

EndoD from S. pneumoniae is a large multimodular virulence factor that contains a putative carbohydrate-binding module (CBM). Here, the structure of the putative CBM is described.
Keywords: Streptococcus pneumoniae, carbohydrate binding, EndoD, N-glycans
Abstract
EndoD is an architecturally complex endo-β-1,4-N-acetylglucosamidase from Streptococcus pneumoniae that cleaves the chitobiose core of N-linked glycans and contributes to pneumococcal virulence. Although the glycoside hydrolase family 85 catalytic module has been structurally and functionally characterized, nothing is known about the ancillary modules and how they contribute to the overall function of the enzyme. Presented here is the 2.0 Å resolution structure of a family 32 carbohydrate-binding module of EndoD, SpCBM32, solved by single-wavelength anomalous dispersion. The putative binding site of this protein is a charge-neutral relatively flat region on the protein surface that contains one prominently exposed tryptophan residue that extends into the solvent. These topographical features are discussed in the biological context of EndoD activity and a hypothesis is made about the complex structure of its potential carbohydrate ligand.
1. Introduction
Carbohydrate-binding modules (CBMs) are generally defined as independently folding noncatalytic components that are found within the primary structure of carbohydrate-active enzymes (CAZymes). These modules possess the ability to bind carbohydrates and thus function to target and concentrate the enzyme in the proximity of its substrate, processes that enhance the efficiency of the overall enzyme (Boraston et al., 2004 ▶). However, more specialized roles for CBMs have also been described, such as the disruption of crystalline polysaccharide structure (Vaaje-Kolstad et al., 2005 ▶), retention of substrate within a defined cellular compartment (Abbott et al., 2007 ▶) and anchoring enzymes onto the surface of bacterial cells (Montainier et al., 2009 ▶).
The biological role of CBMs is best understood in the context of CAZymes that are involved in the utilization of complex structural polysaccharides. Indeed, the majority of functionally and structurally characterized CBMs within the CAZy database (http://www.cazy.org; Cantarel et al., 2009 ▶) are associated with cellulases and hemicellulases and correspondingly interact with carbohydrates that are components of plant cell-wall fibres such as cellulose, xylan and mannan (Boraston et al., 2004 ▶; Gilbert, 2010 ▶). What is emerging in the literature more recently, however, is the biological role of CBMs from CAZymes produced by bacteria that process complex eukaryotic glycans. The metabolism of such host glycans by both commensal and pathogenic bacteria has important implications for the host–microbe relationship in both colonization and infection processes (Shelburne et al., 2008 ▶). Perhaps not surprisingly, the extracellular CAZymes found in pathogens such as Streptococcus pneumoniae contain CBMs that are found to recognize an increasingly wide variety of complex glycans such as the blood-group A/B antigens (Gregg et al., 2008 ▶), Lewis antigens (Boraston et al., 2006 ▶), N-acetyllactosamine (Ficko-Blean & Boraston, 2006 ▶), N-acetylglucosamine (Ficko-Blean & Boraston, 2009 ▶), sialic acid and galactose (Boraston et al., 2007 ▶).
S. pneumoniae is a common but serious human pathogen that is responsible for several debilitating diseases including otitis media, pneumonia and sepsis. Recent work has highlighted the contribution of CAZymes to the full virulence of S. pneumoniae (Hava & Camilli, 2002 ▶; Shelburne et al., 2008 ▶) and dissecting the functions of CBMs from this microbe has provided some of the first insights into the relationship between CBMs and host-glycan catabolism (Boraston et al., 2006 ▶; Lammerts van Bueren et al., 2007 ▶). One unique member of this unique group of virulence factors is EndoD, a glycoside hydrolase family 85 endo-β-1,4-N-acetylglucosaminase that cleaves the chitobiose core of N-linked glycans (Umekawa et al., 2008 ▶; Abbott et al., 2009 ▶; Yin et al., 2009 ▶). The structure of the catalytic module of EndoD has been determined; however, nothing is known about the structure of the remaining modules in this architecturally complex multimodular enzyme (Fig. 1 ▶ a). Here, we describe the 2.0 Å resolution X-ray crystal structure of an internal EndoD module, SpCBM32, that shows structural identity to family 32 CBMs. A detailed analysis of its structure and comparison with other closely related CBM32 structures provides insight into its potential for carbohydrate recognition.
Figure 1.

The architecture of EndoD from S. pneumoniae (Sp_0498; 183.2 kDa) and the structure of SpCBM32. (a) The modular arrangement of EndoD. The enzyme is secreted to the extracellular surface, where its C-terminus becomes (L = LPXTG) covalently attached to the peptidoglycan in the cell wall through a sortase-mediated reaction. The catalytic GH85 module (172–816) is flanked by an N-terminal unknown domain (UNK, 38–170) and the SpCBM32 module (CBM32, 817–945), which lies towards the C-terminus. Several other modules are also predicted in the extensive C-terminal region of the molecule, including tandem bacterial Ig-like or Big3 domains (B3, 1067–1139, 1157–1225) and a FIVAR motif (FV, 1243–1287), which are commonly observed within bacterial surface proteins. The function of these C-terminal modules remains unknown. (b) Primary and secondary structure of SpCBM32 (1) in comparison with known structures: C. perfringens exo-α-sialidase appended CBM32 (2; PDB entry 2v72), B. thetaiotaomicron α-l-fucosidase appended CBM32 (3; PDB entry 3eyp) and M. viridifaciens sialidase CBM32 (4). The black triangles indicate the location of the unqiue Trp948 residue in SpCBM32 (above; filled) and the amino acids involved in calcium coordination (below; filled, side chain; open, main-chain carbonyl). (c) The three-dimensional structure of SpCBM32 in wall-eyed stereo cartoon format with a blue (N-terminal) to red (C-terminal) colour gradient and the structural calcium shown as a magenta sphere. The nine β-strands and two α-helices are labelled. (d) The calcium-binding site of SpCBM32. The pentagonal bipyramidal geometry of the desolvated coordination pocket is displayed with the weighted maximum-likelihood (Murshudov et al., 1997 ▶)/σA (Read, 1986 ▶) 2F obs − F calc map (cyan) contoured at 1.0σ (0.35 e− Å−3).
2. Materials and methods
2.1. Cloning, recombinant production and purification of SpCBM32
The gene fragment encoding the putative CBM32 from S. pneumoniae EndoD, SpCBM32, was amplified from S. pneumoniae genomic DNA (TIGR strain BAA-334D) using the oligonucleotide primers CBMF (5′-TATACATATGAATATCGTTCCAGGTGCA-3′) and CBMR (5′-GCGCCTCGAGTTATCCATTGTCAGAAGT-3′). The amplified DNA product encoding amino acids 817–945 of GH85 was cloned into a pET28a plasmid vector (Novagen, catalogue No. 69864) via engineered 5′ NheI and 3′ XhoI restriction sites to generate pCBM32, which encodes the CBM fused to an N-terminal His6 tag (MGSSHHHHHHSSGLVPRGSHM) via a thrombin protease cleavage site.
The SpCBM32 protein was labelled with selenomethonine by recombinant production in the methionine auxotroph Escherichia coli strain B834 (DE3) (Novagen, catalogue No. 69041) using selenomethionine-supplemented minimal medium prepared as per the instructions of the manufacturer (Athena Enzyme Systems, Baltimore, Maryland, USA). Kanamycin was added to a final concentration of 50 µg ml−1 to select for transformed cells. Cultures were grown at 310 K until they reached an optical density of 0.5–0.7 at 600 nm, whereupon protein production was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation after 4 h of additional growth at 310 K and were then disrupted using a French pressure cell in 20 mM Tris–HCl pH 8.0, 0.5 M NaCl. Polypeptides were purified from clarified cell lysate by immobilized metal-affinity chromatography using 2 ml Ni2+-affinity resin (GE Healthcare, catalogue No. 17-5318-06). IMAC-purified protein was further purified by size-exclusion chromatography (SEC) using a Sephacryl S-200 column (GE Biosciences, catalogue No. 17-0584-05) and 20 mM Tris–HCl pH 8.0. Purified protein was concentrated in a stirred-cell ultrafiltration device with a 5000 molecular-weight cutoff membrane (Millipore, catalogue No. PLCC02510). Protein concentration was determined by UV absorbance at 280 nm using a calculated extinction coefficient (Gasteiger et al., 2005 ▶) of 24 980 M −1 cm−1.
2.2. Crystallization, data collection and structure solution
Crystals of SpCBM32 protein were grown in 30% PEG 4000, 0.2 M ammonium acetate and 0.1 M sodium citrate pH 6.5 by the hanging-drop vapour-diffusion method at 291 K. Crystals were flash-cooled with liquid nitrogen in crystallization solution supplemented with 20–30%(v/v) ethylene glycol. Data were collected on beamline 9.2 at Stanford Synchrotron Radiation Lightsource and were processed using HKL-2000. Data-collection and processing statistics are shown in Table 1 ▶.
Table 1. Data-collection and structure statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength (Å) | 0.9791 |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 134.11, b = 73.51, c = 34.60, β = 96.52 |
| Resolution (Å) | 35.00–2.00 (2.07–2.00) |
| Rmerge† | 0.136 (0.310) |
| 〈I/σ(I)〉 | 18.5 (7.2) |
| Completeness (%) | 98.1 (97.4) |
| Multiplicity | 14.6 (14.3) |
| Mosaicity (°) | 0.9 |
| No. of crystals | 1 |
| Temperature (K) | 100 |
| Oscillation (°) | 1 |
| Rotation range (°) | 680 |
| Exposure per frame (s) | 4 |
| Crystal-to-detector distance (mm) | 200 |
| Detector | MAR325 |
| Refinement | |
| No. of reflections | 20817 |
| Rwork/Rfree | 0.22/0.26 |
| No. of atoms | |
| Protein | 1142 [chain A], 1142 [chain B] |
| Ligand/ion | 2 Ca, 4 ACT |
| Water | 288 |
| B factors (Å2) | |
| Protein | 17.3 [chain A], 18.3 [chain B] |
| Ligand/ion | 30.3 [Ca], 31.4 [ACT] |
| Water | 31.2 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.252 |
| Ramachandran plot (%) | |
| Favoured | 86.9 |
| Allowed | 13.1 |
| Outliers | 0.0 |
R
merge = 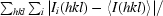
 , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
The structure of SpCBM32 was solved by a single-wavelength anomalous dispersion (SAD) experiment using an X-ray wavelength of 0.9791 Å. The positions of all of the eight Se atoms expected for the two molecules in the asymmetric unit were determined using SHELXC/D (Sheldrick, 2008 ▶) with data extending to 2.5 Å resolution. Initial phases were produced with SHELXE using data to 2.0 Å resolution. PROFESS was used to determine the noncrystallographic symmetry (NCS) operators, followed by density modification with NCS averaging using DM and a solvent content of 54% (Cowtan & Zhang, 1999 ▶). Using the phases output from DM, ARP/wARP (Perrakis et al., 1999 ▶) was able to build a nearly complete model with docked side chains. The model was then completed using Coot (Emsley & Cowtan, 2004 ▶) followed by refinement using REFMAC (Murshudov et al., 1997 ▶). Water molecules were added using the REFMAC implementation of Coot Find Waters and inspected visually prior to deposition. 5% of the observations were flagged as ‘free’ and were used to monitor refinement procedures (Brünger, 1992 ▶). Model validation was performed with SFCHECK (Vaguine et al., 1999 ▶) and PROCHECK (Laskowski et al., 1996 ▶). Model statistics are given in Table 1 ▶. Coordinates and structure factors have been deposited with the PDB under code 2xqx.
3. Results and discussion
3.1. The structure of SpCBM32
The selenomethionine-labelled construct crystallized with two molecules in the asymmetric unit, which overlaid with an r.m.s.d. of 0.19 Å2 over 146 residues, indicating that the module is quite rigid (see Table 1 ▶ for statistics). The monomers were related by nearly perfect twofold NCS, suggesting the possibility of a biological dimer. However, PISA analysis of the assembly indicated that it is likely to not be stable in solution (not shown); indeed, the protein behaved as a monomer in solution when purified by gel-permeation chromatography.
The SpCBM32 monomer adopts the conventional β-sandwich fold that is common to many families of CBMs. This comprises nine β-strands organized into two opposing four-stranded and five-stranded antiparallel β-sheets with an overall jelly-roll topology (Figs. 1 ▶ b and 1 ▶ c). The strands are composed of contiguous stretches of amino-acid residues that are numbered as follows: β1, 822–827; β2, 852–855; β3, 860–865; β4, 870–878; β5, 895–903; β6, 906–915; β7, 921–932; β8, 934–941; β9, 951–959. Two short α-helical regions are also present between β1 and β2 (α1, amino acids 838–843) and β4 and β5 (α2, amino acids 880–883). The first of these α-helices is secured to the protein surface through its participation in coordinating a buried metal ion (Fig. 1 ▶ d). The ion is bound with pentagonal bipyramidal coordination geometry by three Oδ atoms donated from the side chains of Asn843 (2.6 Å), Asp851 (2.9 Å) and Asn955 (2.7 Å) and the four peptide-bond carbonyl O atoms from Gly840 (2.5 Å), Thr845 (2.4 Å), Asp851 (2.6 Å) and Tyr954 (2.3 Å). These distances and the ligation geometry are consistent with other reports of calcium coordination (Harding, 2001 ▶). The identity of this ion as calcium is further supported by the refined B factor when it is modelled as a calcium, which is similar to the B factors of the ligating atoms. A metal ion at this position is structurally well conserved in a large number of CBM families (Boraston et al., 2004 ▶). However, the coordinated metal is likely to play only an indirect role in potential ligand binding as these interactions are distal to the binding site and buried within the protein, thereby contributing to maintaining the structural integrity of the CBM.
A search for structural homologues of SpCBM32 using the DALI server (Holm & Rosenstrom, 2010 ▶; see Table 2 ▶ for statistics) revealed that the structure has highest structural identity to the human heat-shock protein HSPB11 (PDB entry 1tvg; Ramelot et al., 2009 ▶) and to a putative C-terminal CBM32 from the Bacillus thetaiotaomicron GH29-containing CAZyme Bt2192 (PDB entry 3eyp; J. B. Bonanno, J. Freeman, K. T. Bain, S. Hu, R. Romero, S. Wasserman, J. M. Sauder, S. K. Burley & S. C. Almo, unpublished work). Both of these structures are not currently classified in the CAZY database as CBMs. Although the role of the B. thetaiotaomicron module as a CBM appears to be likely considering that it is appended to a carbohydrate-active catalytic module, the biological role of HSPB11 is unknown and analysis of the binding-site location reveals that it lacks any surface-exposed aromatic residues, which are a hallmark of carbohydrate-binding sites. The next two similar structures belong to characterized CBMs from family 32, both of which are in complexes with galactose: the CBM32 from a Clostridium perfringens exo-α-sialidase (Ficko-Blean & Boraston, 2006 ▶; PDB entry 2v72) and the CBM32 from a Micromonospora viridifaciens sialidase (Newstead et al., 2005 ▶; PDB entry 2bzd). These data provide strong support for the SpCBM32 protein functioning as a CBM.
Table 2. DALI search statistics of structural neighbours.
3.2. The putative binding site of SpCBM32
The amino-acid sequence comparison and X-ray crystal structure analysis of SpCBM32 clearly reveal it to be a member of CBM family 32. Unfortunately, despite much effort using glycan microarray screening through the Consortium for Functional Glycomics and qualitative UV difference assays, at present there is no functional evidence to suggest that SpCBM32 displays carbohydrate-binding activity; this is a common observation as CBM32s tend to display very low affinities for their target carbohydrate ligands (Ficko-Blean & Boraston, 2006 ▶, 2009 ▶; Boraston et al., 2007 ▶). However, the binding sites of many CBM32s have been identified and characterized using various biophysical and structural approaches, which have illuminated several common features, including the generally conserved location on the β-sandwich scaffold of the binding site and the presence of solvent-exposed aromatic amino-acid side chains. Thus, comparing the SpCBM32 model with ligand-bound complexes of related CBM32 structures provides valuable insight into its carbohydrate-binding potential (Fig. 2 ▶). Such a comparison immediately reveals the presence of a solvent-exposed tryptophan residue (Trp948) that is found adjacent to what is the conserved location of the family 32 CBM binding site (Fig. 2 ▶ a). This residue is located at the end of SpCBM32 opposite to that where the N- and C-termini are present, suggesting that neighbouring modules, even those which are closely associated, are unlikely to occlude this site. These features make the region surrounding Trp935 an excellent candidate for a carbohydrate-binding site despite the absence of any other structurally conserved functional residues.
Figure 2.

Comparison of the SpCBM32 binding site with other CBM32 structures. (a–c) Side views, with the amino acids involved in carbohydrate recognition shown as sticks. Where appropriate, galactose is displayed as grey sticks and hydrogen-bond contacts are indicated by yellow dashed lines: (a) SpCBM32 (yellow), (b) C. perfringens CBM32 (cyan, PDB entry 2v72) and (c) M. viridifaciens CBM32 (green, PDB entry 2bzd). Electrostatic surface potentials are shown for (d) SpCBM32, (e) C. perfringens CBM32 (PDB entry 2v72) and (f) B. thetaiotaomicron CBM32 (PDB entry 3eyp). Structures are superimposed and positioned in the same orientations with the binding sites facing outward. W948 and the putative branching point are labelled.
There are two definitive structural features within the putative binding site of SpCBM32. The first, as previously mentioned, is Trp948, which projects outwards towards the solvent at an angle of approximately 121° to the base of the putative binding site. The back face of the indole ring stacks against a cis-configured proline residue that loops the backbone back into the protein core. Trp948 is located on an extended loop that is unique to SpCBM32 and is orientated perpendicular to the aromatic residues observed in the C. perfringens CBM32 and M. viridifaciens CBM32 sialidases (Figs. 2 ▶ a–2 ▶ c). The solvent-accessible face is appropriately poised to ‘stack’ against, or CH–π bond to, the apolar surface of a sugar ring ligand. This binding mechanism is a common specificity determinant for CBM–carbohydrate interactions. Interestingly, Trp948 may serve as an anchoring point for an internal residue as the potential binding site appears to accommodate branching on either side (Fig. 2 ▶ d). The second key feature of the putative binding site is that this region is remarkably flat and displays a neutral charge distribution. A comparison with the electrostatic potential of the solvent-accessible surfaces of structurally and functionally characterized CBM32s reveals the uniquely open nature of the putative CBM32 binding site (Figs. 2 ▶ d–2 ▶ f). This observation supports the emerging principle that although CBM32s share overall fold identity and are grouped within sequence-based families, individual binding sites can display great variability and have signature shapes and charge distributions that are tailored to recognize the three-dimensional structure of target carbohydrate ligands (Abbott et al., 2007 ▶, 2008 ▶). The functional diversity that has been described for CBM32 is consistent with the abundance of unique enzyme activities that these modules are found to be appended to [selected examples include the CBM32s from the DALI alignment: endo-β-1,4-N-acetylglucosamidase, exo-α-sialidase (PDB entry 2v72; Ficko-Blean & Boraston, 2006 ▶) and α-l-fucosidase (PDB entry 3eyp); see http://www.cazy.org/CBM32.html for a more exhaustive list]. Thus, the overall topology of the putative binding site and the chemistry of the side chains lining the surface of SpCBM32 are likely to reflect the signature shape of its potential carbohydrate ligand.
3.3. Implications for EndoD function
Although there are exceptions, CBMs typically display a carbohydrate specificity that parallels the specificity of the catalytic module of the parent enzyme. The EndoD catalytic module is active on the chitobiose core of high-mannose N-linked glycans that contain five or fewer mannose residues. Therefore, we suggest that the putative carbohydrate ligand for SpCBM32 is an N-linked glycan or a fragment thereof. The open nature of the putative SpCBM32 binding site is consistent with the crystal structure of a Man-α1,6-(α1,3-Man)-Man trisaccharide bound to the unrelated Galanthus nivalis agglutinin (PDB entry 1jpc; Wright & Hester, 1996 ▶), which highlights the propensity of a branched mannooligosaccharide to adopt a planar conformation and suggests the possibility of binding internal motifs in the glycan rather than the nonreducing terminal ends. Thus, it appears that the SpCBM32 module may function either in substrate capture for the catalytic modules, as is the general proposed role for carbohydrate-binding modules (Boraston et al., 2004 ▶), or in carbohydrate-mediated pneumococcal adhesion to host tissue.
Supplementary Material
PDB reference: carbohydrate-binding module of EndoD, 2xqx
Acknowledgments
This work was supported by a Canadian Institutes for Health Research Operating Grant (FRN 86610).
References
- Abbott, D. W., Eirín-López, J. M. & Boraston, A. B. (2008). Mol. Biol. Evol. 25, 155–167. [DOI] [PubMed]
- Abbott, D. W., Hrynuik, S. & Boraston, A. B. (2007). J. Mol. Biol. 367, 1023–1033. [DOI] [PubMed]
- Abbott, D. W., Macauley, M. S., Vocadlo, D. J. & Boraston, A. B. (2009). J. Biol. Chem. 284, 11676–11689. [DOI] [PMC free article] [PubMed]
- Boraston, A. B., Bolam, D. N., Gilbert, H. J. & Davies, G. J. (2004). Biochem. J. 382, 769–781. [DOI] [PMC free article] [PubMed]
- Boraston, A. B., Ficko-Blean, E. & Healey, M. (2007). Biochemistry, 46, 11352–11360. [DOI] [PubMed]
- Boraston, A. B., Wang, D. & Burke, R. D. (2006). J. Biol. Chem. 281, 35263–35271. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed]
- Cowtan, K. D. & Zhang, K. Y. (1999). Prog. Biophys. Mol. Biol. 72, 245–270. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Ficko-Blean, E. & Boraston, A. B. (2006). J. Biol. Chem. 281, 37748–37757. [DOI] [PubMed]
- Ficko-Blean, E. & Boraston, A. B. (2009). J. Mol. Biol. 390, 208–220. [DOI] [PMC free article] [PubMed]
- Gasteiger, E. H. C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Gilbert, H. J. (2010). Plant Physiol. 153, 444–455. [DOI] [PMC free article] [PubMed]
- Gregg, K. J., Finn, R., Abbott, D. W. & Boraston, A. B. (2008). J. Biol. Chem. 283, 12604–12613. [DOI] [PMC free article] [PubMed]
- Harding, M. M. (2001). Acta Cryst. D57, 401–411. [DOI] [PubMed]
- Hava, D. L. & Camilli, A. (2002). Mol. Microbiol. 45, 1389–1406. [PMC free article] [PubMed]
- Holm, L. & Rosenstrom, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Lammerts van Bueren, A., Higgins, M., Wang, D., Burke, R. D. & Boraston, A. B. (2007). Nature Struct. Mol. Biol. 14, 76–84. [DOI] [PubMed]
- Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. (1996). J. Biomol. NMR, 8, 477–486. [DOI] [PubMed]
- Montanier, C. et al. (2009). Proc. Natl Acad. Sci. USA, 106, 3065–3070.
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Newstead, S. L., Watson, J. N., Bennet, A. J. & Taylor, G. (2005). Acta Cryst. D61, 1483–1491. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Ramelot, T. A., Raman, S., Kuzin, A. P., Xiao, R., Ma, L.-C., Acton, T. B., Hunt, J. F., Montelione, G. T., Baker, D. & Kennedy, M. A. (2009). Proteins, 75, 147–167. [DOI] [PMC free article] [PubMed]
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Shelburne, S. A., Davenport, M. T., Keith, D. B. & Musser, J. M. (2008). Trends Microbiol. 16, 318–325. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Umekawa, M., Huang, W., Li, B., Fujita, K., Ashida, H., Wang, L.-X. & Yamamoto, K. (2008). J. Biol. Chem. 283, 4469–4479. [DOI] [PubMed]
- Vaaje-Kolstad, G., Horn, S. J., Van Aalten, D. M., Synstad, B. & Eijsink, V. G. (2005). J. Biol. Chem. 280, 28492–28497. [DOI] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Wright, C. S. & Hester, G. (1996). Structure, 4, 1339–1352. [DOI] [PubMed]
- Yin, J., Li, L., Shaw, N., Li, Y., Song, J. K., Zhang, W., Xia, C., Zhang, R., Joachimiak, A., Zhang, H.-C., Wang, L.-X., Liu, Z.-J. & Wang, P. (2009). PLoS One, 4, e4658. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: carbohydrate-binding module of EndoD, 2xqx