhead) is observed in perivascular cells in both acute (A & B) and chronic active (C &D) lesions. (E & F) Whereas CD3+ cells do not co-localize with IL- 17 staining within the internal region of a chronic lesion, the fibrillary pattern of IL- 17 immuno-reactivity is representative of possible astrocyte production of IL-17. Minimal staining is observed in tissue from an inactive lesion (G & H). Sparse Th17 cells are observed in NAWM in MS or within control tissue (arrow heads, I-L). Scale bar = 30 μm. Reprinted from Am J Pathol 2008, 172: 146-155 130 with permission from the American Society for Investigative Pathology.
How might Th17 cells contribute to the pathogenesis of MS? In vitro mobilization studies suggest that Th17 cells cross the blood brain barrier (BBB) more efficiently than other T cells. IL-17-secreting CD4 T cells are capable of eliciting damage to the BBB 65, which would promote greater influx of other inflammatory cells. Once present in the CNS parenchyma, Th17 cells are capable of mediating injury via recruitment of PMNs 70 and monocytes, along with co-production of other cytokines, such as IL-22 and IL-21, 120. Additionally, Th17 cells are more adept at killing human neurons in vitro than un-activated T cells, providing enticing data to suggest that Th17 cells are potential mediators of axonal and neuronal damage in MS lesions 65. These initial studies support the idea that Th17 cells may be critical mediators of immune destruction of myelin and axons during MS.
Recent data highlight the potential for targeting Th17 cells in MS. CD4 T cells from healthy individuals skewed to produce IL-17 are more responsive to one current form of treatment for MS, IFNβ, than those from MS patients 41. Further, IFNβ can inhibit the differentiation of naive CD4 T cells into Th17 cells 107. Patient responses to IFNβ may be related to the level of IL-17 prior to the onset of therapy 5. This result re-emphasizes the complexity and heterogeneity of the disease; in particular, the response to current therapies may signify differences in the pathogenesis of MS between patients 67. The Th17 pathway offers a new therapeutic target, including the molecular pathways that regulate IL-17 40. However, consistent with the complexity of cytokine networks and regulation, a singular stance that IL-17 is purely pathogenic is likely to be an oversimplification. In particular, IL-17 may participate in limiting tissue destruction during an inflammatory response 93. Overall, evidence is mounting to suggest that Th17 cells are present during the inflammatory destruction of tissue in MS, but questions remain about the direct contribution of this newly identified lineage of CD4 T cells. At this point, it is reasonable to be inclusive of Th17 cells in the pathogenesis of MS (FIGURE 4), but to what degree they are involved remains to be shown.
Figure 4.

Cellular and molecular factors involved in the immuno-pathogenesis of MS. Rather than representing an all-inclusive summary of the immuno-pathologic features of MS, this diagram highlights recent advances in the understanding of the neural-immune interactions in MS, including factors involved in leukocyte trafficking, axonal injury and antigen presentation. APC = antigen presenting cell; B = B lymphocyte; T = T lymphocyte; Mφ = macrophage, PVMφ = perivascular macrophage; PC = plasma cell; FDC = follicular DC; μglia = microglia; pDC = plasmacytoid DC.
B cell-mediated CNS damage in MS
Evidence gathered from examination of CNS tissue implicates the role of antibodies during the pathogenesis of MS. As noted, the presence of plasma cells, Ig and complement is a typical feature of the MS plaque 47,79. Naturally, this observation has prompted consideration over whether Ig present within plaques specifically targets myelin antigens. Molecular features of B cells found within MS plaques demonstrate that B cell receptor genes are modified in a specific way that indicates their evolving response toward specific targets 10,96. Further specificity of Ig from MS tissue has been described. MBP-reactive 135,137 and MOG-reactive 92 Ig has been isolated from CNS tissue of patients with MS. Additionally, in situ deposition of MOG-specific antibodies has been detected in MS lesions, along with MOG- and MBP-specific Ig complexed with myelin within macrophages 49. In contrast, detection of Ig appeared non-specifically dispersed throughout cellular constituents of plaques and NAWM, without significant differences compared to control tissues, suggesting that Ig presence may be secondary to white matter injury rather than antigen-specific B cell activation unique to MS 11. Thus, while controversy persists as to the antigen-specific nature of B cell involvement in MS, the presence of plasma cells and Ig continues to fuel investigation into their involvement during MS plaque development.
As a representation of B cell involvement in CNS parenchymal damage of MS patients, CSF analysis has afforded several clues on the role of Ig in the pathogenesis of MS. The localized intrathecal production of Ig, referred to as oligoclonal bands (OCB), is detected in over 90% of patients with RRMS 46, and their absence is associated with reduced severity of disease 4. While OCB are thought to be a product of clonally-expanded B cells within the CSF compartment, their potential targets remain elusive. Plasma cells from the CSF of patients with inflammatory demyelinating disease produce antibodies that are capable of binding myelin 134 and recognizing MBP 73. However, a more recent study of the CSF of patients with MS failed to detect IgG from CSF clonal B cells that bound to MBP, PLP or MOG 95. Analysis of OCB has also included investigation of IgM, which is found in a subset of MS patients 133. CSF-restricted IgM isolated from MS patients have been found to target a variety of lipid antigens, predominantly phosphatidylcholine 133. These persistent lipid-specific IgM OCBs are associated with more aggressive disease 20,133, and might be associated with a poor response to interferon therapy 21. Taken as a whole, a multitude of potential targets, including those from myelin as well as other CNS antigens83, have been proposed and investigated in patients with MS (Reviewed by Reindl and colleagues 110).
Modulation of T cell function may be an equally important function of B cells in the immune dysregulation in MS patients. As noted, anti-CD20 monoclonal antibody targeted depletion of B cells is efficacious for the treatment of RR-MS 59. This appears independent of antibody effects, since anti-CD20 treatment does not directly target Ig-secreting plasma cells 46; results in early efficacy in relapse reduction and inflammatory MRI lesions 59; and does not alter CSF Ig levels 33. B cells may promote neuro-inflammation in MS via direct and indirect effects on T cells, such as the secretion of the pro-inflammatory cytokines. For example, B cells from MS patients produce more TNF-α and lymphotoxin in the presence of the T cell-derived pro-inflammatory cytokine IFN-γ compared to healthy controls 8. Following anti-CD20-mediated B cell depletion in RRMS patients T cells produced less IFN-γ and were less proliferative in response to TCR engagement 8. Conversely, B cells are likely to also have immune-suppressive traits that are important in the immuno-pathogenesis of MS. For example, IL-10 secretion by B cells can serve to limit pro-inflammatory auto-reactive CD4 T cell responses 45.
Finally, recent evidence suggests a role for B cells in the generation of ectopic follicles in MS. Building on observations of Prineas 105, Aloisi and colleagues have described the presence of B cell follicles within the meninges in patients with SPMS 115. These are characterized by features of germinal centers, including the presence of B cells, follicular dendritic cells (FDCs), and CXCL13 (a chemokine involved in genesis of lymphoid organs). Found in approximately 40% of autopsy specimens only from patients with SPMS 80, the presence of these follicles was purported to be a maker for cortical lesions immediately adjacent to the ectopic follicle within the meninges. This finding has been disputed in subsequent work that failed to detect meningeal follicles in 12 patients with SPMS 68. Interestingly, there may be an association between the presence of EBV infectious material and secondary lymphoid follicles observed in MS patients 114. The implication of this finding is that latent infection of B cells with EBV drives the expansion and maturation of B cells along with ectopic follicle generation within the CNS, promoting intrathecal Ig production and the targeted destruction of underlying myelin in this region 46. While only observed in patients with SPMS, it is possible that the generation of these lymphoid organ-like structures begins during the relapsing-remitting phase and evolves over time. Whether these structures are inhibited by B cell-depleting therapy in MS remains to be investigated.
Mechanisms of leukocyte entry during MS
Immune access to the CNS is generally considered restricted. In practice, the traditional view of the CNS as an immune privileged site has been replaced with the more appropriate characterization of the CNS as an immune specialized organ 108. Thus, one key element to immune-mediated damage within the CNS during MS is the process by which immune cells are able to gain access to this specialized compartment. Within the context of universal processes governing immune cell trafficking, there are features that are relatively specific for leukocyte migration to, and within, the brain and spinal cord in health and disease. The molecular components, location, and timing of migration are all important factors during the immuno-pathogenesis of MS.
The BBB serves to actively restrict cellular and macromolecular movement between the blood and CNS tissue. Adequate function of the BBB depends upon several unique anatomic and cellular features, including tight junctions between endothelial cells, specialized expression of molecular transporters, and placement of immune cells within the CNS relative to the vasculature 35. Only by engaging in a critically timed sequence of events are auto-reactive lymphocytes able to enter the CNS compartment. Initially, leukocytes engage in rolling, activation and arrest to the endothelium of the BBB. This is greatly facilitated by up-regulation of adhesion molecules by the vasculature, including ICAM1 and VCAM1 102. While a trigger for vascular change remains unclear in MS, hypothetically changes in the vascular endothelium could result from pro-inflammatory mediators circulating within the vasculature, including TNF and/or LPS. Subsequently, migration of cells through and between endothelial cells takes place 63. Eventually, concentrated extravasation of immune cells in perivascular cuffs within the CNS parenchyma culminates in a breach of the BBB that is an essential component to the process of inflammatory destruction of white matter in MS 54.
The complex set of molecules that leukocytes depend upon for entry into CNS tissues involves integrins. Integrins are hetero-dimeric cell surface molecules that mediate adhesion between cells. Out of a panel of leukocyte adhesion receptors, the α4 subunit of VLA-4 was identified as a crucial factor for encephalitogenic T cell binding to CNS endothelium. Blockade of α4β1 engagement with one of its binding partners, VCAM-1, successfully abrogated disease in an animal model of MS 138 Clinical trials of a humanized monoclonal antibody targeting the α4 subunit of VLA- 4, called natalizumab, also demonstrated efficacy in the treatment of MS 104,112. Hence, selective inhibition of specific adhesion molecules are effective at reducing leukocyte entry into the CNS. Of note, natalizumab reduces the influx of a wide range of leukocytes, including T cells and dendritic cells (DCs) 37. In addition to VLA-4, other trafficking molecules impart specificity of migration into the CNS. Recently, ALCAM-1 was shown to be localized to the BBB and up-regulated in active MS lesions 26. In experimental animal systems, blockade of ALCAM-1 delayed disease 26. In addition, osteopontin also serves as a binding partner for VLA-4 and potentially serves as a separate target for reducing leukocyte migration into the CNS of patients with MS 121. Hence, a multitude of adhesion molecules participate in effective leukocyte trafficking to and within the CNS and serve as potential targets for therapies in MS.
Chemokines, a broad class of cytokines mediating chemotaxis, also contribute to leukocyte migration to the CNS. Several chemokines and their receptors have been implicated in leukocyte influx to the CNS in MS 63. For example, CXCL12, constitutively expressed in the CNS, is typically localized to the basolateral aspect of the CNS microvasculature and functions to retain leukocytes within the perivascular space. Redistribution of CXCL12 to the luminal aspect of vessels was observed in autopsy specimens from MS patients, which would allow for the dissemination of lymphocytes into the CNS parenchyma 84. Other chemokines are thought to participate in the recruitment of lymphocytes into the CNS in MS. Recent work in the EAE system has demonstrated that the initial wave of T cell infiltration into the CNS prior to disease onset is CCR6-dependent. 109. Furthermore, in EAE, the initial wave of inflammatory CD4 T cells express IL-17 and are potentially recruited specifically via CCR6 into the CNS via the choroids plexus that expresses the CCR6 ligand, CCL20. These results have yet to be convincingly replicated in human samples. Several other chemokine and chemokine receptors expressed by various cell types in MS show dysregulation including CCR7, CCL19 and CCL21, CCR5 97,127. Thus, there may be a unique chemokine signature at different phases of disease and in different regions of the CNS in order for various leukocytes to localize to the CNS during MS.
The role of antigen presenting cells in MS
Antigen presenting cells (APCs) process and present antigens to T cells, and in the context of MHC, co-stimulatory signals and cytokine secretion, drive adaptive immune responses 30. Experimental evidence based on animal models has shown that antigen-specific encounters within the CNS between T cells and APCs is crucial to the unfolding of MS. In EAE, without newly generated myelin antigens from the CNS by APCs, inflammatory demyelination does not proceed, even in the presence of myelin-reactive CD4 T cells 116,128. Thus, CD4 T cell-mediated disease is thought to be a multi-stage process, involving the initial activation of auto-reactive CD4 T cells, as well as reactivation within the CNS immune compartment. DCs are thought to be the major APC during the secondary phase of cognate interactions with CD4 T cells within the CNS 14,56. Perivascular spaces within areas bordering edges of active lesions contain immnostaining for CD209, a marker for a subset of DCs, suggesting that antigen presentation by DCs at the interface of the BBB contributes to the earliest inflammatory processes promoting lesion formation 61. Visualization of APC interactions with encephalitogenic CD4 T cells using intravital microscopy in a rat model of MS suggests that primed CD4 T cells actively engage with these perivascular APCs en route to entry within the CNS 13.
In addition to DCs, other APCs likely play an important role in antigen presentation during the pathogenesis of MS. Microglia are hematogenously-derived resident APCs within the CNS. Upon activation, they express greater amounts of MHCII and co-stimulatory molecules 2, signifying a greater capacity to promote pro-inflammatory T cell responses within the CNS. Activated microglia are localized to active plaques 76. Experimentally, impeding microglial function attenuates EAE 62. However, relative to other professional APCs, microglia are not as potent at inducing auto-reactive T cell responses 85 and may even down-regulate CD4 T cell functions 94. Thus, as resident APCs within the CNS, microglia are capable of performing APC functions, but likely are not the lynchpin for driving autoimmunity of the CNS in MS. Another APC potentially involved in driving myelin-reactive CD4 T cells in MS is the B cell. As already mentioned, recent work suggests that B cells play a prominent role in the pathogenesis of MS 46,59, and potentially play important roles in antigen presentation to T cells. In addition to effects on T cells, MHCII-dependent interactions with B cells promotes Ig class switching from IgM to IgG. Thus, antigen-specific interactions between B cells and T cells represent a critical step in the generation of Ig responses in MS. It is important to acknowledge that not all interactions between APCs and T cells promote inflammation. In addition to the effects of regulatory B cells mentioned above, APCs can also engender anti-inflammatory responses 48. For example, suppressor myeloid cells are generated after EAE induction and are capable of suppressing T cell function 140. In MS, the process of myeloid suppression is thought to be regulated in part by TREM-2, a trans-membrane signaling protein expressed by microglial cells, macrophages, monocytes and DCs 101. This mechanism may be dysregulated by secretion of soluble TREM-2, which could act as a decoy receptor and prevent inhibitory function of transmembrane TREM-2 101.
Axonal and neuronal damage in MS
Inflammatory CNS injury in MS has increasingly been associated with axonal damage. Although MS has classically been described as a disease marked by the loss of myelin in greater proportion to the loss of axons, axonal damage was noted in the earliest pathological descriptions of MS lesions 29. Modern techniques have allowed for precise demonstrations of axonal damage. Antibodies directed at amyloid precursor protein show damaged axons in active areas of MS lesions 42. Representing a major advance in the field of MS pathology, Trapp et al 129 were able to directly view and quantify transected axons using confocal microscopy by counting axonal ovoids at the ends of transected axons. The active areas of MS lesions were found to contain more transected axons than inactive areas in more chronic lesions. Of note, comparisons of biopsy and autopsy samples from patients with relapsing-remitting, secondary progressive, and primary progressive MS suggest that axonal pathology is greatest within the first year of disease onset, particularly in patients with RRMS 71. These studies propose that axonal pathology occurs in areas of active inflammatory demyelination and early during the course of disease.
In addition, a slower rate of axonal damage is also thought to occur and contribute to the clinical decline observed in MS patients. Trapp et al 129 and Kornek et al 69 showed that axonal ovoids are more common in inactive demyelinated lesions and in NAWM than in the white matter of control patients. However, remyelinated inactive lesions, or shadow plaques, have the same number of abnormal axons as control tissue 69. Patients with higher levels of motor disability have fewer surviving corticospinal axons traveling through their spinal cord, demonstrating a direct correlation of axonal damage and disease progression 125.
The mechanisms involved in axonal damage in MS are under intense investigation. CD8+ T-lymphocytes can cause axonal damage via the release of cytotoxic granules, induction of apoptosis through activation of surface receptors such as Fas, the release of cytokines such as TNF-α, or direct transection of axons 86,91. Macrophages/microglia are also found in close proximity to damaged neurons. Release of toxic molecules by these cells such as proteases and reactive nitrogen species can cause oligodendrocyte injury, demyelination, and axonal degeneration, disrupt the blood-brain barrier, and contribute to the loss of axonal conduction. 118. Axonal damage also occurs by activation of other components of the innate immune response such as Toll-like receptors 43. Toll-like receptor 2 is over-expressed by oligodendrocytes in MS lesions where it inhibits remyelination 117. Antibody-mediated injury to axonal components, e.g. neurofascin, can result in axonal and neuronal dysfunction 83. As a consequence of immune injury to myelin, higher energy demands on demyelinated axons and glutamate-mediated excitotoxicity may further impart unsustainable damage 81 103. Overall, axonal injury in MS is likely mediated by multiple mechanisms at play in both active and chronic lesions.
Neuronal loss in MS can be severe and occurs throughout the brain. Neuronal loss in the range of 18–35% has been reported in the cortex, hippocampus, thalamus, and spinal cord (reviewed in 18). Damage to axons and neurons has been evaluated in vivo using MRI techniques such as quantitative proton MR spectroscopy. In patients presenting with their first clinical attack of MS, the amount of NAA in the whole brain is already decreased, indicating early neuronal damage 111. This reduction is still present one year later and was independent of whether the patients had progressed to develop MS. Other MRI techniques, such as measurement of brain volume or diffusion tensor imaging, have provided more variable results in evaluating axonal and neuronal damage especially in short term studies as factors such as demyelination and edema can confound the results. However, examination of neuronal integrity using OCT shows a loss of macular volume in patients with progressive forms of MS, which correlates with poor visual acuity especially in patients with a history of optic neuritis 25. Similar to axonal injury, the processes resulting in neuronal loss in MS are likely several-fold. Direct immune injury to the gray matter can result in a loss of neurons, as within gray matter lesions a significant increase in apoptotic neurons was observed primarily in large pyramidal cortical neurons 99. However, in layer II of primary motor cortex from NAWM, paralbumin interneurons were more affected than other neurons that are relatively spared in MS patients 31. This differential susceptibility of neurons exposed to the same insult is part of a key consideration in how clinical deterioration, particularly with secondary progression, is related to repeated accumulation of axonal or neuronal damage to various neuronal populations. Further, axonal and neuronal survival may be directly tied to the trophic support provided by myelin, which may be particularly relevant during a high metabolic demand state of neurons exposed to inflammatory stressors 90.
Conclusions
In summary, several new features of cellular and molecular immunity have added to the understanding of the pathology of MS. These include the role of B cells, including antibody-dependent and antibody-independent mechanisms; the extent of axonal and neuronal injury; the contribution of a new lineage of CD4 T cells identified by the production of IL-17; leukocyte trafficking mechanisms to the CNS; and new lymphocyte targets during disease. These stand out among many other recent developments that due to space limitations were not able to be covered in this review. Topics involving resolution of inflammation in MS lesions, suppressor cells (Treg, CD8 T cells, etc.) and remyelination are bound to be important in driving toward a more comprehensive understanding of the pathogenesis of MS. Overall, an attempt has been made not to detail every mechanism involved in the pathology of MS, but rather highlight the features of disease that are under current study (FIGURE 4).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allen IV, McQuaid S, Mirakhur M, et al. Pathological abnormalities in the normal-appearing white matter in multiple sclerosis. Neurol Sci. 2001;22:141. doi: 10.1007/s100720170012. [DOI] [PubMed] [Google Scholar]
- 2.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 3.Amato MP, Portaccio E, Goretti B, et al. Association of neocortical volume changes with cognitive deterioration in relapsing-remitting multiple sclerosis. Arch Neurol. 2007;64:1157. doi: 10.1001/archneur.64.8.1157. [DOI] [PubMed] [Google Scholar]
- 4.Avasarala JR, Cross AH, Trotter JL. Oligoclonal band number as a marker for prognosis in multiple sclerosis. Arch Neurol. 2001;58:2044. doi: 10.1001/archneur.58.12.2044. [DOI] [PubMed] [Google Scholar]
- 5.Axtell RC, de Jong BA, Boniface K, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 16:406. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bajramovic JJ, Plomp AC, Goes A, et al. Presentation of alpha B-crystallin to T cells in active multiple sclerosis lesions: an early event following inflammatory demyelination. J Immunol. 2000;164:4359. doi: 10.4049/jimmunol.164.8.4359. [DOI] [PubMed] [Google Scholar]
- 7.Baker RW, Thompson RH, Zilkha KJ. Fatty-acid composition of brain lecithins in multiple sclerosis. Lancet. 1963;1:26. doi: 10.1016/s0140-6736(63)91152-8. [DOI] [PubMed] [Google Scholar]
- 8.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 67:452. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 9.Bar-Or A, Vollmer T, Antel J, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol. 2007;64:1407. doi: 10.1001/archneur.64.10.nct70002. [DOI] [PubMed] [Google Scholar]
- 10.Baranzini SE, Jeong MC, Butunoi C, et al. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol. 1999;163:5133. [PubMed] [Google Scholar]
- 11.Barnett MH, Parratt JD, Cho ES, et al. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Ann Neurol. 2009;65:32. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- 12.Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- 13.Bartholomaus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462:94. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- 14.Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84:532. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 15.Berthelot L, Laplaud DA, Pettre S, et al. Blood CD8+ T cell responses against myelin determinants in multiple sclerosis and healthy individuals. Eur J Immunol. 2008;38:1889. doi: 10.1002/eji.200838023. [DOI] [PubMed] [Google Scholar]
- 16.Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 17.Bielekova B, Sung MH, Kadom N, et al. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172:3893. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- 18.Bo L. The histopathology of grey matter demyelination in multiple sclerosis. Acta Neurol Scand. 2009;Suppl:51. doi: 10.1111/j.1600-0404.2009.01216.x. [DOI] [PubMed] [Google Scholar]
- 19.Bo L, Vedeler CA, Nyland HI, et al. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol. 2003;62:723. doi: 10.1093/jnen/62.7.723. [DOI] [PubMed] [Google Scholar]
- 20.Bosca I, Magraner MJ, Coret F, et al. The risk of relapse after a clinically isolated syndrome is related to the pattern of oligoclonal bands. J Neuroimmunol. 226:143. doi: 10.1016/j.jneuroim.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 21.Bosca I, Villar LM, Coret F, et al. Response to interferon in multiple sclerosis is related to lipid-specific oligoclonal IgM bands. Mult Scler. 16:810. doi: 10.1177/1352458510371961. [DOI] [PubMed] [Google Scholar]
- 22.Breij EC, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63:16. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 23.Brink BP, Veerhuis R, Breij EC, et al. The pathology of multiple sclerosis is location-dependent: no significant complement activation is detected in purely cortical lesions. J Neuropathol Exp Neurol. 2005;64:147. doi: 10.1093/jnen/64.2.147. [DOI] [PubMed] [Google Scholar]
- 24.Brucklacher-Waldert V, Stuerner K, Kolster M, et al. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009;132:3329. doi: 10.1093/brain/awp289. [DOI] [PubMed] [Google Scholar]
- 25.Burkholder BM, Osborne B, Loguidice MJ, et al. Macular volume determined by optical coherence tomography as a measure of neuronal loss in multiple sclerosis. Arch Neurol. 2009;66:1366. doi: 10.1001/archneurol.2009.230. [DOI] [PubMed] [Google Scholar]
- 26.Cayrol R, Wosik K, Berard JL, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. 2008;9:137. doi: 10.1038/ni1551. [DOI] [PubMed] [Google Scholar]
- 27.Ceccarelli A, Rocca MA, Falini A, et al. Normal-appearing white and grey matter damage in MS. A volumetric and diffusion tensor MRI study at 3.0 Tesla. J Neurol. 2007;254:513. doi: 10.1007/s00415-006-0408-4. [DOI] [PubMed] [Google Scholar]
- 28.Chabas D, Baranzini SE, Mitchell D, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 29.Charcot J-M. Lecons sur les maladies du systeme nerveux faites a la salpetriere. Vol. 189. 1880. [Google Scholar]
- 30.Chastain EM, Duncan DS, Rodgers JM, et al. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clements RJ, McDonough J, Freeman EJ. Distribution of parvalbumin and calretinin immunoreactive interneurons in motor cortex from multiple sclerosis post-mortem tissue. Exp Brain Res. 2008;187:459. doi: 10.1007/s00221-008-1317-9. [DOI] [PubMed] [Google Scholar]
- 32.Compston A, McDonald IR, Noseworthy J, et al. McAlpine's Multiple Sclerosis. Philadelphia: Churchill Livingstone; 2006. [Google Scholar]
- 33.Cross AH, Stark JL, Lauber J, et al. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180:63. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 35.Daneman R, Rescigno M. The gut immune barrier and the blood-brain barrier: are they so different? Immunity. 2009;31:722. doi: 10.1016/j.immuni.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 36.de Rosbo NK, Kaye JF, Eisenstein M, et al. The myelin-associated oligodendrocytic basic protein region MOBP15-36 encompasses the immunodominant major encephalitogenic epitope(s) for SJL/J mice and predicted epitope(s) for multiple sclerosis-associated HLA-DRB1*1501. J Immunol. 2004;173:1426. doi: 10.4049/jimmunol.173.2.1426. [DOI] [PubMed] [Google Scholar]
- 37.del Pilar Martin M, Cravens PD, Winger R, et al. Decrease in the numbers of dendritic cells and CD4+ T cells in cerebral perivascular spaces due to natalizumab. Arch Neurol. 2008;65:1596. doi: 10.1001/archneur.65.12.noc80051. [DOI] [PubMed] [Google Scholar]
- 38.Derfuss T, Parikh K, Velhin S, et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci U S A. 2009;106:8302. doi: 10.1073/pnas.0901496106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dolgin E. The inverse of immunity. Nat Med. 2010;16:740. doi: 10.1038/nm0710-740. [DOI] [PubMed] [Google Scholar]
- 40.Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10:1252. doi: 10.1038/ni.1798. [DOI] [PubMed] [Google Scholar]
- 41.Durelli L, Conti L, Clerico M, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009;65:499. doi: 10.1002/ana.21652. [DOI] [PubMed] [Google Scholar]
- 42.Ferguson B, Matyszak MK, Esiri MM, et al. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120 ( Pt 3):393. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 43.Fernandez M, Montalban X, Comabella M. Orchestrating innate immune responses in multiple sclerosis: Molecular players. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Filippi M, Campi A, Dousset V, et al. A magnetization transfer imaging study of normal-appearing white matter in multiple sclerosis. Neurology. 1995;45:478. doi: 10.1212/wnl.45.3.478. [DOI] [PubMed] [Google Scholar]
- 45.Fillatreau S, Sweenie CH, McGeachy MJ, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 46.Franciotta D, Salvetti M, Lolli F, et al. B cells and multiple sclerosis. Lancet Neurol. 2008;7:852. doi: 10.1016/S1474-4422(08)70192-3. [DOI] [PubMed] [Google Scholar]
- 47.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 48.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genain CP, Cannella B, Hauser SL, et al. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 50.Geurts JJ, Pouwels PJ, Uitdehaag BM, et al. Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging. Radiology. 2005;236:254. doi: 10.1148/radiol.2361040450. [DOI] [PubMed] [Google Scholar]
- 51.Gilmore CP, Donaldson I, Bo L, et al. Regional variations in the extent and pattern of grey matter demyelination in multiple sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J Neurol Neurosurg Psychiatry. 2009;80:182. doi: 10.1136/jnnp.2008.148767. [DOI] [PubMed] [Google Scholar]
- 52.Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 53.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenfield JG, Love S, Louis DN, et al. Greenfield's neuropathology. 8. London: Hodder Arnold; 2008. [Google Scholar]
- 55.Greer JM, Csurhes PA, Cameron KD, et al. Increased immunoreactivity to two overlapping peptides of myelin proteolipid protein in multiple sclerosis. Brain. 1997;120 ( Pt 8):1447. doi: 10.1093/brain/120.8.1447. [DOI] [PubMed] [Google Scholar]
- 56.Greter M, Heppner FL, Lemos MP, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 57.Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 59.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 60.Hellings N, Baree M, Verhoeven C, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63:290. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 61.Henderson AP, Barnett MH, Parratt JD, et al. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66:739. doi: 10.1002/ana.21800. [DOI] [PubMed] [Google Scholar]
- 62.Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- 63.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanter JL, Narayana S, Ho PP, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- 65.Kebir H, Kreymborg K, Ifergan I, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keegan M, Konig F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet. 2005;366:579. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 67.Koike F, Satoh J, Miyake S, et al. Microarray analysis identifies interferon beta-regulated genes in multiple sclerosis. J Neuroimmunol. 2003;139:109. doi: 10.1016/s0165-5728(03)00155-3. [DOI] [PubMed] [Google Scholar]
- 68.Kooi EJ, Geurts JJ, van Horssen J, et al. Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol. 2009;68:1021. doi: 10.1097/NEN.0b013e3181b4bf8f. [DOI] [PubMed] [Google Scholar]
- 69.Kornek B, Storch MK, Weissert R, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kroenke MA, Carlson TJ, Andjelkovic AV, et al. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuhlmann T, Lingfeld G, Bitsch A, et al. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- 72.Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 73.Lambracht-Washington D, O'Connor KC, Cameron EM, et al. Antigen specificity of clonally expanded and receptor edited cerebrospinal fluid B cells from patients with relapsing remitting MS. J Neuroimmunol. 2007;186:164. doi: 10.1016/j.jneuroim.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lanz TV, Ding Z, Ho PP, et al. Angiotensin II sustains brain inflammation in mice via TGF-beta. J Clin Invest. 120:2782. doi: 10.1172/JCI41709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7:115. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- 77.Lassmann H, Bruck W, Lucchinetti C, et al. Remyelination in multiple sclerosis. Mult Scler. 1997;3:133. doi: 10.1177/135245859700300213. [DOI] [PubMed] [Google Scholar]
- 78.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 79.Lucchinetti CF, Bruck W, Lassmann H. Evidence for pathogenic heterogeneity in multiple sclerosis. Ann Neurol. 2004;56:308. doi: 10.1002/ana.20182. [DOI] [PubMed] [Google Scholar]
- 80.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 81.Mahad DJ, Ziabreva I, Campbell G, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 83.Mathey EK, Derfuss T, Storch MK, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCandless EE, Piccio L, Woerner BM, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172:799. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McMahon EJ, Bailey SL, Castenada CV, et al. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 86.Medana I, Martinic MA, Wekerle H, et al. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. 2001;159:809. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 88.Moore GR, Laule C, Mackay A, et al. Dirty-appearing white matter in multiple sclerosis: preliminary observations of myelin phospholipid and axonal loss. J Neurol. 2008;255:1802. doi: 10.1007/s00415-008-0002-z. [DOI] [PubMed] [Google Scholar]
- 89.Munz C, Lunemann JD, Getts MT, et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- 91.Neumann H, Medana IM, Bauer J, et al. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- 92.O'Connor KC, Appel H, Bregoli L, et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J Immunol. 2005;175:1974. doi: 10.4049/jimmunol.175.3.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O'Connor W, Jr, Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol. 2010;11:471. doi: 10.1038/ni.1882. [DOI] [PubMed] [Google Scholar]
- 94.Ortler S, Leder C, Mittelbronn M, et al. B7-H1 restricts neuroantigen-specific T cell responses and confines inflammatory CNS damage: implications for the lesion pathogenesis of multiple sclerosis. Eur J Immunol. 2008;38:1734. doi: 10.1002/eji.200738071. [DOI] [PubMed] [Google Scholar]
- 95.Owens GP, Bennett JL, Lassmann H, et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol. 2009;65:639. doi: 10.1002/ana.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Owens GP, Burgoon MP, Anthony J, et al. The immunoglobulin G heavy chain repertoire in multiple sclerosis plaques is distinct from the heavy chain repertoire in peripheral blood lymphocytes. Clin Immunol. 2001;98:258. doi: 10.1006/clim.2000.4967. [DOI] [PubMed] [Google Scholar]
- 97.Pashenkov M, Teleshova N, Kouwenhoven M, et al. Elevated expression of CCR5 by myeloid (CD11c+) blood dendritic cells in multiple sclerosis and acute optic neuritis. Clin Exp Immunol. 2002;127:519. doi: 10.1046/j.1365-2249.2002.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Perry A, Brat DJ. Practical surgical neuropathology : a diagnostic approach. Philadelphia, PA: Churchill Livingstone/Elsevier; 2010. [Google Scholar]
- 99.Peterson JW, Bo L, Mork S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 100.Pette M, Fujita K, Wilkinson D, et al. Myelin autoreactivity in multiple sclerosis: recognition of myelin basic protein in the context of HLA-DR2 products by T lymphocytes of multiple-sclerosis patients and healthy donors. Proc Natl Acad Sci U S A. 1990;87:7968. doi: 10.1073/pnas.87.20.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piccio L, Buonsanti C, Cella M, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131:3081. doi: 10.1093/brain/awn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piccio L, Rossi B, Scarpini E, et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric G(i)-linked receptors. J Immunol. 2002;168:1940. doi: 10.4049/jimmunol.168.4.1940. [DOI] [PubMed] [Google Scholar]
- 103.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 104.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 105.Prineas JW. Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science. 1979;203:1123. doi: 10.1126/science.424741. [DOI] [PubMed] [Google Scholar]
- 106.Raine CS. Multiple sclerosis: classification revisited reveals homogeneity and recapitulation. Ann Neurol. 2008;63:1. doi: 10.1002/ana.21314. [DOI] [PubMed] [Google Scholar]
- 107.Ramgolam VS, Sha Y, Jin J, et al. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009;183:5418. doi: 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- 108.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 109.Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6- regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 110.Reindl M, Khalil M, Berger T. Antibodies as biological markers for pathophysiological processes in MS. J Neuroimmunol. 2006;180:50. doi: 10.1016/j.jneuroim.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 111.Rovaris M, Gambini A, Gallo A, et al. Axonal injury in early multiple sclerosis is irreversible and independent of the short-term disease evolution. Neurology. 2005;65:1626. doi: 10.1212/01.wnl.0000184493.06254.a6. [DOI] [PubMed] [Google Scholar]
- 112.Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta- 1a for relapsing multiple sclerosis. N Engl J Med. 2006;354:911. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- 113.Salvetti M, Giovannoni G, Aloisi F. Epstein-Barr virus and multiple sclerosis. Curr Opin Neurol. 2009;22:201. doi: 10.1097/WCO.0b013e32832b4c8d. [DOI] [PubMed] [Google Scholar]
- 114.Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Serafini B, Rosicarelli B, Magliozzi R, et al. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Slavin AJ, Soos JM, Stuve O, et al. Requirement for endocytic antigen processing and influence of invariant chain and H-2M deficiencies in CNS autoimmunity. J Clin Invest. 2001;108:1133. doi: 10.1172/JCI13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sloane JA, Batt C, Ma Y, et al. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107:11555. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 119.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 120.Spolski R, Leonard WJ. Cytokine mediators of Th17 function. Eur J Immunol. 2009;39:658. doi: 10.1002/eji.200839066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 122.Steinman L. Antigen-specific therapy of multiple sclerosis: the long-sought magic bullet. Neurotherapeutics. 2007;4:661. doi: 10.1016/j.nurt.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suzuki K, Kamoshita S, Eto Y, et al. Myelin in multiple sclerosis. Composition of myelin from normal-appearing white matter. Arch Neurol. 1973;28:293. doi: 10.1001/archneur.1973.00490230029002. [DOI] [PubMed] [Google Scholar]
- 124.Talbot PJ, Paquette JS, Ciurli C, et al. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann Neurol. 1996;39:233. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tallantyre EC, Bo L, Al-Rawashdeh O, et al. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler. 16:406. doi: 10.1177/1352458510364992. [DOI] [PubMed] [Google Scholar]
- 126.Tejada-Simon MV, Zang YC, Hong J, et al. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol. 2003;53:189. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- 127.Teleshova N, Pashenkov M, Huang YM, et al. Multiple sclerosis and optic neuritis: CCR5 and CXCR3 expressing T cells are augmented in blood and cerebrospinal fluid. J Neurol. 2002;249:723. doi: 10.1007/s00415-002-0699-z. [DOI] [PubMed] [Google Scholar]
- 128.Tompkins SM, Padilla J, Dal Canto MC, et al. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:4173. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- 129.Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 130.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valli A, Sette A, Kappos L, et al. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest. 1993;91:616. doi: 10.1172/JCI116242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van Horssen J, Brink BP, de Vries HE, et al. The blood-brain barrier in cortical multiple sclerosis lesions. J Neuropathol Exp Neurol. 2007;66:321. doi: 10.1097/nen.0b013e318040b2de. [DOI] [PubMed] [Google Scholar]
- 133.Villar LM, Sadaba MC, Roldan E, et al. Intrathecal synthesis of oligoclonal IgM against myelin lipids predicts an aggressive disease course in MS. J Clin Invest. 2005;115:187. doi: 10.1172/JCI22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.von Budingen HC, Harrer MD, Kuenzle S, et al. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. Eur J Immunol. 2008;38:2014. doi: 10.1002/eji.200737784. [DOI] [PubMed] [Google Scholar]
- 135.Warren KG, Catz I. Autoantibodies to myelin basic protein within multiple sclerosis central nervous system tissue. J Neurol Sci. 1993;115:169. doi: 10.1016/0022-510x(93)90221-j. [DOI] [PubMed] [Google Scholar]
- 136.Wilson HC, Scolding NJ, Raine CS. Co-expression of PDGF alpha receptor and NG2 by oligodendrocyte precursors in human CNS and multiple sclerosis lesions. J Neuroimmunol. 2006;176:162. doi: 10.1016/j.jneuroim.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 137.Wucherpfennig KW, Catz I, Hausmann S, et al. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLADR2- restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B-cell and T-cell epitopes. J Clin Invest. 1997;100:1114. doi: 10.1172/JCI119622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 139.Zhang J, Markovic-Plese S, Lacet B, et al. Increased frequency of interleukin 2- responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994;179:973. doi: 10.1084/jem.179.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhu B, Bando Y, Xiao S, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- 141.Zivadinov R, Stosic M, Cox JL, et al. The place of conventional MRI and newly emerging MRI techniques in monitoring different aspects of treatment outcome. J Neurol. 2008;255 (Suppl 1):61. doi: 10.1007/s00415-008-1009-1. [DOI] [PubMed] [Google Scholar]