

Abstract
A series of sterically hindered (methoxylated) polychlorinated biphenyl derivatives was synthesized using the Suzuki and the Ullmann coupling reaction. The Suzuki coupling with Pd(dba)2/2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (DPDB) gave better yields (65–98%) compared to the classic Ullmann coupling reaction (20–38%). Despite the reactive catalyst system, no significant coupling with aromatic chlorine substituents was observed. Crystal structure analysis of four PCB derivatives revealed solid state dihedral angles ranging from 69.7° to 81.0°, which indicates that these highly ortho substituted PCB derivatives have some conformational flexibility.
Keywords: biaryls, palladium, Suzuki cross-coupling, Ullmann cross-coupling, dihedral angle
Polychlorinated biphenyls (PCBs) are a group of ubiquitous environmental contaminants that were manufactured until the 1970s in the United States and are still in use in enclosed applications, such as transformers and capacitors.1,2 Recent studies demonstrate that many PCB congeners are also formed inadvertently as byproducts of industrial processes and can be found as byproducts in paints.3 Laboratory and epidemiologic studies have implicated PCBs in adverse human health effects such as cardiovascular disease, obesity and cancer. One particular health concern are cognitive deficits in laboratory animals and children that have been linked to in utero exposure to PCB congeners with multiple ortho chlorine substituents4,5 and may be mediated by the Ryanodine receptor.6–9 Structure-activity relationship studies demonstrate that sterically hindered PCB congeners with three or four ortho chlorine substituents, specifically congeners with a 2,3,6-trichloro substitution pattern in one phenyl ring, are potent sensitizers of the Ryanodine receptor.9
One major obstacle towards investigating the developmental neurotoxicity of PCBs is the unavailability of pure PCB congeners and their respective metabolites, both as analytical standards for studies on disposition and in vitro and in vivo toxicity. Unsymmetrical PCB derivatives with multiple ortho chlorine substituents can be synthesized using the Ullmann10,11 or the Cadogan diaryl coupling reaction.12 These approaches have significant drawbacks, including poor selectivity, low yields, and the formation of toxic byproducts.12–15 Lower chlorinated PCB derivatives with up to two ortho chlorine substituents can be synthesized with good selectivity and in high yields using the Suzuki coupling of chlorinated iodo- or bromobenzenes with benzene boronic acids.11,16–19 Although sterically hindered Suzuki coupling reactions are well established for the preparation of biaryls with multiple ortho methyl groups,20–23 the available coupling procedures have not been applied to the synthesis of PCB derivatives with three or four ortho chlorine substituents, partly because the catalyst systems employed in these studies will also catalyze the coupling with chloro substituents.
Here we explore strategies for the synthesis of suitable chlorinated precursors of methoxylated and hydroxylated PCB congeners containing a 2,3,6-trichloro substitution pattern in one phenyl ring and their coupling with chlorinated benzene boronic acids using Pd(dba)2/DPDB as an improved approach to multi- ortho substituted PCB congeners, such as PCB 91 (2,2′,3,4′,6-pentachlorobiphenyl), PCB 95 (2,2′,3,5′,6-pentachlorobiphenyl), PCB 132 (2,2′,3,3′,4′,6-hexachlorobiphenyl) and PCB 149 (2,2′,3,4′,5′,6-hexachlorobiphenyl) derivatives. In addition, the molecular structure of selected PCB derivatives in the solid state was determined to aid in our understanding of their three-dimensional structure and, ultimately, their toxicity.
Iodinated chloroanisoles for the preparation of derivatives of environmentally relevant, neurotoxic PCB congeners are not readily available. Multistep syntheses of several suitable precursors, including 2,3,5-trichloro-4-iodoanisole (1), 2,4,5-trichloro-3-iodoanisole (2) and 3,5,6-trichloro-4-iodoveratrole (3) (Figure 1), have been reported previously.10 In particular the synthesis of 1 their large scale synthesis is challenging because key synthesis steps have poor regioselectivity and/or low yields. The following section briefly describes the strategies employed in this study for the synthesis of 1 and the corresponding bromide 4.
Figure 1.

Structure of target intermediates 2,3,5-trichloro-4-iodoanisole (1), 2,4,5-trichloro-3-iodoanisole (2), 3,5,6-trichloro-4-iodoveratrole (3) and 4-bromo-2,3,5-trichloro-anisole (4).
4-Amino-3,5-dichloroanisole (5a) is a key intermediate for the synthesis of 1 (Scheme 1) and has been used by Waller and co-authors for the synthesis of PCB 136 (2,2′,3,3′,6,6′-hexachlorobiphenyl) metabolites.10 Because of the poor yield of the approach employed by Waller and co-authors, we initially investigated the following two approaches for the synthesis of 5a: The first approach is based on the work of Kenny and co-workers, who synthesized 5b in two steps via benzoquinone monoxime 6 from phenol (7).24 In our hands reaction of benzoquinone monoxime 6 with anhydrous HCl/MeOH/Et2O yielded 5a and 6-amino-5-chloro-1,3-dimethoxybenzene in a 1: 19 ratio, as determined by 1H NMR spectroscopy. Similarly, 5a was only a minor product when benzoquinone monoxime 6 was reacted with trimethylsilyl chloride/MeOH instead of anhydrous HCl. The second approach investigated the synthesis of 5a via the regioselective formation of an azo compound, such as 8, from the corresponding halogenated phenol.25,26 Contrary to the literature, the formation of 8 from 3,5-dichlorophenol (9) was not regioselective and, based on TLC analysis, yielded approximately a 1:1 mixture of both possible regioisomers. Consequently, the methylation of this mixture with dimethyl sulfate followed by reduction with Fe/AcOH/H2O yielded a mixture of 2- (5b) and 4-amino-3,5-dichloroanisole (5a). Overall, neither approach offers a more straightforward access to 5a compared to published procedures.10
Scheme 1.

Syntheses of 4-amino-3,5-dichloroanisole (4b), a possible key intermediate for the preparation of 2,3,5-trichloro-4-iodoanisole (1). Pure 11a and 5a were synthesized from 12a, which was synthesized from 9, followed by separation of 12a and 12b by column chromatography (see text for details).
In subsequent attempts we employed strategies similar to the approach described by Waller and co-authors to synthesize 5a (Scheme 1).10 In their approach, the nitration of 3,5-dichloroanisole (10) resulted in a mixture of 3,5-dichloro-4- and 3,5-dichloro-2-nitroanisole (11a+11b), with the undesired 3,5-dichloro-2-nitroanisole (11b) being the major isomer.10 Since the separation of 11a from 11b is difficult, Waller and co-authors reduced the mixture with Na2S2O4 to 4- and 2-amino-3,5-dichloroanisole (5a and 5b) and separated 5a from the undesired byproduct 5b by column chromatography. In our hands the nitration of anisole 10 to 11 was typically incomplete. Furthermore, we were unable to separate 5a from 5b on a preparative scale. Since Hartz and coworkers have successfully used the direct nitration of 3,5-dichlorophenol 9 followed by fractional crystallization to prepare 12a,27 we ultimately synthesized 5a via the nitration of 3,5-dichlorophenol (9) (Scheme 1). Although we were unable to separate 12a from 12b by fractional crystallization, we successfully separated the two regioisomers by column chromatography in a ratio of 12a: 12b of 1: 1.6. The structure of the desired 3,5-dichloro-4-nitrophenol (12a) was verified by x-ray crystal structure analysis (Figure 2). Subsequent methylation of 12a followed by reduction of 11a with Na2S2O4 provided access to 5a. 2,3,5-Trichloro-4-iodoanisole (1) was synthesized from 5a as outlined by Waller et al. using a Sandmeyer reaction and chlorination with HCl/H2O2.
Figure 2.
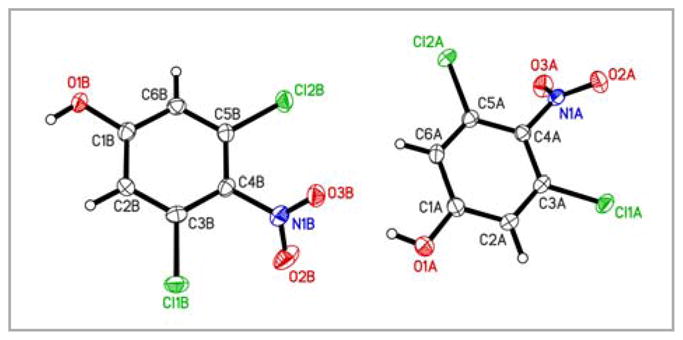
Molecular structure of 3,5-dichloro-4-nitrophenol (11b). Displacement ellipsoids are drawn at the 50% probability level.
The halogenation of 3,5-dichloroanisole (10) in the 4-position offers a direct approach to benzene derivatives with a 3,5-dichloro-4-methoxy substitution pattern, such as 14. We were able to synthesize 4-bromo-3,5-dichloroanisole 14 from 10 by bromination with NBS/HCl (Scheme 2).28 Although this reaction results in the formation of small quantities of the ortho bromination product (as determined by GC-MS analysis), pure 14 could be obtained in 27% yield by recrystalization from methanol. Subsequent chlorination with H2O2/HCl yielded 4, the brominated analogue of 1. Although the iodination of 2,5-dichlorophenol with AgSO4/I2 in dichloromethane appears to occur preferentially in the 4-position,29 iodination of 9 or 10 using the same conditions yielded a mixture of the corresponding 2-iodo-, 4-iodo-, and 2,4-di-iodo products. Similar to the corresponding nitro-phenols or anisoles, the separation of the iodinated products was challenging.
Scheme 2.

Synthesis of 4-bromo-2,3,5-trichloroanisole (4) by bromination of 3,5-dichloroanisole (10) with NBS/HCl28 followed by chlorination of 14 with HCl/H2O2.10
Since bromides are known to be less reactive in Suzuki coupling reactions compared to iodides,30 initial experiments to utilize 4 for the synthesis of PCB derivatives focused on its conversion to the corresponding boronic acids or pinacolborate esters. Attempts to convert 4 to the corresponding boronic acid, using n-BuLi at −78 °C followed by quenching with triisopropyl borate, or the pinacolboronate ester, using bispinacolatodiboron/PdOAc/KOAc in DMSO at 100 °C, were not successful. Synthesis of 1 from 4 via the corresponding aniline, followed by Sandmeyer reaction looked like a promising approach; however, conversion of 4 to the corresponding aniline using LHMDS as ammonia equivalent under palladium catalyzed Buchwald amination conditions (LHMDS, DPDB, anhydrous THF, 65 °C, 15–18 h) was also not successful. This was not entirely surprising because a similar amination reaction with 14 also did not give the expected aniline.31
A variety of catalysts and ligands have been employed for the synthesis of sterically hindered biaryls.20–23 We investigated the preparation of PCB derivatives using Pd(dba)2 and the commercially available ligand DPDB as a well established catalyst system. Table 1 shows a comparison of the yields of PCB derivatives synthesized from 1, 2, 3 or 4 using either the Suzuki (Pd(dba)2, DPDB, K3PO4, toluene, 100°C, 24 h) or Ullmann coupling reaction (230°C, 7d, Cu bronze). The yields of the Suzuki coupling reaction (65–98%) with iodides 1, 2 and 3 were superior to the yields of the corresponding Ullmann coupling reaction (20–38%). Better yields were typically obtained when fresh benzene boronic acids were used as starting materials in the Suzuki coupling reaction. Although the Pd(dba)2/DPDB catalyst system allows the Suzuki coupling of chlorinated benzenes,22 the difference in the reactivity of the iodo versus chloro groups allowed the selective formation of the desired coupling products 15–18. The lower yields of the Ullmann coupling reaction were, in part, due to homo-coupling of the respective iodo starting materials.
Table 1.
Yields [%] of PCB derivatives synthesized using the Suzuki and Ullmann coupling reactions.1
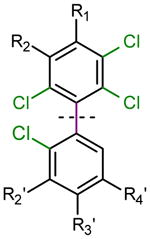 PCB Derivative |
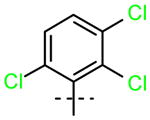 15 (R1=R2=H) |
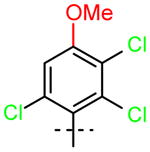 16 (R1=OCH3, R2=H) |
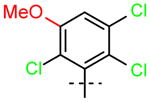 17 (R1=H, R2=OCH3) |
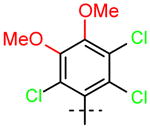 18 (R1=R2=OCH3) |
||||
|---|---|---|---|---|---|---|---|---|
| Suzuki | Ullmann2 | Suzuki | Ullmann2 | Suzuki | Ullmann2 | Suzuki | Ullmann3 | |
 a a
|
76 | 27 | 93 | 28 (14) | 69 | 38 | 71 | - |
 b b
|
75 | 29 | 98 | 20 (10) | 65 | 38 | 71 | - |
 c c
|
78 | 26 | 91 | 27 (5) | 50 | 34 | 66 | - |
Values in parentheses are the yields obtained with the corresponding bromide 4.
Gas chromatographic yields.
The Ullmann coupling reaction yielded a black material and no traces of the starting material were observed by gas chromatographic analysis.
Bromide 4, which is less reactive compared to the corresponding iodide 1,30 reacted in the Suzuki coupling according to GC-MS analysis. However, the reaction was not clean and it was impossible to obtain the coupled product (e.g., 16b) in pure form, even after repeated chromatographic purification followed by crystallization. The yields of the Ullmann coupling reactions with bromide 4 were also inferior to its iodo counterpart 1 (Table 1). Overall, Suzuki coupling with iodides 1a, 2 and 3 offers a comparatively straightforward approach to many parent PCBs and PCB derivatives with multiple ortho chlorine substituents. However, the synthesis of some PCB derivatives, such as 2,2′,3,4′,5′,6-hexachloro-5-methoxy-biphenyl (17d), via the Ullmann coupling reaction may be preferable due to the unavailability of the corresponding boronic acids from commercial sources (Scheme 3).
Scheme 3.

Synthesis of PCBs 91, 95, and 132 (15a–c) and the corresponding methoxylated PCB derivatives 16a–c, 17a–d and 18a–c using the Suzuki and the Ullmann coupling reaction. The monomethoxylated PCBs 16a–c and 17a–d were demethylated using an excess of BBr3 to yield the corresponding hydroxylated PCB derivatives 19a–c and 20a–d. DPDB = 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl.
Since PCB congeners with multiple ortho chlorine substituents can be neurotoxic in vitro9 and in vivo,32 milligram quantities of the hydroxylated metabolites of these PCB congeners are needed to assess their role in the developmental neurotoxicity of PCBs. While the demethylation of lower chlorinated methoxylated PCBs with BBr3 is typically straightforward,16,33 this approach was cumbersome in the case of 16a–c and 17a–d and required a large excess of BBr3 and long reaction times (Scheme 3). Although we were able to obtain the hydroxylated PCBs 19a–c and 20a–d in a quantity and purity sufficient for future biological studies, further work is needed to optimize this demethylation reaction, e.g., by using demethylation reagents such as HBr.10
The three-dimensional structure of a PCB is largely determined by the number of ortho chlorine substituents and, thus, the dihedral angle between the phenyl rings of the biphenyl moiety.34 The dihedral angle is an important factor in the interaction of individual PCB congeners or their metabolites with cellular target molecules. For example, PCBs and their hydroxylated metabolites with three or four ortho chlorine substituents are potent sensitizers of the Ryanodine receptor,9 which is thought to play an important role in the developmental toxicity of PCBs. Here we report the crystal structure of PCB 95 (15b) and several structurally related methoxylated PCBs with three ortho chlorine substituents (16b, 17a and 17d) to unambiguously establish the substitution pattern and add to the number of crystal structures available for application in studies on quantitative structure-activity relationships (Figure 3).
Figure 3.

Molecular structure of (A) 2,2′,3,5′,6-pentachlorobiphenyl (15b), (B) 2,2′,3,5′,6-pentachloro-4-methoxy-biphenyl (16b), (C) 2,2′,3,4′,6-pentachloro-5-methoxy-biphenyl (17a) and (D) 2,2′,3,4′,5′,6-hexachloro-5-methoxy-biphenyl (17d) showing the atom labeling scheme. Displacement ellipsoids are drawn at the 50% probability level.
PCB 95 (15b) crystallized in the monoclinic space group C2/c, with a = 13.7060(3), b = 11.4687(2), c = 17.2180(3) Å, and β = 113.313(1)°. The two methoxylated pentachlorobiphenyls 16b and 17a were also monoclinic (P21/n and P21/c, respectively). Similarly, two structurally related biphenyls with three ortho chlorine substituents, 2,2′,3,3′,6-pentachlorobiphenyl (PCB 84) and 2,2′,4,4′,5′,6-hexachloro-3-methoxybiphenyl, also crystallized in the monoclinic space group P21/c.35,36 The dimensions of the unit cell were a = 9.0848(2), b = 12.6058(2), c = 12.3064(3) Å, and β = 92.0452(9)° for 16b and a = 7.1653(2), b = 13.4435(3), c = 14.4816(4) Å, and β = 100.8733(9)° for 17a. In both structures, the MeO containing rings of inversion related molecules make pi-stacked pairs, with interplanar separations of 3.564(3) Å for 16b and 3.529(3) Å for 17a. In contrast to the pentachlorobiphenyl derivatives, the methoxylated hexachlorobiphenyl 17d crystallized in the orthorhombic space group Pbca, with a = 6.7926(1), b = 17.6314(4), c = 24.7070(6) Å.
The solid state dihedral angle of the four PCB derivatives shown in Figure 3 ranged from 69.65(9)° for 17a to 80.96(7)° for 16b. The other two PCB derivatives showed intermediate dihedral angles of 75.30(7)° and 78.38(12)° for 16b and 17d, respectively. The published dihedral angle of PCB 84 and 2,2′,4,4′,5′,6-hexachloro-3-methoxybiphenyl are 81.5° and 82.7°, respectively,35,36 which is slightly larger than the dihedral angle of the PCB derivatives shown in Figure 3. In contrast, the calculated dihedral angle of PCBs with three ortho chlorine substituents in aqueous solution is 90°.35
In comparison to PCBs with three ortho chlorine substituents, the dihedral angles of PCB derivatives with two ortho chlorine substituents are typically smaller and range from 58.3° to 75.3°,37–40 whereas the dihedral angles of PCB derivatives with four ortho substituents are larger and range from 83.92° to 87.3°.41–43 Overall, the fairly large range of solid state dihedral angles is a result of crystal packing effects that allows even PCB derivatives with three or four ortho chlorine substituents to adopt an energetically less favorable conformation to minimize the lattice energy. As a consequence, PCB derivatives with multiple ortho substituents have some conformational flexibility that can be important in their interactions with cellular target molecules, such as the Ryanodine receptor.
In summary, the Suzuki coupling reaction of chlorinated iodobenzenes and iodoanisoles with chlorinated benzene boronic acids using Pd(dba)2/DPDB allows straightforward access to individual PCB congeners and their methoxylated analogues, especially when compared to the classic Ullmann coupling reaction. The resulting methoxylated PCB derivatives can be converted into the corresponding hydroxylated compounds, thus providing access to putative hydroxylated PCB metabolites for toxicological studies. However, the synthesis of some key intermediates, such as 5a or 13, still remains a challenge due to the poor selectivity of the nitration and iodination reactions used in their synthesis. Compared to the calculated solution dihedral angle of 90°, the broad range of solid state dihedral angles of 69.7° to 81.0° suggests that even PCB derivatives with multiple ortho substituents have considerable conformational flexibility.
Silica gel for flash chromatography (40–64 μm) was purchased from Sorbent Technologies (Atlanta, GA, USA). The NMR spectra were recorded on a Bruker Avance-300 or a Bruker Avance DRX-400 spectrometer in the University of Iowa Central NMR Research Facility (Iowa City, IA, USA). Tetramethylsilane (TMS) was used as an internal standard. Combustion analyses were performed by Atlantic Microlab Inc. (Atlanta, GA, USA). HRMS were recorded by the High Resolution Mass Spectrometry Facility of the University of California Riverside (Riverside, CA, USA). Melting points were measured on a Mel-Temp melting point apparatus and are uncorrected. If no solvent is mentioned, melting points of the product after column chromatographic purification are reported. The purity of all PCB congeners and PCB derivatives was determined with an Agilent 6859 Gas Chromatograph (Agilent Technologies, CA, USA) equipped with flame ionization detector (FID) and a HP-1 (Methyl Silicone Gum) column (Hewlett Packard, PA, USA) and calculated based on the relative peak area. The following conditions were used for the gas chromatographic analysis: injector: 250 °C, detector (FID): 300 °C, starting temperature: 50 °C, final temperature: 250 °C, heating rate: 10 °C/min. In addition, GC-MS analysis of all compounds was performed in the electron impact (EI) mode on an Agilent 6890N Gas Chromatograph coupled with an Agilent 5975 Mass spectrometer (Agilent Technologies, CA, USA) using the same GC column and temperature program. Only the isotopic ion with the lowest mass is reported for all fragments observed in the MS spectra. The spectral data of all intermediates reported in Schemes 1–3 was in agreement with literature data.10
Nitration of 3,5-dichlorophenol (9)
A solution of 9 (2.5 g, 15.3 mmol) and sodium nitrite (1.45 g, 27.6 mmol) in water (25 mL) was cooled to 0 °C and sulfuric acid (1.2 mL diluted with 6 mL water) was added over a 15 minute period.27 The reaction mixture was heated under reflux for 6 hours with the incremental addition of additional sodium nitrite (6.33 g, 119.4 mmol). The mixture was allowed to cool to 20 °C, stirred for 17 h and extracted with ethylacetate (2 × 80 mL). The combined organic extracts were washed with water (30 mL), brine (30 mL), dried over sodium sulfate and filtered to yield a crude mixture of 12a and 12b in 44–92% yield. The 2-nitro and 4-nitro derivatives were separated by repeated column chromatography on silica gel with a gradient of hexane to chloroform to methanol-chloroform (5:95, v/v).
3,5-Dichloro-4-nitrophenol (12a)
Brown-yellow solid; yield 12 %; Mp 141–144 °C (hexane-chloroform; Mp 150 °C 44).
Rf = 0.85 (chloroform-methanol, 4:1).
1H NMR (400 MHz, CDCl3): δ = 6.89 (s, 2H).
13C NMR (100 MHz, CDCl3): δ = 115.9, 127.1, 159.2.
MS (EI, 70 eV): m/z (%) = 207 (31) [M]+, 177 (100), 149 (37), 133 (29), 97 (24), 85 (16), 73 (19), 62 (36).
HRMS: m/z [M]+ calculated for C6H3NO3Cl2 206.9485; found 206.9481.
3,5-Dichloro-2-nitrophenol (12b)
Yellow solid; yield 22 %; Mp 44–48 °C (Mp 51 °C 44).
Rf = 0.84 (chloroform-methanol, 4:1).
1H NMR (400 MHz, CDCl3): δ = 7.12 (m, 2H), 9.88 (s, -OH, 1H).
13C NMR (100 MHz, CDCl3): δ = 118.3, 123.9, 130.4, 140.8, 155.3.
MS (EI, 70 eV): m/z (%) = 207 (100) [M]+, 177 (89), 162 (12), 149 (68), 133 (34), 110 (20), 97 (71), 85 (15), 73 (25), 62 (45).
HRMS: m/z [M]+ calculated for C6H3NO3Cl2 206.9485; found 206.9485.
Methylation of 3,5-dichloro-4-nitrophenol (12a) and 3,5-dichloro-2-nitrophenol (12b)
The respective nitro compound (0.31 g, 1.5 mmol) was methylated with dimethyl sulfate 0.2 mL, 2.25 mmol) and K2CO3 (0.42 g, 3 mmol) in DMF (1 mL) at 60 °C for approximately 1 h.45 The reaction mixture was poured into ice-cold water and extracted with ethylacetate (3 × 10 mL). The combined organic extracts were washed with water (15 mL), 2N HCl (3 mL), brine (5 mL) and dried over Na2SO4. The product was purified by column chromatography on silica gel using a hexane-ethylacetate gradient.
4.3.2. 3,5-Dichloro-4-nitroanisole (11a)
Yellow solid; yield 92 %; Mp 56 °C (Mp 70 °C44).
Rf = 0.18 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.87 (s, -OCH3, 3H), 6.92 (s, 2H).
13C NMR (100 MHz, CDCl3): δ = 56.3, 103.5, 114.4, 127.5, 160.3.
MS (EI, 70 eV): m/z (%) = 221 (33) [M]+, 191 (100), 160 (20), 132 (10), 111 (11), 97 (40), 62 (23).
HRMS: m/z [M]+ calculated for C7H5NO3Cl2 220.9641; found 220.9648.
3,5-Dichloro-2-nitroanisole (11b)
Yellow solid; yield 65 %; Mp 65–66 °C (Mp 75 °C44).
Rf = 0.20 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.92 (s, -OCH3, 3H), 6.96 (d, J = 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 57.0, 111.9, 121.6, 137.0, 152.2.
MS (EI, 70 eV): m/z (%) = 221 (100) [M]+, 191 (56), 174 (64), 160 (68), 148 (40), 128 (51), 109 (59), 97 (90), 74 (44), 62 (36).
HRMS: m/z [M]+ calculated for C7H5NO3Cl2 220.9641; found 220.9636.
Bromination of 3,5-dichloroanisole (10)
N-Bromosuccinimide (5.3 g, 29.8 mmol) and HCl (10% v/v, 1 mL) were added to a solution of 10 (5.0 g, 29.8 mmol) in acetone (50 mL) and stirred at 20 °C for 30 minutes.28 The solvent was removed under reduced pressure and the residue was washed with hexane (25 mL). Recrystallization from methanol yielded the desired 4-bromo-3,5-dichloroanisole (14) in 25 % yield.
Mp 59–60 °C.
Rf = 0.24 (hexane).
1H NMR (300 MHz, CDCl3): δ = 3.70 (s, -OCH3, 3H), 6.93 (s, 2H).
13C (75 MHz, CDCl3): δ = 55.9, 114.0, 114.8, 136.5, 158.8.
MS (EI, 70 eV): m/z (%) = 254 (58) [M]+, 239 (8), 224 (8), 211 (20), 97 (21), 62 (15).
HRMS: m/z [M]+ calculated for C7H5BrOCl2 253.8901; found 253.8896.
Chlorination of 4-bromo-3,5-dichloroanisole (14)
Hydrogen peroxide (30%, 2.6 mL) and conc. HCl (6.7 mL) was added to 4-bromo-3,5-dichloroanisole (14) (5.2 g, 20.3 mmol) in acetic acid (50 mL).10 Dichloromethane (6 mL) was added to redissolve 14 and the heterogenous mixture was stirred for 16 h. The reaction mixture was extracted with dichloromethane (3 × 25 mL) and the combined organic extracts were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. Recrystallization of the crude product from methanol gave 4-bromo-2,3,5-trichloroanisole (4) in 60% yield as a white solid.
Mp 107–108 °C.
Rf = 0.17 (hexane).
1H NMR (400 MHz, CDCl3): δ = 3.90 (s, -OCH3, 3H), 6.98 (s, 1H).
13C (100 MHz, CDCl3): δ = 57.0, 111.6, 115.0, 121.6, 134.1, 135.5, 155.1.
MS (EI, 70 eV): m/z (%) = 288 (49) [M]+, 273 (18), 245 (24), 131 (21), 96 (14), 61 (11).
HRMS: m/z [M]+ calculated for C7H4BrOCl3 287.8511; found 287.8509.
General procedure for the Ullmann coupling reaction
A mixture of aryl iodide (3 mmol), methoxy aryl iodide (1.5 mmol) and activated copper bronze (3 gm) in a sealed glass ampoule flushed with nitrogen was heated in a sand bath at 230 °C for 7 days.10 The ampoule was allowed to cool to room temperature, opened and the contents were extracted with boiling CH2Cl2 (3 × 100 mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure to give a dark brown, viscous oil. Repeated column chromatography on silica gel eluted with hexane followed by recrystallization gave the corresponding biphenyl derivatives in moderate yield (Table 1).
General procedure for the Suzuki coupling reaction
Suzuki reactions were carried out in 60 mL sample collection vials (I-Chem, New Castle, DE, USA) with a Teflon rubber septum. The dried glass vial was charged with the methoxy aryl iodide (2 mmol) chlorinated aryl boronic acid (2 mmol) followed by DPDB (45 mg), bis(dibenzylideneacetone) palladium (26 mg), K3PO4·H2O (1.2 g) and toluene (2 mL), evacuated and backfilled with nitrogen. The reaction mixture was stirred at 110 °C for 24 h, diluted with dichloromethane (5 mL), filtered through Celite, and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel using hexane as eluent to provide the desired compounds (Table 1).
2,2′,3,4′,6-Pentachlorobiphenyl (PCB 91, 15a)
Viscous oil (Mp 62–63 °C46); >85% purity by GC-MS.
Rf = 0.59 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 7.12 (d, J = 8.4 Hz, 1H), 7.36–7.39 (m, 2H), 7.47 (d, J = 8.8 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 127.5, 128.3, 129.6, 130.6, 131.5, 131.9, 133.3, 133.7, 134.3, 134.6, 135.3, 137.8.
MS (EI, 70 eV): m/z (%) = 324 (70) [M]+, 289 (23), 254 (62), 184 (19).
HRMS: m/z [M]+ calculated for C12H5Cl5 323.8828; found 323.8837.
2,2′,3,5′,6-Pentachlorobiphenyl (PCB 95, 15b)
Colorless crystalline solid; Mp 91–92 °C (chloroform-methanol) (93–94 °C46).
Rf = 0.59 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 7.20 (d, J = 2.0 Hz, 1H), 7.35–7.38 (m, 2H), 7.46 (pseudo t, J = 8.7 Hz, 2H).
13C NMR (100 MHz, CDCl3): δ = 128.4, 130.1, 130.5, 130.7, 130.8, 131.8, 132.0, 132.8, 133.2, 133.5, 137.4, 137.7.
MS (EI, 70 eV): m/z (%) = 324 (88) [M]+, 289 (42), 254 (86), 184 (38), 127 (26), 109 (19).
Anal. Calcd for C12H5Cl5: C, 44.17; H, 1.53; Found: C, 43.97; H, 1.31.
2,2′,3,3′,4′,6-Hexachlorobiphenyl (PCB 132, 15c)
Colorless crystalline solid; Mp 112–114 °C (chloroform-methanol) (116–118 °C46).
Rf = 0.53 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 7.05 (d, J = 8.3 Hz, 1H), 7.37 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 8.7 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 128.4, 128.6 (2 × C), 130.8, 132.0, 132.5, 133.1, 133.5, 133.7, 134.6, 136.3, 138.2.
MS (EI, 70 eV): m/z (%) = 358 (56) [M]+, 322 (24), 289 (44), 218 (22), 145 (19).
HRMS: m/z [M]+ calculated for C12H4Cl6 357.8439; found 357.8445.
2,2′,3,4′,6-Pentachloro-4-methoxy-biphenyl (16a)
Colorless crystalline solid; Mp 127–128 °C (chloroform-methanol).
Rf = 0.38 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.96 (s, 3H, -OCH3), 7.01 (s, 1H), 7.12 (d, J = 8.2 Hz, 1H), 7.34 (dd, J = 2.1 & 8.2 Hz, 1H), 7.52 (d, J = 2.1 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 56.7, 111.2, 121.0, 127.4, 129.46, 129.49, 132.1, 133.0, 134.46, 134.55, 134.9, 135.1, 156.0.
MS (EI, 70 eV): m/z (%) = 354 (62) [M]+, 311 (14), 240 (26), 171 (12).
Anal. Calcd for C13H7Cl5O: C, 43.75; H, 1.96; Found: C, 44.04; H, 1.98.
2,2′,3,5′,6-Pentachloro-4-methoxy-biphenyl (16b)
Colorless crystalline solid; Mp 129–130 °C (chloroform-methanol).
Rf = 0.39 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 3.97 (s, 3H, -OCH3), 7.01 (s, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.35 (dd, J = 2.5 & 8.6 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H).
13C MR (100 MHz, CDCl3): δ = 56.8, 111.1, 121.0, 129.3, 129.9, 130.6, 131.2, 132.5, 132.6, 132.9, 134.3, 137.4, 156.0.
MS (EI, 70 eV): m/z (%) = 354 (73) [M]+, 311 (34), 286 (16), 241 (44), 207 (27), 171 (20).
Anal. Calcd for C13H7Cl5O: C, 43.75; H, 1.96; Found: C, 43.91; H, 1.78.
2,2′,3,3′,4′,6-Hexachloro-4-methoxy-biphenyl (16c)
Colorless crystalline solid; Mp 168–169 °C (chloroform-methanol).
Rf = 0.34 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.97 (s, 3H, -OCH3), 7.01 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 7.48 (d, J = 8.3 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 56.8, 111.2, 121.2, 128.5, 129.4, 129.7, 132.3, 132.9, 134.3, 134.4, 136.3, 156.2.
MS (EI, 70 eV): m/z (%) = 388 (50) [M]+, 345 (17), 275 (18), 205 (10).
Anal. Calcd for C13H6Cl6O: C, 39.94; H, 1.55; Found: C, 39.68; H, 1.47.
2,2′,3,4′,6-Pentachloro-5-methoxy-biphenyl (17a)
Colorless solid; M.P 118–120 °C (chloroform-methanol).
Rf = 0.39 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3), 7.11 (d, J = 8.2 Hz, 1H), 7.11 (s, 1H), 7.36 (dd, J = 2.1 & 8.2 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 56.7, 113.3, 122.2, 124.5, 127.4, 129.6, 131.4, 131.7, 134.1, 134.7, 135.1, 138.3, 154.1.
MS (EI, 70 eV): m/z (%) = 354 (58) [M]+, 339 (2), 311 (16), 241 (21), 171 (9).
Anal. Calcd for C13H7Cl5O: C, 43.75; H, 1.96; Found: C, 43.77; H, 1.76.
2,2′,3,5′,6-Pentachloro-5-methoxy-biphenyl (17b)
Colorless solid; Mp 40–42 °C (chloroform-methanol).
Rf = 0.38 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3), 7.12 (s, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.35 (dd, J = 2.5 & 8.6 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H)
13C NMR (100 MHz, CDCl3): δ = 56.7, 113.3, 122.0, 124.3, 129.9, 130.4, 130.7, 131.67, 131.74 131.8, 132.7, 137.5, 138.1, 154.1.
MS (EI, 70 eV): m/z (%) = 354 (91) [M]+, 339 (5), 311 (25), 241 (28), 171 (29).
Anal. Calcd for C13H7Cl5O: C, 43.75; H, 1.96; Found: C, 43.80; H, 1.91.
2,2′,3,3′,4′,6-Hexachloro-5-methoxy-biphenyl (17c)
Colorless crystalline solid; Mp 134–135 °C (chloroform-methanol).
Rf = 0.34 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3), 7.03 (d, J = 8.3 Hz, 1H), 7.12 (s, 1H), 7.50 (d, J = 8.3 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 56.7, 113.4, 122.0, 124.2, 128.6, 131.9, 132.4, 133.6, 134.4, 136.4, 138.4, 154.2.
MS (EI, 70 eV): m/z (%) = 388 (44) [M]+, 345 (14), 275 (8), 205 (8).
Anal. Calcd for C13H6Cl6O: C, 39.94; H, 1.55; Found: C, 40.20; H, 1.44.
2,2′,3,4′,5′,6-Hexachloro-5-methoxy-biphenyl (17d)
Colorless solid; yield 43 %; Mp 91–92 °C (chloroform-methanol).
Rf = 0.39 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3,), 7.13 (s, 1H), 7.29 (s, 1H), 7.64 (s, 1H)
13C NMR (75 MHz, CDCl3): δ = 56.7, 113.6, 122.1, 124.3, 131.0, 131.4, 131.8, 131.9, 132.3, 133.6, 135.8, 137.2, 154.3.
MS (EI, 70 eV): m/z (%) = 388 (47) [M]+, 345 (16), 338 (12), 275 (23), 205 (12).
Anal. Calcd for C13H6Cl6O: C, 39.92; H, 1.55; Found: C, 40.09; H, 1.27.
2,2′,3,4′,6-Pentachloro-4,5-dimethoxy-biphenyl (18a)
Colorless solid; Mp 70–72 °C (chloroform-methanol).
Rf = 0.42 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3), 4.00 (s, 3H, -OCH3), 7.12 (d, J = 8.3 Hz, 1H), 7.35 (dd, J = 8.3 & 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 61.22, 61.24, 127.0, 127.4, 127.8, 128.6, 129.6, 131.7, 133.2, 134.5, 135.2, 149.4, 151.2.
MS (EI, 70 eV): m/z (%) = 384 (64) [M]+, 369 (24), 341 (18), 326 (20), 228 (20).
Anal. Calcd for C14H9Cl5O2: C, 43.51; H, 2.35; Found: C, 43.74; H, 2.26.
2,2′,3,5′,6-Pentachloro-4,5-dimethoxy-biphenyl (18b)
Colorless oil.
Rf = 0.44 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 3.95 (s, 3H, -OCH3), 4.00 (s, 3H, -OCH3), 7.19 (d, J = 2.4 Hz, 1H), 7.35 (dd, J = 2.4 & 8.3 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 61.22, 61.24, 127.0, 127.7, 128.5, 130.0, 130.7, 130.8, 132.1, 132.7, 133.0, 137.3, 149.4, 151.2.
MS (EI, 70 eV): m/z (%) = 384 (60) [M]+, 369 (26), 341 (18), 326 (15), 228 (22).
Anal. Calcd for C14H9Cl5O2: C, 43.51; H, 2.35; Found: C, 43.91; H, 2.24.
2,2′,3,3′,4′,6-Hexachloro-4,5-dimethoxy-biphenyl (18c)
Colorless solid; Mp 93–95 °C (chloroform-methanol).
Rf = 0.41 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 3.96 (s, 3H, -OCH3), 4.00 (s, 1H, -OCH3), 7.05 (d, J = 8.3 Hz, 1H), 7.49 (d, J = 8.3 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 61.19, 61.22, 127.1, 127.6, 128.4, 128.6, 128.9, 132.4, 133.2, 134.0, 134.5, 136.3, 149.4, 151.3.
MS (EI, 70 eV): m/z (%) = 418 (56) [M]+, 403 (20), 375 (9), 360 (20), 262 (20).
Anal. Calcd for C14H8Cl6O: C, 39.95; H, 1.92; Found: C, 40.00; H, 1.79.
General Procedure for the demethylation reaction
Boron tribromide (1 M in hexane, 5 equiv.) was added to a solution of the methoxylated biphenyl (0.3 mmol) in anhydrous dichloromethane (5 mL) under a nitrogen atmosphere.33 The reaction mixture was stirred at room temperature for 14 h, cooled with ice/salt, and hydrolyzed with an equal volume of ice-cold water. The aqueous phase was extracted with dichloromethane (2 × 50 mL). The combined organic layers were washed with water (3 × 25 mL), dried over MgSO4, and the solvent was removed under reduced pressure. The product was purified by column chromatography on silica gel with hexane-ethyl acetate (1:1, v/v) as eluent followed by recrystallization using methanol-chloroform (3:1, v/v), yielding 50–55% of corresponding hydroxyl compounds as white solids or colorless oils.
2,2′,3,4′,6-Pentachlorobiphenyl-4-ol (19a)
Colorless oil; yield 62 %.
Rf = 0.21 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 5.85 (br s, 3H, -OH), 7.12 (d, J = 8.2 Hz, 1H), 7.16 (s, 1H), 7.35 (dd, J = 2.5 & 8.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 115.4, 118.5, 127.4, 129.6, 129.8, 132.2, 133.4, 133.8, 134.4, 135.0, 135.2, 152.4.
MS (EI, 70 eV): m/z (%) = 340 (60) [M]+, 270 (31), 241 (11), 171 (20).
HRMS: m/z [M-H]+ calculated for C12H4OCl5 338.8705; found 338.8705.
2,2′,3,5′,6-Pentachlorobiphenyl-4-ol (19b)
Colorless oil; yield 47 %.
Rf = 0.18 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 5.90 (br s, 3H, -OH), 7.16 (s, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.36 (dd, J = 2.5 & 8.6 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 115.4, 118.5, 129.6, 130.0, 130.6, 131.2, 132.5, 132.6, 133.2, 133.6, 137.2, 152.4.
MS (EI, 70 eV): m/z (%) = 340 (68) [M]+, 270 (345), 241 (11), 171 (20).
HRMS: m/z [M-H]+ calculated for C12H4OCl5 338.8705; found 338.8702.
2,2′,3,3′,4′,6-Hexachlorobiphenyl-4-ol (19c)
Colorless crystalline solid; yield 49 %; Mp 94–95 °C (chloroform-methanol).
Rf = 0.19 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 5.86 (br s, 3H, -OH), 7.05 (d, J = 8.3 Hz, 1H), 7.16 (s, 1H) 7.48 (d, J = 8.3 Hz, 1H).
13C NMR (75 MHz, CDCl3): δ = 115.5, 118.6, 128.5, 129.4, 129.9, 132.4, 133.2, 133.6, 134.4, 134.5, 136.2, 152.6.
MS (EI, 70 eV): m/z (%) = 374 (50) [M]+, 304(32), 275 (10), 205 (17).
HRMS: m/z [M-H]+ calculated for C12H3OCl6 372.8315; found 372.8316.
2,2′,3,4′,6-Pentachlorobiphenyl-5-ol (20a)
Colorless oil; yield 50 %.
Rf = 0.19 (hexane-ethylacetate, 9:1).
1H NMR (300 MHz, CDCl3): δ = 5.79 (br s, 1H, -OH), 7.13 (d, J = 8.3 Hz, 1H), 7.26 (s, 1H), 7.38 (dd, J = 2.1 & 8.3 Hz, 1H), 7.55 (d, J = 2.1 Hz, 1H).
13C NMR (100 MHz, CDCl3): δ = 117.5, 119.5, 124.6, 127.5, 129.7, 131.4, 132.5, 134.2, 134.3, 135.5, 137.4, 150.5.
MS (EI, 70 eV): m/z (%) = 340 (63) [M]+, 305 (10), 270 (27), 242 (14), 207 (12), 171 (19).
HRMS: m/z [M]+ calculated for C12H5OCl5 339.8778; found 338.8791.
2,2′,3,5′,6-Pentachlorobiphenyl-5-ol (20b)
Colorless oil; yield 50 %.
Rf = 0.19 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 5.15 (br s, 1H, -OH), 7.19 (d, J = 2.5 Hz, 1H), 7.26 (s, 1H), 7.37 (dd, J = 2.5 & 8.6 Hz, 1H), 7.43 (d, J = 8.6 Hz, 1H).
13C (100 MHz, CDCl3): δ = 117.6, 119.3, 124.5, 130.2, 130.4, 130.8, 131.8, 132.6, 132.9, 137.1, 137.2, 150.6.
MS (EI, 70 eV): m/z (%) = 340 (82) [M]+, 305 (16), 270 (37), 241 (18), 207 (13), 171 (40).
HRMS: m/z [M-H]+ calculated for C12H4OCl5 338.8705; found 338.8707.
2,2′,3,3′,4′,6-Hexachlorobiphenyl-5-ol (20c)
White crystalline solid; yield 50 %; Mp 126–128 °C.
Rf = 0.17 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 5.75 (br s, 1H, -OH), 7.05 (d, J = 8.4 Hz, 1H), 7.28 (s, 1H), 7.51 (d, J = 8.4 Hz, 1H)
13C NMR (100 MHz, CDCl3): δ = 117.7, 119.4, 124.4, 128.6, 128.8, 132.6, 132.7, 133.7, 134.7, 136.0, 137.5, 150.6.
MS (EI, 70 eV): m/z (%) = 374 (50) [M]+, 339 (10), 305 (15), 275 (10), 205 (15).
HRMS: m/z [M]+ calculated for C12H4OCl6 373.8388; found 373.8394.
2,2′,3,4′,5′,6-Hexachlorobiphenyl-5-ol (20d)
Colorless oil; yield 50 %.
Rf = 0.18 (hexane-ethylacetate, 9:1).
1H NMR (400 MHz, CDCl3): δ = 5.72 (br s, 1H, -OH), 7.28 (s, 1H), 7.30 (s, 1H), 7.65 (s, 1H).
13C NMR (100 MHz, CDCl3): δ = 117.8, 119.3, 124.5, 131.2, 131.6, 131.7, 132.4, 132.8, 133.9, 135.4, 136.3, 150.5.
MS (EI, 70 eV): m/z (%) = 374 (48) [M]+, 303 (18).
HRMS: m/z [M-H]+ calculated for C12H3OCl6 372.8315; found 372.8319.
X-ray crystal structure analysis
X-ray diffraction data were collected at 90.0(2) K on a Nonius KappaCCD diffractometer as described previously.34 Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 797054 - 797058. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgments
The project described was supported by grants ES05605, ES013661 and ES017425 from the National Institute of Environmental Health Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
References
- 1.Hansen LG. The ortho side of PCBs: Occurrence and disposition. Kluwer Academic Publishers; Boston: 1999. [Google Scholar]
- 2.Robertson LW, Hansen LG. Recent advances in the environmental toxicology and health effects of PCBs. University Press of Kentucky; Lexington: 2001. [Google Scholar]
- 3.Hu D, Hornbuckle KC. Environ Sci Technol. 2010;44:2822. doi: 10.1021/es902413k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schantz SL, Widholm JJ, Rice DC. Environ Health Perspect. 2003;111:357. doi: 10.1289/ehp.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kodavanti PRS. Intracellular signaling and developmental neurotoxicity. In: Zawia NH, editor. Molecular Neurotoxicology: environmental agents and transcription-transduction coupling. CRC press; Boca Roton, FL: 2004. p. 151. [Google Scholar]
- 6.Wong PW, Brackney WR, Pessah IN. J Biol Chem. 1997;272:15145. doi: 10.1074/jbc.272.24.15145. [DOI] [PubMed] [Google Scholar]
- 7.Wong PW, Joy RM, Albertson TE, Schantz SL, Pessah IN. Neurotoxicology. 1997;18:443. [PubMed] [Google Scholar]
- 8.Wong PW, Pessah IN. Mol Pharmacol. 1996;49:740. [PubMed] [Google Scholar]
- 9.Pessah IN, Hansen LG, Albertson TE, Garner CE, Ta TA, Do Z, Kim KH, Wong PW. Chem Res Toxicol. 2006;19:92. doi: 10.1021/tx050196m. [DOI] [PubMed] [Google Scholar]
- 10.Waller SC, He YA, Harlow GR, He YQ, Mash EA, Halpert JR. Chem Res Toxicol. 1999;12:690. doi: 10.1021/tx990030j. [DOI] [PubMed] [Google Scholar]
- 11.Telu S, Parkin S, Robertson LW, Lehmler H-J. Environ Int. 2009 doi: 10.1016/j.envint.2009.01.013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergman Å, Klasson Wehler E, Kuroki H, Nilsson A. Chemosphere. 1995;30:1921. [Google Scholar]
- 13.Goldstein JA, Hass JR, Linko P, Harvan DJ. Drug Metab Dispos. 1978;6:258. [PubMed] [Google Scholar]
- 14.Shaikh NS, Parkin S, Lehmler HJ. Organometallics. 2006;25:4207. [Google Scholar]
- 15.Moron M, Sundström G, Wachtmeister CA. Acta Chem Scand. 1973;27:3121. doi: 10.3891/acta.chem.scand.27-3121. [DOI] [PubMed] [Google Scholar]
- 16.Lehmler HJ, Robertson LW. Chemosphere. 2001;45:1119. doi: 10.1016/s0045-6535(01)00052-2. [DOI] [PubMed] [Google Scholar]
- 17.Lehmler HJ, Robertson LW. Chemosphere. 2001;45:137. doi: 10.1016/s0045-6535(00)00546-4. [DOI] [PubMed] [Google Scholar]
- 18.Kania-Korwel I, Parkin S, Robertson LW, Lehmler HJ. Chemosphere. 2004;56:735. doi: 10.1016/j.chemosphere.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 19.Luthe GM, Schut BG, Aaseng JE. Chemosphere. 2009;77:1242. doi: 10.1016/j.chemosphere.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Ji HY, Fu XL, Yang Y, Xue YR, Gao GH. Chin Chem Lett. 2009;20:927. [Google Scholar]
- 21.Kang H, Facchetti A, Stern CL, Rheingold AL, Kassel WS, Marks TJ. Org Lett. 2005;7:3721. doi: 10.1021/ol051365x. [DOI] [PubMed] [Google Scholar]
- 22.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 23.Feuerstein M, Doucet H, Santelli M. Tetrahedron Lett. 2001;42:6667. [Google Scholar]
- 24.Kenny JR, Maggs JL, Meng X, Sinnott D, Clarke SE, Park BK, Stachulski AV. J Med Chem. 2004;47:2816. doi: 10.1021/jm030891w. [DOI] [PubMed] [Google Scholar]
- 25.Fryszkowska A, Tilford RW, Guo F, Kaszynski P. Tetrahedron. 2005;61:2327. [Google Scholar]
- 26.Bhalerao NVM, Panse DG, Bapat BV, Ghatge BB. Indian J Chem, Sect B. 1985;24B:327. [Google Scholar]
- 27.Hartz RA, Ahuja VT, Rafalski M, Schmitz WD, Brenner AB, Denhart DJ, Ditta JL, Deskus JA, Yue EW, Arvanitis AG, Lelas S, Li YW, Molski TF, Wong H, Grace JE, Lentz KA, Li J, Lodge NJ, Zaczek R, Combs AP, Olson RE, Mattson RJ, Bronson JJ, Macor JE. J Med Chem. 2009;52:4161. doi: 10.1021/jm900302q. [DOI] [PubMed] [Google Scholar]
- 28.Tietze LF, Vock CA, Krimmelbein IK, Nacke L. Synthesis. 2009:2040. [Google Scholar]
- 29.Springer DM, Luh BY, Goodrich J, Bronson JJ. Bioorg Med Chem. 2003;11:265. doi: 10.1016/s0968-0896(02)00336-x. [DOI] [PubMed] [Google Scholar]
- 30.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 31.Huang X, Buchwald SL. Org Lett. 2001;3:3417. doi: 10.1021/ol0166808. [DOI] [PubMed] [Google Scholar]
- 32.Yang D, Kim KH, Phimister A, Bachstetter AD, Ward TR, Stackman RW, Mervis RF, Wisniewski AB, Klein SL, Kodavanti PRS, Anderson KA, Wayman G, Pessah IN, Lein PJ. Environ Health Perspect. 2009;17:426. doi: 10.1289/ehp.11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bauer U, Amaro AR, Robertson LW. Chem Res Toxicol. 1995;8:92. doi: 10.1021/tx00043a012. [DOI] [PubMed] [Google Scholar]
- 34.Lehmler HJ, Parkin S, Robertson LW. Chemosphere. 2002;46:485. doi: 10.1016/s0045-6535(01)00177-1. [DOI] [PubMed] [Google Scholar]
- 35.Lehmler HJ, Robertson LW, Parkin S. Acta Cryst. 2005;E61:o3025. [Google Scholar]
- 36.Rissanen K, Valkonen J, Mannila B. Acta Cryst. 1988;C44:684. [Google Scholar]
- 37.Vyas SM, Parkin S, Lehmler HJ. Acta Cryst. 2006;E62:o2905. [Google Scholar]
- 38.Rissanen K, Valkonen J, Mannila B. Acta Cryst. 1988;C44:682. [Google Scholar]
- 39.Singh P, Pedersen LG, McKinney JD. Acta Cryst. 1986;C42:1172. [Google Scholar]
- 40.Miao XS, Chu SG, Xu XB, Jin XL. Acta Crystallogr. 1996;C52:2581. [Google Scholar]
- 41.Pedersen BF. Acta Cryst. 1975;B31:2931. [Google Scholar]
- 42.Singh P, McKinney JD. Acta Cryst. 1979;B35:259. [Google Scholar]
- 43.Shaikh NS, Parkin S, Lehmler HJ. Acta Cryst. 2006;E62:o662. [Google Scholar]
- 44.Hodgson HH, Wignall JS. J Chem Soc. 1927:2216. [Google Scholar]
- 45.Waterhouse I. J Labelled Compd Radiopharm. 1999;42:1075. [Google Scholar]
- 46.Bolgar M, Cunningham J, Cooper R, Kozloski R, Hubball J, Miller DP, Crone T, Kimball H, Janooby A, Miller B, Fairless B. Chemosphere. 1995;31:2687. [Google Scholar]