

The α isoform of human autotaxin has been crystallized. Diffraction data were collected to 3.0 Å resolution using synchrotron radiation.
Keywords: autotaxin, lysophosphatidic acid, lysophospholipase D, ectonucleotide pyrophosphatase/phosphodiesterase 2
Abstract
Autotaxin (ATX), which is also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2) or phosphodiesterase Iα (PD-Iα), is an extracellular lysophospholipase D which generates lysophosphatidic acid (LPA) from lysophosphatidylcholine (LPC). ATX stimulates tumour-cell migration, angiogenesis and metastasis and is an attractive target for cancer therapy. For crystallographic studies, the α isoform of human ATX was overproduced in Escherichia coli, purified and crystallized using the hanging-drop vapour-diffusion method. X-ray diffraction data were collected to 3.0 Å resolution from a monoclinic crystal form belonging to space group C2, with unit-cell parameters a = 311.4, b = 147.9, c = 176.9 Å, β = 122.6°.
1. Introduction
Autotaxin (ATX), which is also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2) or phosphodiesterase Iα (PD-Iα), was originally identified as an autocrine motility factor released by a human melanoma A2058 cell line (Stracke et al., 1992 ▶). Various experiments subsequently identified potential roles for ATX in cancer progression, tumour-cell invasion and metastasis, including the promotion of tumour angiogenesis (van Meeteren & Moolenaar, 2007 ▶; Yuelling & Fuss, 2008 ▶; Okudaira et al., 2010 ▶). In 2002, two groups independently identified ATX as the key component of an extracellular pathway for the generation of lysophosphatidic acid (LPA) from lysophosphatidylcholine (LPC) by its lysophospholipase D (lysoPLD) activity (Tokumura et al., 2002 ▶; Umezu-Goto et al., 2002 ▶). To date, three isoforms of ATX have been identified in both human and mouse (Giganti et al., 2008 ▶) and are referred to as the α (915 amino acids), β (863 amino acids) and γ (889 amino acids) isoforms of autotaxin, with the α isoform being identical to the originally discovered ATX from A2058 cells (Stracke et al., 1992 ▶). The amino-acid sequence of ATX is divided into several domains, including two closely located N-terminal somatomedin B-like domains, a central phosphodiesterase domain and a C-terminal nuclease-like domain. ATX is a member of the NPP (ectonucleotide pyrophosphatase/phosphodiesterase) family (Bollen et al., 2000 ▶). The crystal structure of a bacterial NPP, which was identified by a database search using the amino-acid sequence of the catalytic domain of mouse NPP1 as the template, indicates that the central catalytic domain of mammalian NPPs consists of an α/β structure (Zalatan et al., 2006 ▶). Based on its structural similarities to the better characterized NPP1, it was assumed that ATX is also synthesized as a type II integral membrane protein. However, it is now recognized that ATX is actually synthesized as a preproenzyme and that the proteolytically processed protein is secreted (Jansen et al., 2005 ▶; Koike et al., 2006 ▶).
LPA is an extracellular signalling molecule which binds to specific cell-surface receptors to promote cell growth, survival, motility and differentiation. Since ATX is the key component of an extracellular pathway for the generation of LPA, inhibition of ATX is a viable and effective way to interfere with LPA signalling (Federico et al., 2008 ▶). The lack of a three-dimensional structure of ATX makes the application of structure-based inhibitor design very difficult. Thus, structural study of ATX should be useful for the development of novel ATX inhibitors. Recently, the crystallization of rat autotaxin has been reported (Day et al., 2010 ▶; Hausmann et al., 2010 ▶). Here, we report the crystallization and preliminary X-ray crystallographic study of the α isoform of human ATX.
2. Materials and methods
2.1. Overproduction and purification
Since the N-terminal hydrophobic sequence of ATX functions as a signal peptide (Jansen et al., 2005 ▶; Koike et al., 2006 ▶) and the N-terminus of the secreted ATX from a human melanoma cell line (A2058) was reported to be Asp49 (Murata et al., 1994 ▶), the DNA encoding residues Ser48–Ile915 (C-terminus) of the α isoform of human ATX (also known as ATX-T; GenBank ID L46720.1) was amplified from pCD2/Basinger, which is a human foreskin fibroblast cDNA library (provided by the Japanese Collection of Research Bioresources; LI019), by long and accurate PCR (LA PCR) using LA Taq polymerase (TaKaRa) with 5′-CACCATGGACTCCCCCTGGACCAACATCTC-3′ and 5′-AATCTCGCTCTCATATGTATGCAG-3′ as the forward and reverse primers, respectively. The PCR conditions were as follows: 367 K for 5 min for the initial denaturation, 35 cycles of 367 K for 30 s, 328 K for 30 s and 345 K for 2.5 min, and 345 K for 10 min for the final extension. The PCR product was cloned into pET101/D-TOPO expression plasmid (Invitrogen) and transformed into Escherichia coli TOP10 strain (Invitrogen). The construct was verified by sequencing using a CEQ2000XL automated sequence-analysis system (Beckman Coulter).
E. coli BL21 (DE3) pLysS cells (Novagen) harbouring the expression plasmid were grown in LB medium (3 l shake flask containing 1 l medium) at 310 K to an OD600 of 0.6. Overproduction of hATX was induced by adding 0.2 mM IPTG for 8 h at 310 K. After this period, the cells were harvested by centrifugation at 8000g for 15 min and suspended in buffer A (20 mM Tris–HCl pH 7.5, 150 mM NaCl). The cells were stored at 253 K. The frozen cell pellet was dissolved in 25 ml buffer A; lysozyme, Triton X-100 and DNase I were then added to final concentrations of 400 µg ml−1, 0.4%(v/v) and 5 µg ml−1, respectively. The mixture was gently shaken at room temperature (∼298 K) for 30 min and then centrifuged at 20 000g for 60 min. The supernatant was filtrated using a 0.45 µm pore filter and applied onto a 1 ml Zn2+-charged HiTrap Chelating column (GE Healthcare) equilibrated with buffer A. The column was washed with 20 column volumes of wash buffer (10 mM imidazole in buffer A). After washing, hATX was eluted with ten column volumes of elution buffer (500 mM imidazole in buffer A). The hATX was further purified by gel chromatography using a Superdex 200 pg column (GE Healthcare) equilibrated with buffer A. The fractions containing hATX were pooled and concentrated to 7.5 mg ml−1 using an Amicon Ultra-15 (Millipore). The enzymatic (lysoPLD and phosphodiesterase) activities of recombinant hATX were measured according to previous reports (Lee et al., 2001 ▶; Tokumura et al., 2002 ▶).
2.2. Crystallization
Initial sparse-matrix crystal screening (Jancarik & Kim, 1991 ▶) was conducted using Crystal Screen, Crystal Screen Cryo, Index and SaltRX from Hampton Research and Wizard I, II and III and Cryo I and II from Emerald BioSystems. Crystallization was carried out by the hanging-drop method, in which 1 µl protein solution was mixed with the same volume of reservoir solution. The drops were suspended over 200 µl reservoir solution in 48-well plates and incubated at 293 K.
2.3. X-ray data collection
Data collection was performed by the rotation method at 100 K using an ADSC Q210r CCD detector with synchrotron radiation (λ = 1.000 Å on beamline NW12A of PF-AR, Tsukuba, Japan). The Laue group and unit-cell parameters were determined using the HKL-2000 package (Otwinowski & Minor, 1997 ▶). Self-rotation and native Patterson functions were calculated using the programs POLARRFN and FFT from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
3.1. Overproduction, purification and crystallization
The α isoform of hATX was successfully cloned, overproduced and purified to homogeneity. The lysoPLD activity of recombinant hATX (V max = 10.5 µmol min−1 mg−1) was comparable with that reported previously (Haga et al., 2008 ▶) for chaperone-coproduced hATX (V max = 9.6 µmol min−1 mg−1). A similar result has been reported for mammalian cell-expressed rat ATX (V max = 9.0 µmol min−1 mg−1; Umezu-Goto et al., 2002 ▶). SDS–PAGE of the purified enzyme revealed a single 100 kDa protein band on Coomassie Brilliant Blue staining. Initial crystal screening produced several microcrystals within two weeks. Tiny needle-shaped crystals grew from condition No. 40 of Crystal Screen Cryo [19%(v/v) 2-propanol, 19%(m/v) PEG 4000 and 5%(v/v) glycerol in 0.095 M sodium citrate pH 5.6]. Trials to improve the reproducibility and the quality of the crystals were performed by varying the pH, the buffer system and the concentration of the crystallizing agent. To obtain crystals suitable for X-ray analysis, a droplet was prepared by mixing equal volumes (2 µl + 2 µl) of the protein solution and reservoir solution [19%(v/v) 2-propanol, 19%(m/v) PEG 4000 and 5%(v/v) glycerol in 0.25 M sodium citrate pH 5.5] and was suspended over 500 µl reservoir solution in 24-well plates. Plate-shaped crystals with typical dimensions of approximately 0.15 × 0.08 × 0.005 mm grew within two weeks (Fig. 1 ▶).
Figure 1.

A monoclinic crystal of hATX with dimensions of 0.15 × 0.08 × 0.005 mm.
3.2. X-ray analysis
Since the crystallization conditions of hATX described above contained 19%(m/v) PEG 4000, 19%(v/v) 2-propanol and 5%(v/v) glycerol in the reservoir solution, X-ray data collection could be performed under cryogenic conditions without any further addition of cryoprotectant. Crystals from the hanging drop were directly mounted in nylon loops and flash-cooled in a cold nitrogen-gas stream at 100 K just prior to data collection. The Laue group of the hATX crystals was found to be 2/m and the unit-cell parameters were a = 311.4, b = 147.9, c = 176.9 Å, β = 122.6°. Only reflections with h + k = 2n were observed for hkl reflections, indicating the monoclinic space group C2. Assuming the presence of five to nine molecules per asymmetric unit led to an empirically acceptable V M value ranging from 3.43 to 1.91 Å3 Da−1, corresponding to a solvent content ranging from 64.2 to 35.5% (Matthews, 1968 ▶). Calculation of the self-rotation function (Fig. 2 ▶) showed a quite clear (91% of the height of the origin peak) noncrystallographic twofold axis, indicating that the number of molecules in the asymmetric unit of the hATX crystal would be an even number. We thus assume that the asymmetric unit consists of six or eight molecules, with corresponding V M values of 2.86 or 2.14 Å3 Da−1 and solvent contents of 57.1 or 42.7%, respectively. However, judging from the limited resolution of the ATX crystal, tight packing (eight molecules in the asymmetric unit) may be unlikely. Therefore, we presume that the most probable estimation of the number of molecules in the asymmetric unit is six. Neither threefold nor fourfold axes could be detected in the self-rotation search. We next checked for a pseudo-translation vector in the native Patterson map of the hATX crystal; however, no significant peak was found. The current best diffraction data from an hATX crystal were collected to 3.0 Å resolution (Fig. 3 ▶). Although the hATX crystal diffracted to beyond 3.0 Å resolution, a long unit-cell parameter (a = 311.4 Å) and the limited size of the detector area made the collection of the higher resolution data difficult. Data-collection statistics are summarized in Table 1 ▶. A search for heavy-atom derivatives for use in phasing by the multiple isomorphous replacement and the preparation of SeMet-substituted hATX using LeMaster medium (Hendrickson et al., 1990 ▶) and E. coli B834 (DE3) cells for use in phasing by the Se-MAD/SAD method are under way.
Figure 2.

Stereographic projection of the self-rotation function in spherical polar angles at κ = 180° for the hATX crystal. A self-rotation search was carried out with the program POLARRFN from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶) using the data in the 30.0–3.0 Å resolution shell and an integration radius of 30.0 Å. The two strongest peaks [relative height of 100, (ω, ϕ) = (90.0, 90.0°) and (90.0, −90.0°)] indicate the crystallographic twofold symmetry (origin peak). Contour lines are drawn at an increment of 10% of the origin peak. The program produces a constant rotation angle κ for different axis directions defined by ω (the angle from the pole) and ϕ (the angle around the equator). The ϕ angles are marked on the circumference and the ω angles are defined as 0° or 180° at the centre and 90° around the edge. Noncrystallographic twofold rotation axes (91% height of the origin peak) are found at (ω, ϕ) = (57.3, 0.0°) and (147.3, 0.0°).
Figure 3.

X-ray diffraction image from an hATX crystal. The circles indicate resolutions of 13.98, 7.06, 4.78, 3.66 and 3.00 Å.
Table 1. Data-collection statistics for hATX.
Values in parentheses are for the outer shell.
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 311.4, b = 147.9, c = 176.9, β = 122.6 |
| Estimated No. of molecules in asymmetric unit | 6 |
| Solvent content (%) | 57.1 |
| X-ray source | PF-AR NW12A |
| Detector | ADSC Q210r |
| Wavelength (Å) | 1.000 |
| Resolution range (Å) | 50–3.00 (3.11–3.00) |
| No. of observed reflections | 480382 |
| No. of unique reflections | 132049 |
| Multiplicity | 3.6 (2.8) |
| Mean I/σ(I) | 13.0 (3.3) |
| Rmerge† (%) | 12.5 (29.3) |
| Completeness (%) | 97.8 (82.3) |
R
merge = 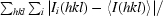
 , where Ii(hkl) is the ith measurement and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
, where Ii(hkl) is the ith measurement and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
Acknowledgments
We thank Drs Y. Yamada, N. Matsugaki and N. Igarashi of Photon Factory for their help with data collection at the synchrotron facility.
References
- Bollen, M., Gijsvers, R., Ceulemans, H., Stalmans, W. & Stefan, C. (2000). Crit. Rev. Biochem. Mol. Biol. 35, 393–432. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Day, J. E., Hall, T., Pegg, L. E., Benson, T. E., Hausmann, J. & Kamtekar, S. (2010). Acta Cryst. F66, 1127–1129. [DOI] [PMC free article] [PubMed]
- Federico, L., Pamuklar, Z., Smyth, S. S. & Morris, A. J. (2008). Curr. Drug Targets, 9, 698–708. [DOI] [PMC free article] [PubMed]
- Giganti, A., Rodriguez, M., Fould, B., Moulharat, N., Coge, F., Chomarat, P., Galizzi, J.-P., Valet, P., Saulnier-Blache, J.-S., Boutin, J. A. & Ferry, G. (2008). J. Biol. Chem. 283, 7776–7789. [DOI] [PubMed]
- Haga, A., Hashimoto, K., Tanaka, N., Nakamura, K. T. & Deyashiki, Y. (2008). Protein Expr. Purif. 59, 9–17. [DOI] [PubMed]
- Hausmann, J., Christodoulou, E., Kasiem, M., De Marco, V., van Meeteren, L. A., Moolenaar, W. H., Axford, D., Owen, R. L., Evans, G. & Perrakis, A. (2010). Acta Cryst. F66, 1130–1135. [DOI] [PMC free article] [PubMed]
- Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. (1990). EMBO J. 9, 1665–1672. [DOI] [PMC free article] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Jansen, S., Stefan, C., Creemers, J. W. M., Waelkens, E., Van Eynde, A., Stalmans, W. & Bollen, M. (2005). J. Cell Sci. 118, 3081–3089. [DOI] [PubMed]
- Koike, S., Keino-Masu, K., Ohto, T. & Masu, M. (2006). Genes Cells, 11, 133–142. [DOI] [PubMed]
- Lee, J., Jung, I. D., Nam, S. W., Clair, T., Jeong, E. M., Hong, S. Y., Han, J. W., Lee, H. W., Stracke, M. L. & Lee, H. Y. (2001). Biochem. Pharmacol. 62, 219–224. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Meeteren, L. A. van & Moolenaar, W. H. (2007). Prog. Lipid Res. 46, 145–160. [DOI] [PubMed]
- Murata, J., Lee, H. Y., Clair, T., Krutzsch, H. C., Arestad, A. A., Sobel, M. E., Liotta, L. A. & Stracke, M. L. (1994). J. Biol. Chem. 269, 30479–30484. [PubMed]
- Okudaira, S., Yukiura, H. & Aoki, J. (2010). Biochimie, 92, 698–706. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Stracke, M. L., Krutzsch, H. C., Unsworth, E. J., Arestad, A., Cioce, V., Schiffmann, E. & Liotta, L. A. (1992). J. Biol. Chem. 267, 2524–2529. [PubMed]
- Tokumura, A., Majima, E., Kariya, Y., Tominaga, K., Kogure, K., Yasuda, K. & Fukazawa, K. (2002). J. Biol. Chem. 277, 39436–39442. [DOI] [PubMed]
- Umezu-Goto, M., Kishi, Y., Taira, A., Hama, K., Dohmae, N., Takio, K., Yamori, T., Mills, G. B., Inoue, K., Aoki, J. & Arai, H. (2002). J. Cell Biol. 158, 227–233. [DOI] [PMC free article] [PubMed]
- Yuelling, L. M. & Fuss, B. (2008). Biochim. Biophys. Acta, 1781, 525–530. [DOI] [PMC free article] [PubMed]
- Zalatan, J. G., Fenn, T. D., Brunger, A. T. & Herschlag, D. (2006). Biochemistry, 45, 9788–9803. [DOI] [PubMed]