

Abstract
Activation of the complement cascade, a powerful effector mechanism of the innate immune system, is associated with neuroinflammation but also with elimination of inappropriate synapses during development. Synthesis of C1q, a recognition component of the complement system, occurs in brain during ischemia/reperfusion and Alzheimer's disease, suggesting that C1q may be a response to injury. In vitro, C1q, in the absence of other complement proteins, improves neuronal viability and neurite outgrowth and prevents β-amyloid-induced neuronal death, suggesting that C1q may have a direct neuroprotective role. Here, investigating the molecular basis for this neuroprotection in vitro, addition of C1q to rat primary cortical neurons significantly upregulated expression of genes associated with cholesterol metabolism, such as cholesterol-25-hydroxylase and insulin induced gene 2, and transiently decreased cholesterol levels in neurons, known to facilitate neurite outgrowth. In addition, the expression of syntaxin-3 and its functional association with synaptosomal-associated protein 25 was increased. C1q also increased the nuclear translocation of cAMP response element-binding protein and CCAAT/enhancer-binding protein-δ (C/EBP-δ), two transcription factors involved in nerve growth factor (NGF) expression and downregulated specific microRNAs, including let-7c that is predicted to target (and thus inhibit) NGF and neurotrophin-3 (NT-3) mRNA. Accordingly, C1q increased expression of NGF and NT-3, and small interfering RNA inhibition of C/EBP-δ, NGF, or NT-3 expression prevented the C1q-dependent neurite outgrowth. No such neuroprotective effect is seen in the presence of C3a or C5a. Finally, the induced neuronal gene expression required conformationally intact C1q. These results show that C1q can directly promote neuronal survival, thereby demonstrating new interactions between immune proteins and neuronal cells that may facilitate neuroprotection.
Introduction
The complement system is a powerful effector mechanism of the innate immune system that contributes to protection from infection and resolution of injury (Köhl, 2006). However, tissue damage can result from dysregulated activation of the complement system as seen in arthritis, age-related macular degeneration, and Alzheimer's disease (AD) (Alexander et al., 2008; Sjöberg et al., 2009). Treatment of mouse models of AD with a C5a receptor antagonist significantly reduced neuropathology (Fonseca et al., 2009), suggesting a detrimental consequence of complement activation. However, during development C1q, a component of the complement initiator C1 complex, is expressed in synaptic regions of developing postnatal CNS, and its absence in knock-out mouse results in the failure of anatomical refinement of retinogeniculate connections and excessive retinal innervation (Stevens et al., 2007) and enhanced synaptic connectivity in neocortical slices that lead to epileptogenesis (Chu et al., 2010). Thus, depending on the timing and local environment, the complement cascade can facilitate proper neuronal development or accelerate chronic inflammatory response contributing to neurodegeneration.
Induced synthesis of C1q in the CNS has been seen in several injury models, such as viral infection (Dietzschold et al., 1995), kainic acid treatment (Goldsmith et al., 1997), stroke (Huang et al., 1999), and in hippocampal organotypic slice cultures stimulated with β-amyloid (Aβ) (Fan and Tenner, 2004). In blood, C1q is normally present in complex with proenzymes C1r and C1s as the C1 macromolecular initiator of the classical complement pathway (Ziccardi and Tschopp, 1982). However, C1q can be synthesized in the absence of C1r and C1s by myeloid cells in vitro (Bensa et al., 1983), consistent with the ability of C1q to function not only as part of the C1 complex initiating the classical complement pathway but independently. C1q in the absence of other complement components has been shown to enhance ingestion of apoptotic cells by phagocytic cells and modulate inflammation (Ogden et al., 2001; Fraser et al., 2009, 2010), suggesting a basis for the strong association of lupus with C1q deficiency in humans (Walport et al., 1998). We recently reported that C1q, in the absence of other complement components, increases neuronal survival and neurite outgrowth compared with untreated neurons and protects against Aβ-induced neurotoxicity (Pisalyaput and Tenner, 2008). Heat-inactivated C1q, C1q tails, and C1q globular “heads” showed complete loss of neuroprotective ability, indicating that protection requires conformationally intact C1q (Pisalyaput and Tenner, 2008). The observed selective upregulation of C1q in the CNS and the demonstrated C1q-dependent neuroprotection suggest that C1q can induce a potent and novel neuroprotective program.
Here, microarray analysis has identified candidate molecular mechanisms underlying these novel neuroprotective effects of C1q. Utilizing in vitro primary immature cortical neurons, C1q upregulated expression of genes associated with cholesterol metabolism and neurite outgrowth and downregulated the expression of genes associated with inflammation, as well as specific microRNAs (miRNAs), including let-7c that is predicted to target nerve growth factor (NGF) and neurotrophin-3 (NT-3). The upregulated expression of several of these genes at the mRNA and protein levels was validated, and suppression of expression of specifically upregulated genes prevented the C1q-mediated neuronal protection. These results uncover a previously unidentified mechanism of a direct protective effect on neurons of this injury-induced protein, thereby revealing novel targets for neuroprotection in the absence of activation of the complement cascade.
Materials and Methods
Reagents.
Serum-free Neurobasal (NB) medium, trypsin–EDTA, N2 supplement, and l-glutamine were obtained from Invitrogen. Poly-l-lysine hydrobromide, anti-β-actin, and microtubule associated protein-2 (MAP-2) antibodies were obtained from Sigma-Aldrich. Anti-neurotrophin-3 (NT-3) and NGF-β antibodies were obtained from Millipore. Anti-syntaxin 3 (stx3) and synaptosomal-associated protein 25 (SNAP25) antibodies were obtained from Abcam. Anti-CCAAT/enhancer-binding protein-δ (C/EBP-δ), phosphorylated cAMP response element-binding protein (pCREB), and CREB1 antibodies were obtained from Santa Cruz Biotechnology. Alexa Fluor 488-conjugated pCREB antibody was obtained from Cell Signaling Technology. Human C1q was isolated from serum as described previously (Tenner et al., 1981) and modified by Young et al. (1991). C1q was heat inactivated for 30 min at 56°C, and C1q tails were obtained by pepsin digestion as described (Reid, 1976). Purified human C3a and C5a were obtained from Complement Technology.
Animals, neuron isolation, and culture.
All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of University of California, Irvine. Cortical neurons were isolated from day 18 Sprague Dawley rat embryos (Charles River Laboratories) or day 15 C57BL/6 mouse embryos as described previously (Li et al., 2004). Neurons were plated on poly-l-lysine (1 mg/ml) at a density of 500 cells/mm2 in NB medium supplemented with N2 and grown for 3 d before stimulation with 10 nm C1q. Mouse neurons were treated with 5 nm AraC (Sigma-Aldrich) 24 h before adding C1q to limit glia proliferation. In some experiments, neurons were transfected with 5 nm silencer select negative control small interfering RNA (siRNA) (Ambion) or silencer select siRNA specific for NGF, NT-3, or C/EBP-δ using the siPORT NeoFX Transfection Agent kit (Ambion), according to the instructions of the manufacturer.
RNA extraction and microarray analysis.
Total RNA from untreated neurons and neurons treated with C1q for 3 h was extracted using the RNeasy Mini kit (Qiagen) as described previously (Benoit et al., 2008). RNA quality and quantity were assessed with the 2100 Bioanalyzer (Agilent Technologies) and the NanoDrop Spectrophotometer (NanoDrop Technologies). Gene expression profiles were studied using the Rat Gene 1.0 ST array (Affymetrix). Briefly, total RNA is retro-transcribed to cDNA, followed by cRNA amplification and biotin labeling. After purification, cRNA are hybridized on imprinted slides, washed, and scanned using the Affymetrix GCOS software (performed by the microarray core facility at University of California, Irvine). Data processing and analysis were performed using JMP Genomics 4.0 software (SAS Institute). Briefly, interarray median correction was used to normalize signal intensities. Then, significant differences in gene expression in C1q-treated neurons compared with untreated neurons were identified by ANOVA test using the Bonferroni's multiple testing method and a false-positive rate (α error) of 0.05. Only 388 genes, including four miRNAs, with a p value < 0.01 and an absolute fold difference ≥ 2, were considered as significantly modulated (supplemental Table 1, available at www.jneurosci.org as supplemental material). The EnsEMBL sequence predicted targets of the miRNAs modulated by C1q were determined using the MicroCosm Targets tool of the miRBase, which uses the miRanda algorithm to determine score, and the statistical model proposed by Rehmsmeier et al. (2004) to calculate p value. Only predicted targeted genes with a p value < 0.001 were included in the analysis. Functional classification and clustering of modulated genes by C1q and miRNA predicted targets were performed using DAVID software (http://david.abcc.ncifcrf.gov/) (Dennis et al., 2003; Huang et al., 2009). All data were entered in the Gene Expression Omnibus database following the MIAME procedure (Brazma et al., 2001) and can be retrieved using the accession number GSE18860.
Reverse transcription, PCR, and quantitative real-time PCR.
The cDNA synthesis was performed with 100 ng of total RNA, 0.5 μg of oligo-dT primer, 40 units of RNaseOUT recombinant ribonuclease inhibitor, and 200 units of Moloney murine leukemia virus reverse transcriptase (RT) (Invitrogen) according to the protocol of the manufacturer. PCR was performed with 200 ng of cDNA using the Taq Master Mix (Qiagen). Quantitative real-time (qRT)-PCR was performed using the 7300 fast real-time PCR system (Applied Biosystems) and the relative quantification method. Briefly, amplification was conducted in a 25 μl volume using 12.5 μl of FastStart SYBR Green Master (ROX) mix (Roche Applied Science), 100 ng of template cDNA, and 0.3 μm each of forward and reverse gene-specific primers. The primers (for primer sequences, see supplemental Table 2, available at www.jneurosci.org as supplemental material) were designed using the primer3 tool (http://frodo.wi.mit.edu/primer3/) and obtained from Integrated DNA Technologies. RT was omitted in negative controls. The fold change (FC) in target gene cDNA relative to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) endogenous control was determined as follows: FC = 2−ΔΔCt, where ΔΔCt = (CtTarget − CtGAPDH)test − (CtTarget − CtGAPDH)control. Ct values were defined as the number of the PCR cycles at which the fluorescence signals were detected (Schmittgen and Livak, 2008).
Cholesterol determination.
Neurons were stimulated with 10 nm C1q for different periods of times, washed in PBS, and harvested with 0.05% trypsin (Invitrogen). After counting to adjust samples to the same cell number, lipids are extracted in 200 μl of chloroform/isopropanol/NP-40 (7:11:0.1). After centrifugation, lipids are dried at 50°C for 30 min, followed by a SpeedVac to evaporate the chloroform. Cholesterol levels were determined using the Amplex Red cholesterol assay (Invitrogen) in accordance with the protocol of the manufacturer. Fluorescence was measured using an excitation wavelength of 544 nm and emission wavelength of 590 nm.
Immunocytochemistry.
Neurons were stimulated with 10 nm C1q for different periods of times, fixed with 3.7% paraformaldehyde, and permeabilized with 0.1% Triton X-100. For filipin staining, neurons are incubated with 125 μg/ml filipin (Sigma-Aldrich) for 24 h and then washed three times. Filipin is a fluorescent polyene antibiotic that forms complexes with cholesterol that can be visualized with ultraviolet light (Schroeder et al., 1971). Immunocytochemistry was performed according to standard procedures adapted from Glynn and McAllister (2006). Briefly, after blocking, neurons were incubated with rabbit polyclonal anti-stx3 antibody (dilution 1:100), rabbit polyclonal anti-C/EBP-δ antibody (dilution 1:500), mouse monoclonal anti-MAP-2 antibody (dilution 1:1000), and/or mouse monoclonal anti-SNAP25 antibody (dilution 1:1000) for 1 h at room temperature. After three washes, slides were incubated with Alexa Fluor 488-conjugated anti-rabbit or anti-mouse IgG antibodies (Invitrogen) and Alexa Fluor 555-conjugated anti-rabbit or anti-mouse IgG antibodies (Invitrogen) (dilution 1:2000) for 1 h at room temperature. For pCREB staining, slides were incubated overnight at 4°C with Alexa Fluor 488-conjugated pCREB antibodies (dilution 1:100). The slides were mounted with 5 μl of Prolong Gold anti-fade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Cells were then examined using the Nikon Eclipse Ti-E fluorescent microscope and the NIS-Element AR 3.00, sp7 software. For confocal study, cells were analyzed using the Carl Zeiss LSM710-META confocal microscope and the ZEN2009 software. Protein expression was quantified using NIH Image J software. The nuclear/cytoplasmic ratios of pCREB and C/EBP-δ were quantified as described (Noursadeghi et al., 2008). The total neurite length and the number of roots were determined using NeuronJ (Meijering et al., 2004).
Coimmunoprecipitation and Western blot.
For coimmunoprecipitation experiments, neurons (2.5 million plated in 100 mm dish) were harvested in 500 μl of extraction buffer and incubated for 15 min on ice. Insoluble fraction is then pelleted by centrifugation at 14,000 rpm for 15 min at 4°C. The soluble fraction is incubated with 2.5 μl of rabbit anti-stx3 polyclonal antibodies overnight at 4°C under agitation. Then, cell lysates are incubated with 20 μl of protein G Sepharose beads (GE Healthcare) for 2 h at 4°C under agitation. Beads and bound antibodies were then pelleted by centrifugation (2000 rpm, 4°C) and washed three times in PBS. Pellets are resuspended in 50 μl of loading buffer and boiled for 5 min. For soluble protein extraction, neurons were washed with 1 ml of cold HBSS and harvested in 200 μl of extraction buffer [1% Triton X-100, 25 mm Tris-HCl, 5 mm EDTA, 250 mm NaCl, 10% glycerol, and 1× protease inhibitor cocktail (Roche Applied Science)]. After 10 min of incubation on ice, neurons were scraped and the lysate was centrifuged for 15 min at 14,000 rpm at 4°C. The protein concentration in the soluble fraction was determined by microBCA assay (Pierce) using bovine serum albumin (BSA) as standards. For Western blot analysis, equal amounts of proteins were separated by 10% SDS-PAGE and then transferred to nitrocellulose membranes (GE Healthcare). The membranes were then incubated in blocking buffer (5% BSA/TBS/0.1% Tween 20) for 1 h at room temperature and incubated overnight at 4°C with mouse monoclonal anti-SNAP25 antibody (dilution 1:1000), rabbit polyclonal anti-NT-3 antibody (dilution 1:500), rabbit polyclonal anti-NGF-β antibody (dilution 1:800), rabbit polyclonal anti-pCREB antibody (dilution 1:600), rabbit polyclonal anti-CREB1 antibody (dilution 1:10,000), or mouse monoclonal anti-β-actin antibody (1:2000). After three washes, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit (1:5000; Jackson ImmunoResearch) or HRP-conjugated anti-mouse (1:10,000; Jackson ImmunoResearch) antibodies for 1 h at room temperature. The proteins were then developed using enhanced chemiluminescence plus (ECL +; GE Healthcare) and analyzed using the Nikon D700 digital SLR camera and NIH Image J software as described (Khoury et al., 2010).
Statistical analysis.
Results were calculated as means ± SD and compared with two-tailed nonparametric Mann–Whitney U test or two-way ANOVA, followed by Bonferroni's post hoc test, α error = 0.05 for all tests. Differences were considered significant when p was <0.05.
Results
Regulation of gene and miRNA expression by C1q in primary cortical neurons
To delineate the C1q-modulated pathways mediating the previously reported in vitro neuroprotection in rat immature neurons (Pisalyaput and Tenner, 2008), we assessed differences in gene expression between untreated and C1q-treated rat primary cortical neurons in vitro by microarray analysis. C1q significantly upregulated the expression of 127 genes while downregulating expression of 261 genes compared with untreated neurons (supplemental Table 1, available at www.jneurosci.org as supplemental material). The microarray results were validated by quantitative real-time PCR (16 tested genes) (Fig. 1C, note the y-axis is a log2 fold change). The coefficient of correlation between the log2 FC ratio obtained from the microarray and that obtained from the qRT-PCR was 0.84 (data not shown), suggesting that the data obtained from the microarray are highly reliable. Using Gene Ontology (GO) annotation and functional gene clustering, the C1q modulated genes could be classified in six major functional groups (Fig. 1A). The first cluster related to cholesterol and lipid metabolism included the upregulated genes encoding PNPLA7 [also known as NTEL-1 (neuropathy target esterase like 1)], which exhibits an hydrolase activity against lysophospholipid substrates, APOF, which is involved in transport and/or esterification of cholesterol, and insulin induced gene 2 (INSIG2) and cholesterol-25-hydroxylase (CH25H), which play a role in the regulation of cholesterol homeostasis (Fig. 1A,C). The second cluster associated with membrane and cytoskeleton functions included the upregulated genes encoding the transmembrane proteins or membrane associated receptors SLC23A2, TMEM79, IL5RA, and NT5E (also known as CD73) and proteins involved in membrane expansion, growth of neurites, and/or neural cell differentiation, such as STX3, SEPT9, CYP26A1, and HPSE. C1q downregulated the expression of genes associated with neurotransmission (MAOB), chemotaxis (CXCL3 and S100A9), inflammation and cell stress (CASQ2), and development and regulation of cell cycle and death (HEY1 and MESDC2) (Fig. 1A,C). In addition to these six main groups, C1q upregulated the expression of the cAMP-inducible transcription factor C/EBP-δ (Fig. 1C).
Figure 1.

Regulation of gene and miRNA expression by C1q in primary cortical neurons. A, B, Functional clustering of GO annotated genes (A) and miRNAs (B) significantly modulated by C1q compared with untreated (UT) neurons. Standardized means shown with a color gradient from blue (downregulated) to yellow (upregulated). C, Comparison of array and qRT-PCR data for 16 significantly modulated genes by C1q. The results are expressed as the means of log2 FC of C1q over untreated. D, Gene expression of miRNA predicted targets studied by qRT-PCR over a 16 h time course study. Results are expressed as the mean ± SD of log2 FC from triplicate samples from two independent experiments. Line color corresponds to the color-coded classifications in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
C1q also modulated the expression of different miRNAs, increasing the expression of the miRNA rno-miR-28 while downregulating the expression of rno-miR-410, rno-miR-497, and rno-let-7c (Fig. 1B) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Predicted targets of these miRNAs are involved in general cellular processes, such as metabolic process, regulation of gene expression, transport, signaling, and protein modification but also in very specific cellular processes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Of particular interest, miR-28 is predicted to target some genes associated with regulation of neurotransmission (SYN1) and gene regulation (CREB-L1 and EIF3D). The miRNA miR-410 is predicted to target genes including those associated with signaling (GPR119 and NTRK2 also known as TrkB), while the predicted targets for miR-497 include genes associated with exocytosis (STX1A), regulation of cell death [TNFRSF5, also known as CD40, and AATF (apoptosis-antagonizing transcription factor)] and with neurogenesis (ATN-1 and MAP2K1). Finally, the miRNA let-7c is predicted to target genes involved in arginine metabolism (ARG2) and regulation of cell death (CLN3) and genes with neurotrophic activities, such as NGF, Netrin-1 (NTN1), and NT-3. Because miR-410, miR-497, and let-7c are downregulated by C1q, the expression of their predicted target genes could be predicted to be subsequently increased in C1q-treated neurons.
To test the role of miRNAs in C1q-dependent gene expression, the mRNA levels of miRNA predicted targets was assessed by qRT-PCR over a 16 h time period (Fig. 1D). As predicted from the C1q-dependent increased expression of miR-28, the expression of the genes predicted to be targeted by miR-28 was generally downregulated in C1q-treated neurons compared with untreated neurons (Fig. 1D). Furthermore, the expression of the genes predicted to be targeted by miR-410, miR-497, or let-7c, miRNAs that were downregulated by C1q, was increased generally over the 16 h time course compared with untreated neurons (Fig. 1D). Together, these data are the first reports demonstrating that C1q modulates expression of genes encoding neurotrophins or involved in regulation of cell death and neurite outgrowth in part through regulation of miRNA expression.
C1q alters cholesterol distribution and decreases cholesterol levels in neurons
C1q modulated the expression of several enzymes associated with cholesterol metabolism and homeostasis (Fig. 1). To assess the effect on the intracellular cholesterol distribution, neurons were treated or not with C1q for 24 h and then stained with filipin, which forms complexes with cholesterol that can be detected with ultraviolet light (Schroeder et al., 1971). Filipin labeling showed that neurons treated with C1q have similar levels of staining as untreated neurons in the cell body but exhibited a decrease in filipin staining in the neurites compared with untreated neurons (Fig. 2A), suggesting that the cholesterol distribution is affected by C1q. Because filipin fluorescence intensity is not necessarily linearly related to cholesterol content (Maxfield and Wüstner, 2002), free cholesterol levels in untreated and C1q-treated neurons was determined by fluorometric assay over a 48 h time course. Neurons treated with C1q showed a very rapid and significant (p < 0.01) decrease in cholesterol content after 4 h of treatment with C1q compared with untreated neurons (Fig. 2B). The cholesterol content then increased slowly over the 48 h time course in C1q-treated neurons, but was still significantly lower than levels observed in untreated neurons at 24 h (Fig. 2B). These results suggest that C1q contributes to regulation of cholesterol distribution and metabolism in neurons.
Figure 2.
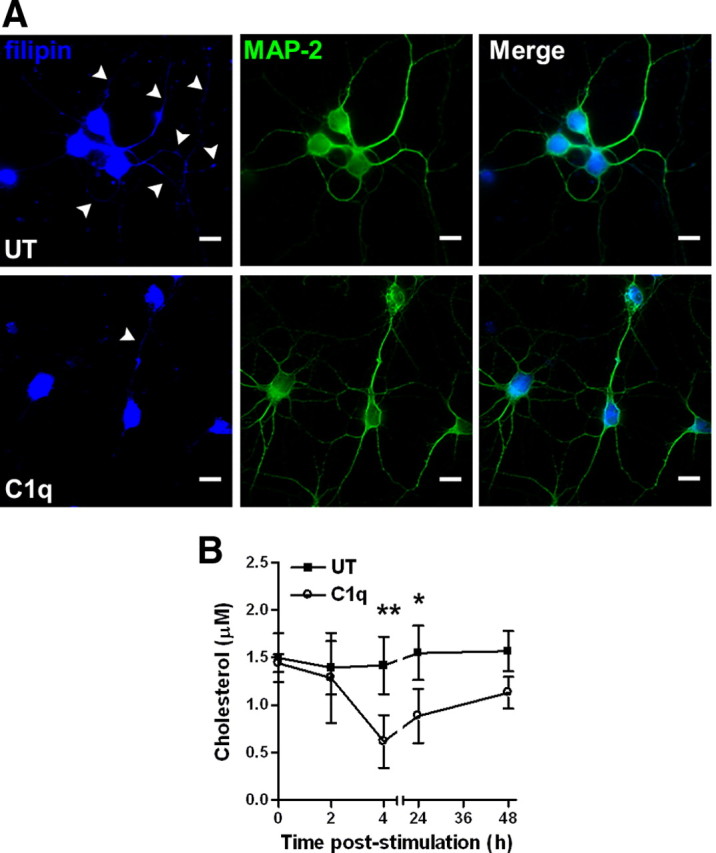
C1q modifies cholesterol distribution and decreases cholesterol levels in neurons. A, C1q alters cholesterol distribution in neurons. Neurons cultured with or without C1q for 24 h were stained with filipin and anti MAP-2 antibodies and analyzed by fluorescence microscopy. Scale bars, 10 μm. B, Quantification of cholesterol content in untreated (UT) and C1q-treated neurons by enzymatic fluorometric assay over a 48 h time course. Results represent means ± SD (n = 3). Two-way ANOVA followed by Bonferroni's post hoc test, *p < 0.05 and **p < 0.01.
C1q enhances stx3 expression and potentiates its interaction with SNAP25
Stx3 was found to be one of the most upregulated genes (log2 FC = 3.0, i.e., eightfold increase) (supplemental Table 1, available at www.jneurosci.org as supplemental material) in C1q-treated neurons (Fig. 1A,C). Immunocytochemical analysis validated the upregulated stx3 protein expression in neurons stimulated for 24 h with C1q (Fig. 3A,B). C1q-treated neurons showed a twofold (p < 0.001) increase in stx3 expression (field area = 14.7 ± 1.0%) (Fig. 3B) compared with untreated neurons (field area = 7.0 ± 1.2%) (Fig. 3B). Stx3 expression remained significantly (p < 0.05) increased per cell in C1q-treated neurons (3.6 ± 0.7) (Fig. 3B) compared with untreated neurons (1.9 ± 0.2) (Fig. 3B), after Stx3 expression was normalized to the DAPI signal to obviate differences attributable to a difference in the number of cells between untreated and C1q-treated neurons.
Figure 3.
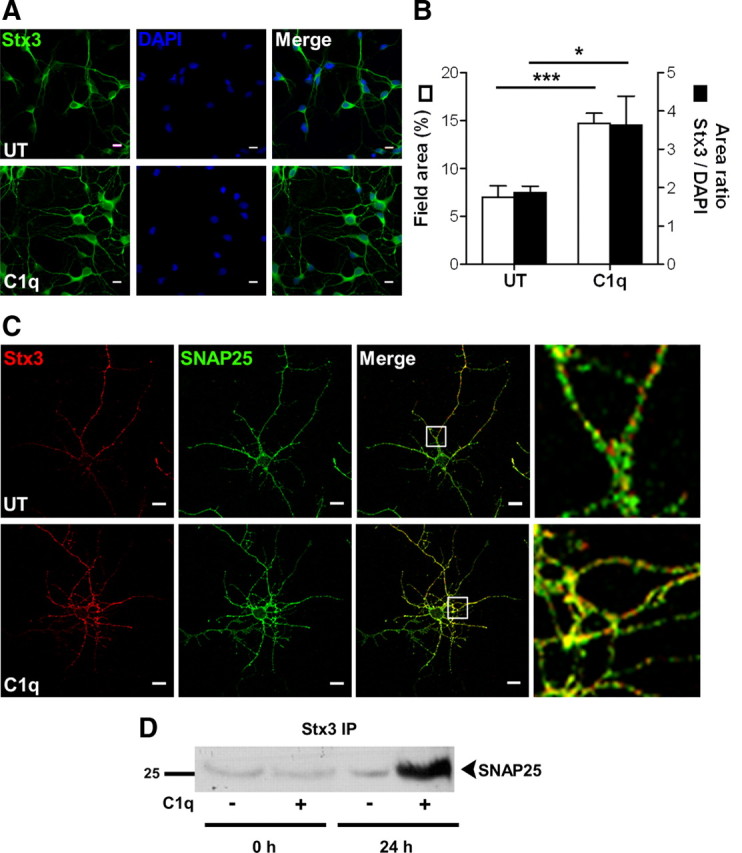
C1q increases stx3 expression and its interaction with SNAP25. A, B, C1q increases the protein levels of stx3 in neurons. Neurons cultured with or without C1q for 24 h were stained with anti-stx3. Nuclei are stained with DAPI. A, Representative micrographs of stx3 staining. B, Quantitative image analysis of stx3 expressed as means ± SD (n = 4, 7 fields per condition) of percentage of field area (□) or area ratio stx3/DAPI (■). Two-tailed nonparametric Mann–Whitney U test, *p < 0.05 and ***p < 0.001. C, Colocalization of stx3 and SNAP25 in C1q-treated neurons. Neurons cultured with or without C1q for 24 h were stained with anti-stx3 (Alexa Fluor 555, red) and anti-SNAP25 (Alexa Fluor 488, green) antibodies and analyzed by confocal microscopy. Representative micrographs of two independent experiments are shown with white boxed section expanded on the right. UT, Untreated. Scale bars, 10 μm. D, SNAP25 immunoprecipitates with stx3 after 24 h of stimulation with C1q. One blot representative of three independent experiments is shown. IP, Immunoprecipitation.
Stx3 stimulates growth of neurites by interacting with SNAP25 (Darios and Davletov, 2006). Although SNAP25 expression was not increased in C1q-treated neurons (supplemental Table 1, available at www.jneurosci.org as supplemental material), dual immunostaining of stx3 and SNAP25 clearly demonstrated a strong colocalization of SNAP25 with stx3 (Fig. 3C), particularly in the neurites (Fig. 3C, right panels). Moreover, C1q increased the physical association of SNAP25 with stx3, as seen by the enhanced immunoprecipitation of SNAP25 with stx3 in C1q-treated neurons compared with untreated neurons (Fig. 3D). These results suggest that C1q increases the expression of stx3 protein in neurons potentiating its interaction with SNAP25, its partner required to promote the growth of neurites.
Intracellular signaling pathways modulated by C1q
C1q increased the expression of the transcription factor C/EBP-δ (Fig. 1C), which is known to act in concert with CREB to induce NGF expression in the CNS (McCauslin et al., 2006). Because it is known that C1q stimulates the phosphorylation of CREB in monocytes (Fraser et al., 2007), Western blot analysis of neuronal extracts were performed, revealing a transient phosphorylation of CREB after 30 min and 1 h of stimulation with C1q (Fig. 4A). Furthermore, using immunocytochemistry (Fig. 4B) and quantitative image analysis (Fig. 4C), we found that the nuclear/cytoplasmic ratio of pCREB was significantly (p < 0.05) increased after 30 min of stimulation with C1q (0.8 ± 0.2) compared with untreated neurons (0.5 ± 0.1). The nuclear translocation of C/EBP-δ in untreated and C1q-treated neurons was similarly assessed over a 6 h time course (Fig. 4D,E). C1q significantly increased the nuclear/cytoplasmic C/EBP-δ ratio after 30 min (2.3 ± 0.9 vs 1.1 ± 0.3, p < 0.05), 1 h (2.6 ± 1.1 vs 1.0 ± 0.4, p < 0.01), and 6 h (2.3 ± 0.4 vs 1.1 ± 0.3, p < 0.05) of stimulation compared with untreated neurons (Fig. 4E). Together, these results show that C1q increased phosphorylation of CREB and nuclear translocation of both pCREB and C/EBP-δ. Dual immunostaining of the two proteins similarly demonstrated that the nuclear colocalization of pCREB and C/EBP-δ is increased after 30 min of stimulation with C1q compared with untreated neurons (Fig. 4F), consistent with the cotranslocation of pCREB and C/EBP-δ to the nucleus.
Figure 4.

Signaling through pCREB and C/EBP-δ in C1q-stimulated neurons. A, Expression of pCREB and total CREB in C1q-stimulated neurons assessed by Western blot. One blot representative of three independent experiments is shown. B–E, Nuclear translocation of pCREB and C/EBP-δ in C1q-treated neurons. Neurons treated with C1q for 30 min to 6 h were stained with anti-pCREB (Alexa Fluor 488, green) or anti-C/EBP-δ (Alexa Fluor 555, red) antibodies. Nuclei are stained with DAPI (blue). Representative micrographs of neurons stimulated with C1q for 30 min and stained for pCREB are shown in B. Representative micrographs of neurons stimulated with C1q for 1 h and stained for C/EBP-δ are shown in D. C, E, Quantitative image analysis of nuclear translocation of pCREB (C) and C/EBP-δ (E). Results represent means ± SD (n = 3, 5 fields per condition). Two-way ANOVA, followed by Bonferroni's post hoc test, *p < 0.05 and **p < 0.01. F, Nuclear colocalization of pCREB and C/EBP-δ in C1q-treated neurons after 30 min of stimulation with C1q. Representative micrographs of three independent experiments are shown. Arrows indicate overlapping of pCREB and C/EBP-δ. G–I, C/EBP-δ inhibition prevents C1q-dependent neuroprotection. Knocked down expression of C/EBP-δ after siRNA transfection was assessed by PCR after 6 h of stimulation with C1q (G) and the effect of C/EBP-δ inhibition on C1q-dependent neuroprotection was assessed by MAP-2 immunocytochemistry (H) and image analysis for quantification of total neurite length and number of roots (I) after 24 h of stimulation with C1q. Results represent means ± SD (n = 3, 7 fields per condition). Scale bars, 10 μm. UT, Untreated; px, pixels. Two-tailed nonparametric Mann–Whitney U test, *p < 0.05 and **p < 0.01.
Finally, specific siRNA that inhibited the expression of C/EBP-δ (Fig. 4G) abrogated the C1q-dependent neuroprotection. As demonstrated by MAP-2 immunocytochemistry (Fig. 4H) and quantitative image analysis (Fig. 4I), C1q significantly (p < 0.05) increased the total neurite length and the number of roots in neurons after 24 h of stimulation. Importantly, inhibition of C/EBP-δ expression in C1q-stimulated neurons significantly decreased both the total neurite length (p < 0.01) and the number of roots (p < 0.05) to levels observed in untreated neurons (Fig. 4I). Together, these data suggest that C/EBP-δ plays a critical role in C1q-triggered neuroprotective pathways and may act in concert with pCREB.
Role of neurotrophins in C1q-dependent neuroprotection
C1q downregulated the expression of let-7c miRNA, which is predicted to target and thus inhibit the expression of neurotrophic factors, but enhanced C/EBP-δ expression that can bind and activate neurotrophic factor promoters such as NGF. To determine whether neurotrophic factors are indeed modulated in C1q-treated neurons, the expression of NGF and NT-3 in untreated and C1q-treated neurons was specifically assayed by qRT-PCR over a 16 h time course (Fig. 1D). C1q increased the gene expression of NGF (log2 FC = 1.8 ± 0.1) after 6 h of stimulation (Figs. 1D, 5B) and the gene expression of NT-3 after 6 h (log2 FC = 1.9 ± 1.1) (Figs. 1D, 5B) and 16 h of stimulation (log2 FC = 2.0 ± 0.6) (Fig. 1D) compared with untreated neurons.
Figure 5.
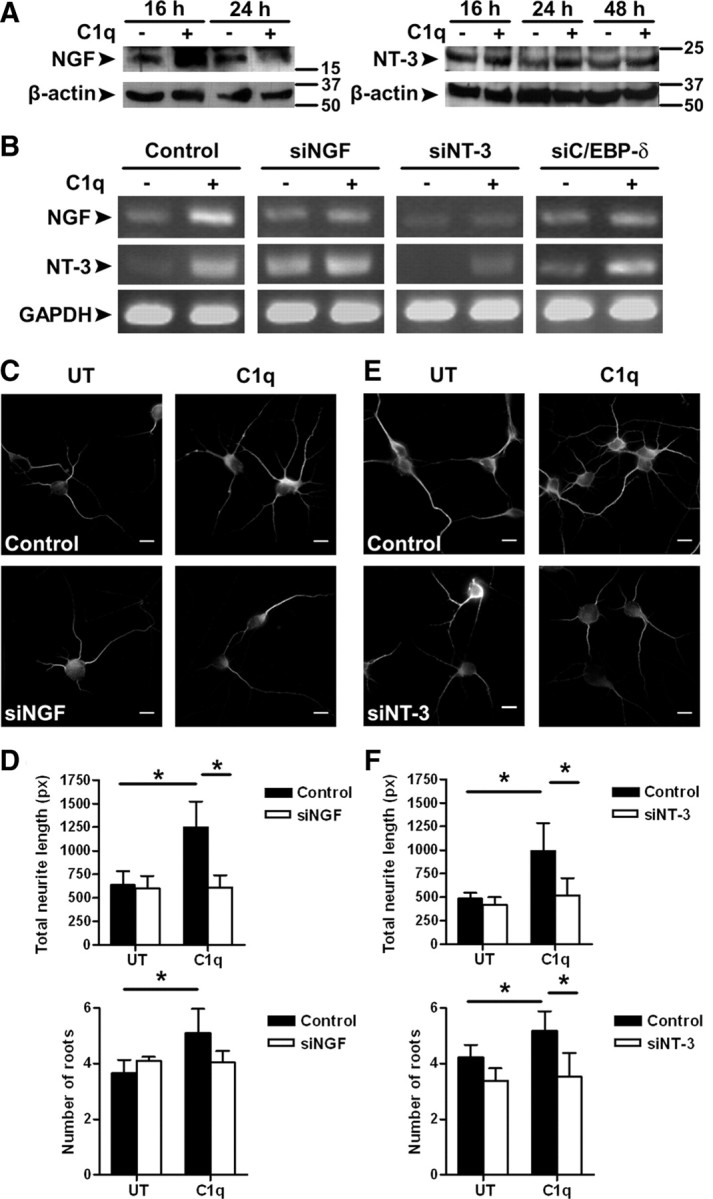
Role of neurotrophins in C1q-dependent neuroprotection. A, Expression of NGF and NT-3 in untreated and C1q-treated neurons assessed by Western blot. Representative blots of three independent experiments are shown. B, Expression of NGF, NT-3, and GAPDH after transfection with negative control siRNA or siRNA specific for NGF, NT-3, or C/EBP-δ was assessed by PCR after 6 h of incubation with or without C1q. Gels representative of three independent experiments are shown. C–F, Inhibition of NGF and NT-3 expression prevents C1q-dependent neuroprotection. The effect of NGF and NT-3 inhibition on C1q-dependent neuroprotection was assessed by MAP-2 immunocytochemistry (C, E) and image analysis for quantification of total neurite length and number of roots (D, F) after 24 h of stimulation with C1q. Results represent means ± SD (n = 3, 7 fields per condition). Scale bars, 10 μm. UT, Untreated; px, pixels. Two-tailed nonparametric Mann–Whitney U test, *p < 0.05.
Protein expression of NGF and NT-3 was then assessed by Western blot (Fig. 5A). C1q increased the protein levels of the mature form of NGF after 16 h compared with untreated neurons, but this increase was transient as NGF levels decreased to untreated levels after 24 h (Fig. 5A). The expression of the NT-3 protein precursor is increased after 24 and 48 h of culture with C1q (Fig. 5A). These results show that C1q sequentially increased expression of NGF and NT-3 in neurons.
We then assessed the effect of inhibition of NGF and NT-3 on neurotrophin expression and C1q-dependent neuroprotection. Treatment of neurons with specific siRNA targeting NGF resulted in a decrease of NGF mRNA expression in C1q-treated neurons as expected, with no change in NT-3 expression compared with C1q-stimulated neurons transfected with control siRNA (Fig. 5B). The total neurite length was significantly (p < 0.05) decreased in C1q-stimulated neurons transfected with siNGF compared with control neurons (Fig. 5C,D) at 24 h. The C1q-dependent increase in the number of roots showed a downward trend during treatment with siNGF but did not reach significance (p = 0.1) (Fig. 5D). Interestingly, inhibition of C/EBP-δ expression also prevented the increase in NGF expression after stimulation with C1q (Fig. 5B), suggesting that C/EBP-δ plays a major role in the C1q-dependent upregulation of NGF. Furthermore, inhibition of NT-3 decreased NT-3 expression in C1q-treated neurons as expected but also resulted in decreased expression of NGF mRNA (Fig. 5B). A blast of siNT-3 sequence against the RNA reference sequence database did not find any match for NGF mRNA sequence (data not shown), ruling out the possibility that the siNT-3 also targeted NGF mRNA and suggesting that NGF expression in C1q-treated neurons is probably attributable to a direct effect of NT-3. In addition, NT-3 inhibition significantly (p < 0.05) decreased the total neurite length and the number of roots compared with control neurons (Fig. 5E,F). Together, these results suggest that C1q increased the expression of neurotrophins in neurons and that NGF, under the control of C/EBP-δ and NT-3, plays a critical role in C1q-dependent neuroprotection.
C3a and C5a have no direct neuroprotective effect and reduce the neuroprotective effect of C1q
Because C3a and C5a have reported neuroprotective effects in different developmental or environmental contexts (Heese et al., 1998; Osaka et al., 1999; Jauneau et al., 2006; Bénard et al., 2008), we evaluated the capacity of C3a and C5a to directly promote neurite outgrowth in this neuron-only culture system (Fig. 6). Neither C3a nor C5a promoted neurite outgrowth (regardless of the dose used) (Fig. 6), suggesting that they have no direct neuroprotective effect. Moreover, the neuroprotective effect of C1q is reduced in the presence of C3a or C5a in a dose-dependent manner (Fig. 6). These results suggest that the direct neuroprotective pathways stimulated by C1q in neurons are fundamentally different from the effects that other complement proteins may have directly or by acting through glia cells.
Figure 6.

C3a and C5a have no direct neuroprotective effect and reduce the neuroprotective effect of C1q. The effect of C3a and C5a on C1q-dependent neuroprotection was assessed by MAP-2 immunocytochemistry (A) and image analysis for quantification of total neurite length (B) after 24 h of stimulation. Results represent means ± SD (n = 3, 5 fields per condition). Scale bars, 10 μm. px, Pixels. Two-tailed nonparametric Mann–Whitney U test, *p < 0.05 and ***p < 0.001.
C1q modulates mouse neuronal survival and gene expression
To validate the generality of what is seen in rat neurons and to enable movement to in vivo mouse models, we determined the effect of C1q on neuronal survival and gene expression in mouse neurons (Fig. 7A,B). As assessed by MAP-2 staining, C1q promoted growth of neurites in mouse neurons (Fig. 7A) as seen in rat neurons (Pisalyaput and Tenner, 2008). In addition, qRT-PCR analysis of gene expression demonstrated that, for all but one of the tested genes, C1q similarly modulates gene expression levels in both rat and mouse neurons (Fig. 7B), implicating similar C1q-stimulated pathways in mouse and rat neurons. Finally, similar to the need for structurally native C1q for the neuroprotective effect (Pisalyaput and Tenner, 2008), the induced gene expression program stimulated by C1q in neurons required a conformationally intact molecule because C1q tails or heat-inactivated C1q failed to promote the induction of STX3, CH25H, or C/EBP-δ (Fig. 7C).
Figure 7.
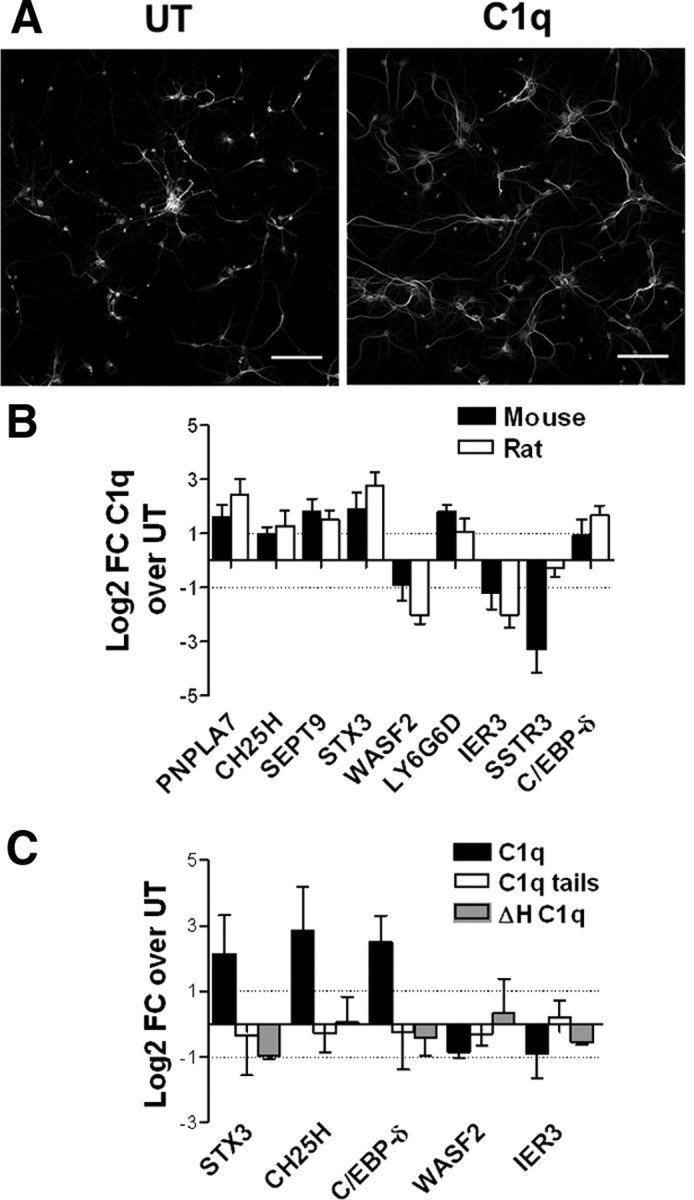
C1q modulates mouse neuronal survival and gene expression. A, C1q promotes neurite outgrowth in primary cortical mouse neurons. Mouse neurons were incubated with or without C1q for 24 h, fixed, and stained with MAP-2 antibodies. Scale bars, 100 μm. B, Mouse and rat primary cortical neurons were incubated with or without C1q for 3 h, and gene expression was assessed by qRT-PCR. C, Mouse primary cortical neurons were incubated with 10 nm C1q, C1q tails, or heat-inactivated C1q (ΔHC1q) for 3 h, and gene expression was assessed by qRT-PCR. Results are expressed as the mean ± SD of log2 FC from triplicate samples from two independent experiments. UT, Untreated.
Discussion
In this study, the gene expression and the signaling pathways triggered by C1q in neurons associated with neuroprotection in vitro have been identified by microarray analysis. C1q upregulated the expression of genes associated with cholesterol and lipid metabolism and membrane and cytoskeleton processes (Figs. 1, 8), two major functions that intrinsically affect organization of neural cell membranes (Piomelli et al., 2007).
Figure 8.

Pathways modulated by C1q in neurons. C1q rapidly upregulates the expression of genes associated with membrane and cytoskeleton function, including neurite outgrowth (stx3) and cholesterol and lipid metabolism, suggesting that C1q may modulate a complex system involved in membrane stabilization and growth of neurites. In addition, C1q increases the phosphorylation of CREB and the expression of C/EBP-δ, which both stimulate NGF production, as well as their nuclear translocation, while downregulating the expression of miRNAs predicted to target several neurotrophic factors, including NGF, NT-3, and NTN1. Yellow, Upregulated; blue, downregulated.
C1q modulated the expression of several enzymes associated with cholesterol homeostasis (Fig. 1). Brain-derived neurotrophic factor, which regulates synaptic function and development, also regulates cholesterol metabolism (Suzuki et al., 2007). Cholesterol depletion in neurons enhances neurite outgrowth (Ko et al., 2005), and treatment of neurons with statins, which are cholesterol-lowering drugs through inhibition of hydroxymethylglutaryl (HMG)-CoA reductase, enhances the number of neurites and the neurite length and branching (Pooler et al., 2006). C1q upregulated the expression of CH25H, an enzyme that catalyzes the conversion of cholesterol to 25-hydroxycholesterol, and the expression of INSIG2. Interestingly, INSIG2 and 25-hydroxycholesterol prevent the translocation of sterol regulatory element binding proteins to the nucleus and thus decrease the transcription of the HMG-CoA reductase, the central enzyme in cholesterol synthesis (Radhakrishnan et al., 2007; Ikonen, 2008). Consistent with the hypothesis that C1q may regulate cholesterol synthesis, both the distribution and levels of cholesterol are affected by C1q in neurons (Fig. 2). The very rapid decrease in cholesterol content after C1q treatment suggests a rapid efflux of cholesterol. Additional investigations are needed to determine how C1q modulates cholesterol efflux in neurons, but interestingly C1q bound to oxidized (atherogenic) low-density lipoprotein also increases cholesterol efflux in human macrophages (Fraser and Tenner, 2010). C1q upregulated stx3 expression at the mRNA and protein levels and increased its colocalization and interaction with SNAP25 (Fig. 3), a syntaxin partner necessary to promote neurite outgrowth. PC12 neuronal cells completely lacking stx3 are unable to grow neurites, even after stimulation with NGF (Darios and Davletov, 2006). Together, these data suggest that C1q may enhance neurite outgrowth indirectly through regulation of cholesterol levels and directly through upregulation of different proteins that promote neurite outgrowth.
C1q treatment resulted in increased phosphorylation of CREB, similar to that seen in monocytes (Fraser et al., 2007), and increased expression of C/EBP-δ (Fig. 4). CREB is a transcription factor critical for long-term memory and synaptic plasticity (Josselyn and Nguyen, 2005). C/EBP proteins have been shown to play a role in synaptic plasticity in the brain (Alberini, 2009). C/EBP-δ and CREB have been shown to both contribute to the inducible expression of NGF in the CNS (Colangelo et al., 1998; McCauslin et al., 2006). Here, addition of C1q increased the nuclear translocation of pCREB and C/EBP-δ with these two factors colocalizing in the nucleus of C1q-treated neurons (Fig. 4). Inhibition of C/EBP-δ expression by siRNA prevented C1q-dependent NGF upregulation and C1q-dependent neuroprotection (Figs. 4, 5), consistent with a direct role for a C/EBP-δ-NGF cascade in the C1q neuroprotective pathway, with C/EBP-δ as a critical transcription factor in this pathway.
Several miRNAs were also modulated by C1q in neurons (Fig. 1B). miRNAs function as “guide” molecules in posttranscriptional gene silencing by base pairing with target mRNAs, leading to mRNA cleavage or translational repression (Kim, 2005). C1q downregulated miR-410 [a CNS-specific miRNA mainly expressed during embryogenesis (Wheeler et al., 2006)] and miR-497 and let-7c, both of which are predicted to target genes involved in regulation of cell death and/or neuronal development and survival. Different miRNAs have been involved in synaptic development, plasticity mechanisms, and memory (Kosik, 2006). Members of the murine let-7 family may play a conserved role in synapse development in mammals (Corbin et al., 2009). These results suggest that gene regulation by miRNAs in C1q-treated neurons may be involved in neuronal morphogenesis and/or survival.
As expected from both the downregulation of let-7c and the nuclear translocation of pCREB and C/EBP-δ, NGF and NT-3 mRNA and protein expression are increased in C1q-treated neurons. The mature form of NGF regulates cell survival, whereas proNGF selectively induces apoptosis (Nykjaer et al., 2004). By increasing protein levels of the mature form of NGF, C1q selectively promotes neuronal survival. Knockdown experiments of NGF and NT-3 using siRNA showed that inhibition of NGF or NT-3 prevents the C1q-dependent neuroprotection (Fig. 5), indicating a critical role for these trophic factors in the C1q-stimulated pathway. We also observed that, in the presence of siNGF, untreated neurons showed an increase expression of NT-3. NGF may specifically block NT-3 signaling during sympathetic neuron development (Kuruvilla et al., 2004) and reduce NT-3 protein levels in vivo (Randolph et al., 2007), suggesting that the absence of NGF may result in NT-3 increased expression in neurons. Interestingly, inhibition of NT-3 prevented the C1q-induced upregulation of NGF (Fig. 5B). It has been shown that treatment of Schwann cells with NT-3 increases the expression of NGF, and the NGF expression is significantly reduced when cells are cultured in medium containing high glucose, which is associated with decreased CREB expression (Suzuki et al., 2004). Thus, NT-3 in concert with CREB, which is phosphorylated and thus activated by C1q in neurons (Fig. 4), contributes to NGF expression.
In AD, the presence of the complete C1 complex (C1q plus C1r2C1s2) and the other complement proteins (which are synthesized in the activated/disease state in the CNS) in addition to fibrillar Aβ deposits lead to the activation of the classical complement cascade, resulting in the generation of the chemotactic factors C3a and C5a, which can recruit (Yao et al., 1990) and activate glial cells, and likely synergize with Toll-like receptor engagement at the plaque (Reed-Geaghan et al., 2009) to stimulate a robust inflammatory reaction, including glial secretion of proinflammatory cytokines, reactive oxygen species, and nitric oxide, all of which is detrimental to neurons (Zhou et al., 2008, the model in their Fig. 8). In contrast, other studies have provided evidence for a beneficial role for complement activation products (Wyss-Coray et al., 2002; Rus et al., 2005; Maier et al., 2008), and more recent studies have demonstrated an unexpected role of the early complement cascade C1 through C3 in removing unwanted synapses (i.e., elimination of inappropriate multiple innervation of cortical and retinal neurons) during development (Stevens et al., 2007; Chu et al., 2010). In addition to considering the influence of the local environment on the nature and intensity of the response, it is important to distinguish the activities that result from the activation of the entire or even part of the complement cascade (such as C1 through C3 or C5), from the activities that are induced by the presence of C1q only, i.e., in the absence of any “C1” protease activity (C1r2C1s2) and thus no C3 or C5 convertase activity or other downstream complement activation products (such as C5a or the membrane attack complex). The higher association of C1q deficiency with lupus (>90%) versus any other complement component in humans (Walport et al., 1998) suggests that there is substantial physiologic consequences of a C1q deficiency, independent of its function as part of the multicomponent initiator of the classical complement cascade.
Interestingly, C3a, which is produced after complement activation via any of the three activation pathways, has also been shown to induce neuroprotection through NGF expression but mediated through a microglia cell line (Heese et al., 1998) rather than directly on neurons as shown here. More recently, C3a and C5a have been shown to act in synergy with the proinflammatory cytokine interleukin-1β to upregulate NGF protein in astrocytes (Jauneau et al., 2006), suggesting that C3a and C5a may have indirect neuroprotective effects in vivo, in different developmental or environmental contexts in addition to their noted inflammatory properties in the CNS (Sewell et al., 2004; Fonseca et al., 2009). We showed here that C3a and C5a do not have direct neuroprotective effects, and, additionally, they reduce the neuroprotective effect of C1q (Fig. 6). Therefore, the ability to increase NGF and NT-3 protein levels directly in this neuron-only culture system with purified C1q protein (no other serum/complement proteins) is fundamentally different from indirect activities of the complement activation products C3a and C5a on neurons/glial cells (for review, see Klos et al., 2009; Woodruff et al., 2010) and is consistent with a previously unappreciated direct neuroprotective response of C1q in the absence of other complement proteins.
In summary, our data demonstrate that C1q triggers a complex program of gene expression that enhances neurite outgrowth and limits neuronal stress and inflammation in vitro (Fig. 8). The induction of C1q synthesis in the absence of other downstream complement proteins both in vitro and early after injury in vivo in several CNS injury models coupled with the subsequent program of gene expression induced by C1q seen here in both rat and mouse neurons, as well as the known ability of C1q alone to enhance clearance of apoptotic cells and suppress inflammatory cytokines (Fraser et al., 2010) suggest a measured, programmed, and effective response to injury and a balance between protection and robust inflammation to counteract injury. Thus, although much of the literature has focused on a detrimental effect of immune molecules in neurodegenerative diseases and CNS inflammation, the data presented here emphasize the role of neuroimmune interactions that could be beneficial for neuroprotection. Additional analysis of the pathways induced by C1q in response to various in vivo injury models should lead to the discovery of new therapeutic targets to facilitate neuroprotection, proper neurodevelopment, and/or restore normal function of the nervous system in AD and other neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Grants AI 41090 and AG 00538, and the Cypress College STEM Summer Bridge Program funded by the U.S. Department of Education. We thank Rahasson Ager for help and advice with neuron culture preparation and Lindsey Weiner and Ji Young Jang for excellent technical help.
References
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin-Yang in neuroinflammation—neuro-protection and -degeneration. J Neurochem. 2008;107:1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénard M, Raoult E, Vaudry D, Leprince J, Falluel-Morel A, Gonzalez BJ, Galas L, Vaudry H, Fontaine M. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol Immunol. 2008;45:3767–3774. doi: 10.1016/j.molimm.2008.05.027. [DOI] [PubMed] [Google Scholar]
- Benoit M, Ghigo E, Capo C, Raoult D, Mege JL. The uptake of apoptotic cells drives Coxiella burnetii replication and macrophage polarization: a model for Q fever endocarditis. PLoS Pathog. 2008;4:e1000066. doi: 10.1371/journal.ppat.1000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensa JC, Reboul A, Colomb MG. Biosynthesis in vitro of complement subcomponents C1q, C1s and C1 inhibitor by resting and stimulated human monocytes. Biochem J. 1983;216:385–392. doi: 10.1042/bj2160385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colangelo AM, Johnson PF, Mocchetti I. beta-adrenergic receptor-induced activation of nerve growth factor gene transcription in rat cerebral cortex involves CCAAT/enhancer-binding protein delta. Proc Natl Acad Sci U S A. 1998;95:10920–10925. doi: 10.1073/pnas.95.18.10920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin R, Olsson-Carter K, Slack F. The role of microRNAs in synaptic development and function. BMB Rep. 2009;42:131–135. doi: 10.5483/bmbrep.2009.42.3.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Davletov B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature. 2006;440:813–817. doi: 10.1038/nature04598. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Dietzschold B, Schwaeble W, Schäfer MK, Hooper DC, Zehng YM, Petry F, Sheng H, Fink T, Loos M, Koprowski H, Weihe E. Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. J Neurol Sci. 1995;130:11–16. doi: 10.1016/0022-510x(94)00269-t. [DOI] [PubMed] [Google Scholar]
- Fan R, Tenner AJ. Complement C1q expression induced by Abeta in rat hippocampal organotypic slice cultures. Exp Neurol. 2004;185:241–253. doi: 10.1016/j.expneurol.2003.09.023. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol. 2009;183:1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DA, Tenner AJ. Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol. 2010;185:3932–3939. doi: 10.4049/jimmunol.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DA, Arora M, Bohlson SS, Lozano E, Tenner AJ. Generation of inhibitory NFκB complexes and phosphorylated cAMP response element-binding protein correlates with the activity of complement protein C1q in human monocytes. J Biol Chem. 2007;282:7360–7367. doi: 10.1074/jbc.M605741200. [DOI] [PubMed] [Google Scholar]
- Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol. 2009;183:6175–6185. doi: 10.4049/jimmunol.0902232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- Goldsmith SK, Wals P, Rozovsky I, Morgan TE, Finch CE. Kainic acid and decorticating lesions stimulate the synthesis of C1q protein in adult rat brain. J Neurochem. 1997;68:2046–2052. doi: 10.1046/j.1471-4159.1997.68052046.x. [DOI] [PubMed] [Google Scholar]
- Heese K, Hock C, Otten U. Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem. 1998;70:699–707. doi: 10.1046/j.1471-4159.1998.70020699.x. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang J, Kim LJ, Mealey R, Marsh HC, Jr, Zhang Y, Tenner AJ, Connolly ES, Jr, Pinsky DJ. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Jauneau AC, Ischenko A, Chatagner A, Benard M, Chan P, Schouft MT, Patte C, Vaudry H, Fontaine M. Interleukin 1b and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J Neuroinflammation. 2006;3:8. doi: 10.1186/1742-2094-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges. Curr Drug Targets CNS Neurol Disord. 2005;4:481–497. doi: 10.2174/156800705774322058. [DOI] [PubMed] [Google Scholar]
- Khoury MK, Parker I, Aswad DW. Acquisition of chemiluminescent signals from immunoblots with a digital single-lens reflex camera. Anal Biochem. 2010;397:129–131. doi: 10.1016/j.ab.2009.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46:2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Zou K, Minagawa H, Yu W, Gong JS, Yanagisawa K, Michikawa M. Cholesterol-mediated neurite outgrowth is differently regulated between cortical and hippocampal neurons. J Biol Chem. 2005;280:42759–42765. doi: 10.1074/jbc.M509164200. [DOI] [PubMed] [Google Scholar]
- Köhl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol. 2006;586:71–94. doi: 10.1007/0-387-34134-X_6. [DOI] [PubMed] [Google Scholar]
- Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7:911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118:243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Li M, Pisalyaput K, Galvan M, Tenner AJ. Macrophage colony stimulatory factor and interferon-gamma trigger distinct mechanisms for augmentation of beta-amyloid-induced microglia-mediated neurotoxicity. J Neurochem. 2004;91:623–633. doi: 10.1111/j.1471-4159.2004.02765.x. [DOI] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Wüstner D. Intracellular cholesterol transport. J Clin Invest. 2002;110:891–898. doi: 10.1172/JCI16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauslin CS, Heath V, Colangelo AM, Malik R, Lee S, Mallei A, Mocchetti I, Johnson PF. CAAT/enhancer-binding protein delta and cAMP-response element-binding protein mediate inducible expression of the nerve growth factor gene in the central nervous system. J Biol Chem. 2006;281:17681–17688. doi: 10.1074/jbc.M600207200. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-kappaB nuclear translocation in primary human macrophages. J Immunol Methods. 2008;329:194–200. doi: 10.1016/j.jim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem. 1999;73:303–311. [PubMed] [Google Scholar]
- Piomelli D, Astarita G, Rapaka R. A neuroscientist's guide to lipidomics. Nat Rev Neurosci. 2007;8:743–754. doi: 10.1038/nrn2233. [DOI] [PubMed] [Google Scholar]
- Pisalyaput K, Tenner AJ. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem. 2008;104:696–707. doi: 10.1111/j.1471-4159.2007.05012.x. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Xi SC, Wurtman RJ. The 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitor pravastatin enhances neurite outgrowth in hippocampal neurons. J Neurochem. 2006;97:716–723. doi: 10.1111/j.1471-4159.2006.03763.x. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph CL, Bierl MA, Isaacson LG. Regulation of NGF and NT-3 protein expression in peripheral targets by sympathetic input. Brain Res. 2007;1144:59–69. doi: 10.1016/j.brainres.2007.01.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid KB. Isolation, by partial pepsin digestion, of the three collagen-like regions present in subcomponent C1q of the first component of human complement. Biochem J. 1976;155:5–17. doi: 10.1042/bj1550005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rus H, Cudrici C, Niculescu F. C5b-9 complement complex in autoimmune demyelination and multiple sclerosis: dual role in neuroinflammation and neuroprotection. Ann Med. 2005;37:97–104. doi: 10.1080/07853890510007278. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schroeder F, Holland JF, Bieber LL. Fluorometric evidence for the binding of cholesterol to the filipin complex. J Antibiot (Tokyo) 1971;24:846–849. doi: 10.7164/antibiotics.24.846. [DOI] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155:55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83–90. doi: 10.1016/j.it.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kiyosue K, Hazama S, Ogura A, Kashihara M, Hara T, Koshimizu H, Kojima M. Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J Neurosci. 2007;27:6417–6427. doi: 10.1523/JNEUROSCI.0690-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Sekido H, Kato N, Nakayama Y, Yabe-Nishimura C. Neurotrophin-3-induced production of nerve growth factor is suppressed in Schwann cells exposed to high glucose: involvement of the polyol pathway. J Neurochem. 2004;91:1430–1438. doi: 10.1111/j.1471-4159.2004.02824.x. [DOI] [PubMed] [Google Scholar]
- Tenner AJ, Lesavre PH, Cooper NR. Purification and radiolabeling of human C1q. J Immunol. 1981;127:648–653. [PubMed] [Google Scholar]
- Walport MJ, Davies KA, Botto M. Clq and systemic lupus erythematosus. Immunobiology. 1998;199:265–285. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- Wheeler G, Ntounia-Fousara S, Granda B, Rathjen T, Dalmay T. Identification of new central nervous system specific mouse microRNAs. FEBS Lett. 2006;580:2195–2200. doi: 10.1016/j.febslet.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med. 2010;12:179–192. doi: 10.1007/s12017-009-8085-y. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- Young KR, Jr, Ambrus JL, Jr, Malbran A, Fauci AS, Tenner AJ. Complement subcomponent C1q stimulates immunoglobulin production by human B lymphocytes. J Immunol. 1991;146:3356–3364. [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease. J Neurochem. 2008;106:2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziccardi RJ, Tschopp J. The dissociation properties of native C1. Biochem Biophys Res Commun. 1982;107:618–623. doi: 10.1016/0006-291x(82)91536-4. [DOI] [PubMed] [Google Scholar]