

Abstract
Mutations in GJB2, encoding connexin 26 (Cx26), cause both autosomal dominant and autosomal recessive nonsyndromic hearing loss at the DFNA3 and DFNB1 loci, respectively. Most of the over 100 described GJB2 mutations cause autosomal recessive nonsyndromic hearing loss. Only a minority has been associated with autosomal dominant hearing loss. In this study, we present two families with autosomal dominant nonsyndromic hearing loss caused by a novel mutation in GJB2 (p.Asp46Asn). Both families were ascertained from the same village in northern Iran consistent with a founder effect. This finding implicates the D46N missense mutation in Cx26 as a common cause of deafness in this part of Iran mandating mutation screening of GJB2 for D46N in all persons with hearing loss who originate from this geographic region.
Keywords: connexin 26, D46N, autosomal dominant nonsyndromic hearing loss, DFNA3, Iran
Introduction
Approximately one in 1,000 newborns has severe-to-profound hearing loss and in more than half of this group of babies the etiology is genetic [Smith et al. 2005]. The pattern of inheritance is most frequently autosomal recessive, while autosomal dominant, X-linked and mitochondrial inheritances occur less frequently. Mutations in one gene, GJB2, have been found to cause up to half of autosomal recessive nonsyndromic hearing loss (ARNSHL) cases in developed countries [Kelley et al, 1998; Estivill et al, 1998; Green et al, 1999].
GJB2 encodes Connexin 26 (Cx26), a member of a highly conserved protein family found throughout the animal kingdom [van Steensel et al, 2004]. Six connexins oligomerize to form pore-like plasma membrane hemichannels called connexons. Connexons in adjacent cells then align to form gap junction channels, which facilitate electrical and biochemical coupling between cells. Although the function of gap junction channels in the inner ear has not been definitively determined, a role in maintaining cochlear homeostasis by recycling K+ ions following hair cell depolarization has been postulated [Kikuchi et al, 1995].
The majority of the over 100 deafness-causing mutations described in GJB2 causes ARNSHL, however 29 GJB2 mutations have been associated with autosomal dominant hearing loss (ADHL) (Table I). About two-thirds of these mutations cause a syndrome in which hearing loss is associated with distinct skin manifestations. Examples of these syndromes include Keratitis-Ichthyosis-Deafness (KID) syndrome [OMIM 148210], Vohwinkel syndrome [OMIM 124500], Bart-Pumphrey syndrome [OMIM 149200] and palmoplantar keratoderma (PPK) with deafness [OMIM 148350]. The remaining one-third of dominant GJB2 mutations causes autosomal dominant nonsyndromic hearing loss (ADNSHL). In this study, we present a novel GJB2 mutation responsible for ADNSHL in two families from a village in northern Iran.
Table I.
AD nonsyndromic and syndromic GJB2 mutations
| Mutation | Protein domain | Number of affecteds | Phenotype | Reference | |
|---|---|---|---|---|---|
| cDNA level | Protein level | ||||
| c.131G>C | W44S | E1 | - | Nonsyndromic | [http://davinci.crg.es/deafness/] |
| c.132G>C | W44C | E1 | 2 French families | Nonsyndromic | [Denoyelle et al. 1998] |
| c.223C>T | R75W | M2 | - | Nonsyndromic | [Janecke et al. 2001; Mani et al. 2008] |
| c.224G>A | R75Q | M2 | 4 | Nonsyndromic | [http://davinci.crg.es/deafness/] |
| c.428G>A | R143Q | M3 | 1 Austrian family | Nonsyndromic | [Löffler et al. 2001] |
| c.487A>C | M163L | E2 | Nonsyndromic | [Matos et al, 2008] | |
| c.535G>A | D179N | E2 | 1 Italian family | Nonsyndromic | [http://davinci.crg.es/deafness/] |
| c.551G>A | R184Q | E2 | - | Nonsyndromic | [Hamelmann et al, 2001] |
| c.605G>T | C202F | M4 | A large French family | Nonsyndromic | [Morlé et al. 2000] |
| c.34G>C | G12R | NT | 1 | Syndromic (Keratitis-Ichthyosis-Deafness) | [Richard et al. 2002] |
| c.42C>G | N14K | NT | 1 | Hypotrichosis-deafness | [van Steensel et al. 2004] |
| c.40A>C | N14Y | NT | 1 | Syndromic (Keratitis-Ichthyosis-Deafness) | [Arita et al. 2006] |
| c.50C>T | S17F | NT | 1 | Syndromic (Keratitis-Ichthyosis-Deafness) | [Richard et al. 2002] |
| c.119C>T | A40V | M1 | 1 | Syndromic (Keratitis-Ichthyosis-Deafness and the follicular occlusion triad) | [Montgomery JR et al. 2004] |
| 125_127delAGG | E42del | M1 | 1 family | Syndromic (Palmoplantar keratoderma with deafness) | [Rouan et al. 2001] |
| c.134G>A | G45E | E1 | - | Syndromic (Keratitis-Ichthyosis-Deafness) | [Janeck et al. 2005; Jonard et al. 2008] |
| c.148G>A | D50N | E1 | 7 | Syndromic (Keratitis-Ichthyosis-Deafness; Hystrix-like Ichthyosis-Deafness) | [van Steensel et al. 2002] |
| c.148G>T | D50Y | E1 | 1 | Syndromic (Keratitis-Ichthyosis-Deafness) | [Yotsumoto et al. 2003] |
| c.160A>C | N54H | E1 | 1 | Syndromic (Bart-Pumphrey) | [Akiyama et al. 2007] |
| not described | N54K | E1 | 1 family | Syndromic (Bart-Pumphrey) | [Richard et al. 2000] |
| c.175G>C | G59R | E1 | 1 | Syndromic (Palmoplantar keratoderma with deafness) | [Leonard et al. 2005] |
| c.175G>A | G59S | E1 | 1 | Syndromic (Palmoplantar keratoderma with deafness) | [Alexandrino et al. 2005] |
| c.176C>G | G59A | E1 | 1 family | Syndromic (Palmoplantar keratoderma with deafness) | [Heathcote et al. 2000] |
| c.196G>C | D66H | E1 | 10 | Syndromic (Vohwinkel) | [Maestrini et al. 1999] |
| c.219A>G | H73R | M2 | 1 family | Syndromic (Palmoplantar keratoderma with deafness) | [de Zwart-Storm et al. Mar. 2008] |
| c.223C>T | R75W | M2 | - | Syndromic (Palmoplantar keratoderma with deafness) | [Richard et al. 1998; Yuan et al. 2008] |
| c.224G>A | R75Q | E1 | 1 Turkish family | Syndromic (Palmoplantar keratoderma with deafness) | [Uyguner et al. 2002] |
| c.389G>T | G130V | CL | 1 family | Syndromic (Vohwinkel) | [Snoeckx et al. 2005] |
| c.424T>C | F142L | M3 | 1 | Syndromic (Mucositis-deafness) | [Brown et al. 2003] |
| c.548C>T | S183F | E2 | 1 family | Syndromic (Palmoplantar keratoderma with deafness) | [de Zwart-Storm et al. 2008] |
Materials and Methods
Subjects
This study was approved by the Institutional Review Boards of the University of Social Welfare and Rehabilitation Sciences in Tehran, Iran and the University of Iowa, Iowa City, IA, United States. Informed consent was obtained from all family members wishing to participate in this research. For each consenting person, clinical assessment and genetic counseling were performed. Audiometric testing was performed by measuring air and bone conduction thresholds (Fig 1).
Figure 1.

Audiograms associated with GJB2 D46N hearing loss (better hearing ear is shown). Top: Sequential audiograms from two persons in family L-1301 showing progression to severe-to-profound hearing loss. Middle: Audiograms from five persons in family L-1301 at different ages showing relatively uniform audioprofiles. Bottom: Audiograms from four persons in family L-3057 at different ages showing variably in hearing thresholds by age.
Haplotyping
Genomic DNA was isolated from peripheral blood using established techniques [Miller et al., 1988] and used to complete haplotype reconstruction for microsatellite markers that map to chromosome 13q12, the genomic region that includes GJB2. Markers were PCR amplified and products were resolved on the ABI Prism 3130 Genetic Analyzer. Alleles were assigned using GeneMapper 3.0 software (Applied Biosystems, Inc.), with haplotypes reconstructed using custom-made software.
GJB2 Screening
Mutation screening was completed on an Applied Biosystems (ABI) model 3700 automated sequencer. Sequence data were compared to published sequence for GJB2 using the Sequencher 4.1.2 software program package (Gene Codes, MI, USA). The putative structural and functional significance of observed mutations was assessed using the ConSeq web server (http://conseq.tau.ac.il/). Conseq scores obtained using this alignment method range from 1 (variable) to 9 (conserved).
Results
Subjects
Family members from two families, L-1301 and L-3057, segregating ADNSHL were recruited for this study (Fig 2). Although most persons consented to participate, some individuals (shown with asterisk in Fig 2) declined to have audiograms. For those persons with hearing loss, audiometry confirmed the loss to be sensorineural. Figure 2 shows hearing thresholds for the better hearing ear in six persons from Family L-1301 and in four persons from Family L-3057. In most persons, the hearing loss was postlingual with an onset in the first decade, however VI:13 and VI:9 had prelingual hearing loss that quickly progressed to become severe to profound in VI:13 (Figure 2: L-1301:1 and L-1301:2). Intrafamilial variation was observed (Fig 2). Pure tone audiograms by age group are shown in Figure 3. No features of syndromic hearing loss were observed in any person with hearing loss in these families (Fig 4).
Figure 2.
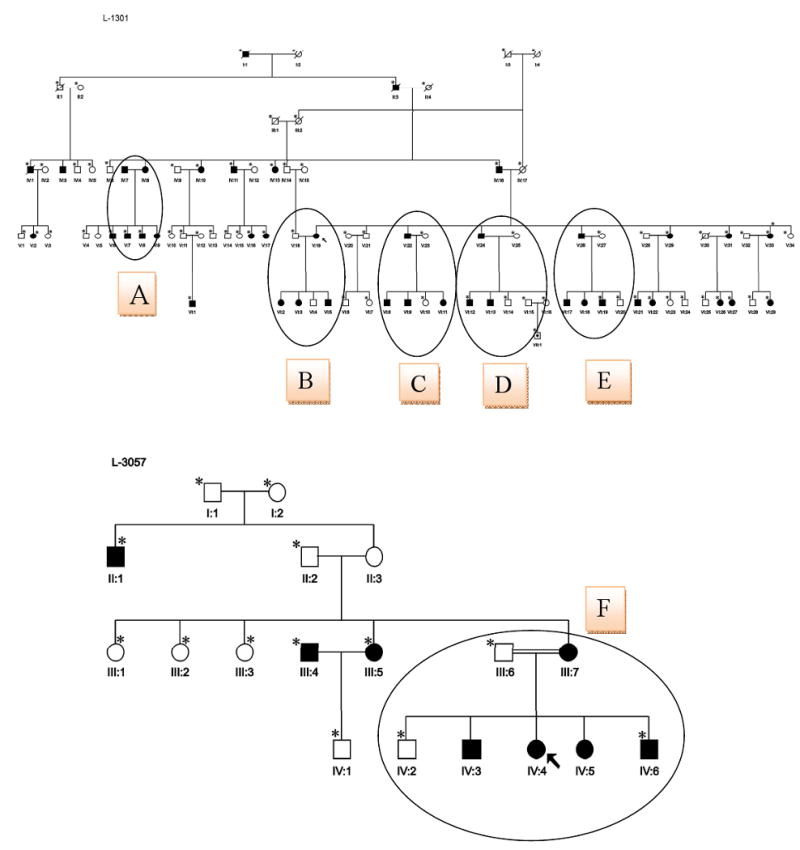
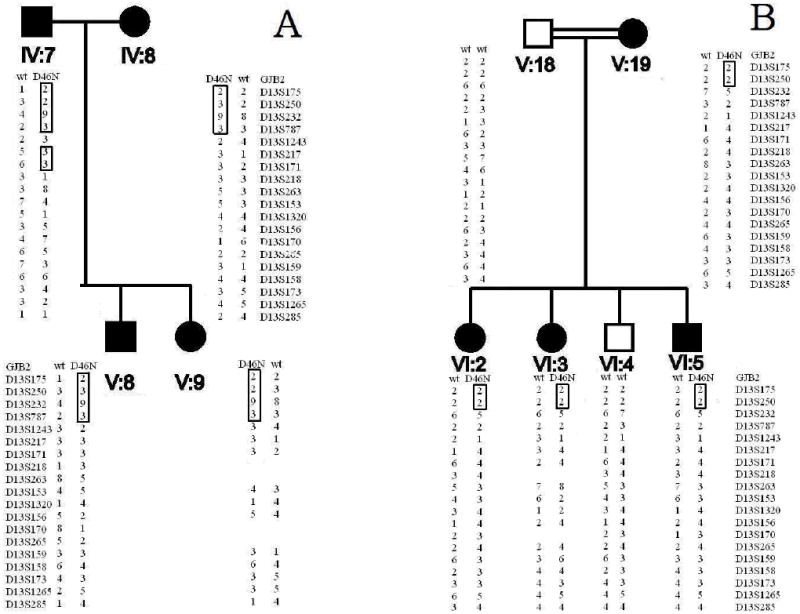
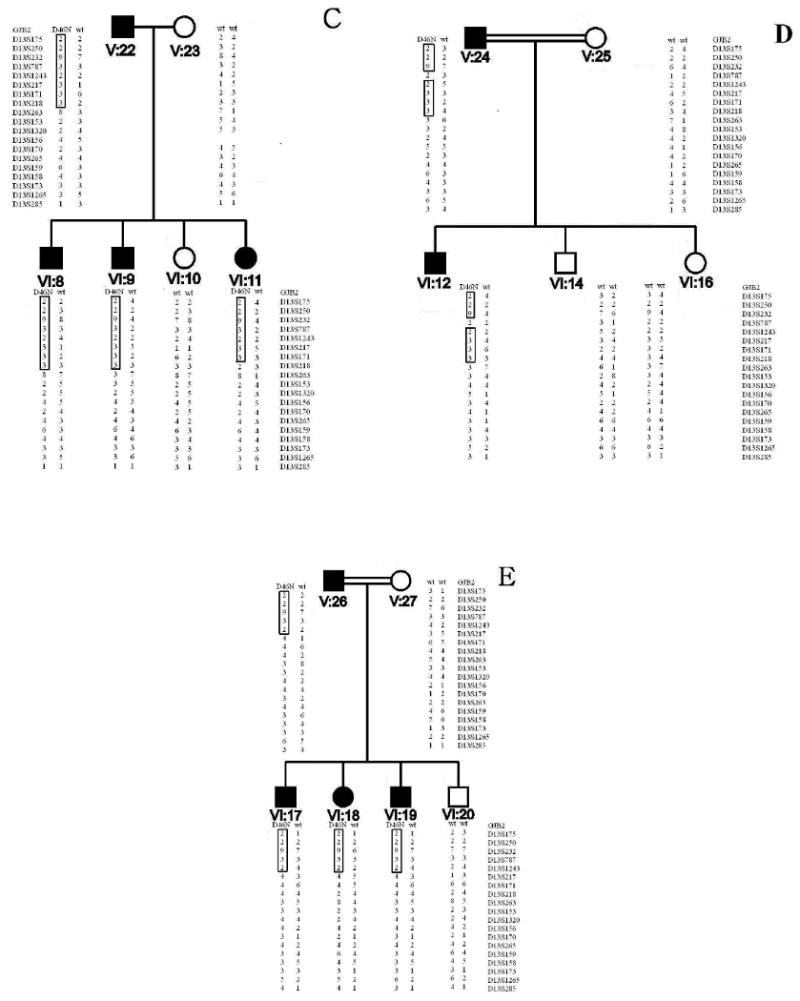
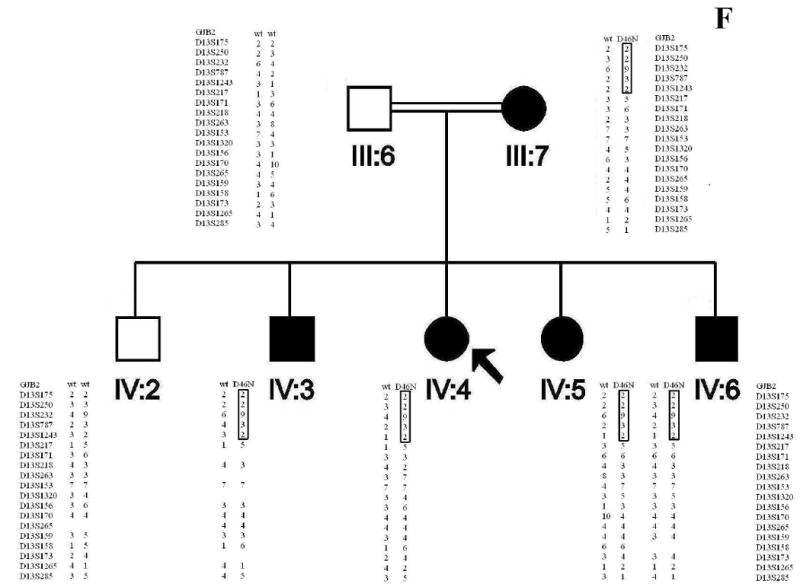
Figure 2 A. Pedigrees of two families, L-1301 (upper pedigree) and L-3057 (lower pedigree) with hearing loss caused by a novel mutation in GJB2, D46N. The proband of each family is indicated by an arrow. All individuals who did not have audiograms are indicated with an asterisk. Studied branches are shown with capital letters. Figure 2 B. All genotypes are indicated on pedigrees beneath the studied individuals.
Figure 3.

Audioprofiles showing average audiograms by age group for both families.
Figure 4.
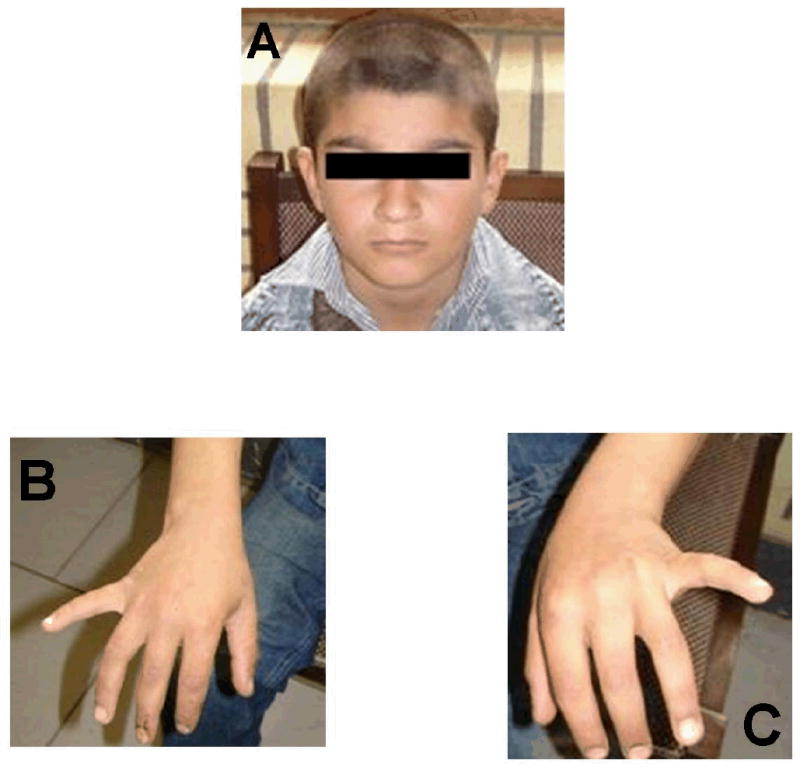
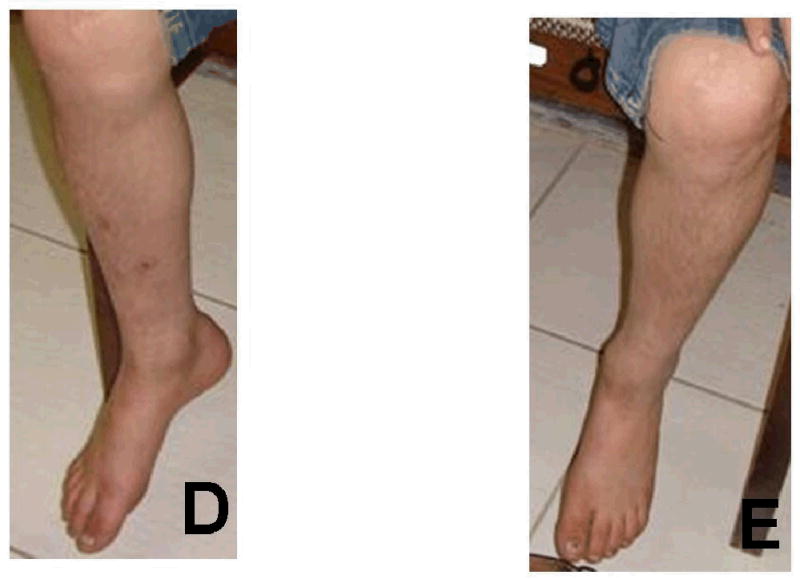
Photos of one affected individual showing absence of skin symptoms.
Haplotype Analysis and Mutation Screening
Haplotype reconstruction was consistent with linkage to the DFNA3 locus on chromosome 13q12. Mutation analysis of GJB2 identified a novel mutation, 351G>A, in affected subjects (Fig 2). This missense mutation replaces the acidic aspartate residue at position 46 with the neutral asparagine residue (D46N). The mutation was not present in non-affected family members and was similarly absent in 200 Iranian control samples. D46 is located in the first extracellular loop (E1) of Cx26, a region known to be highly conserved among connexin family members and to play an important role in voltage gating and in facilitating docking between opposing connexons [Meşe et al, 2007; White et al, 1995; Rubin et al, 1992; Martin and Evans, 2004]. The ConSeq alignment confirmed conservation of this amino acid in multiple species (ConSeq score = 9; Figure 5). Based on these data, D46N is believed to be the disease-causing mutation in both families segregating ADNSHL.
Figure 5.
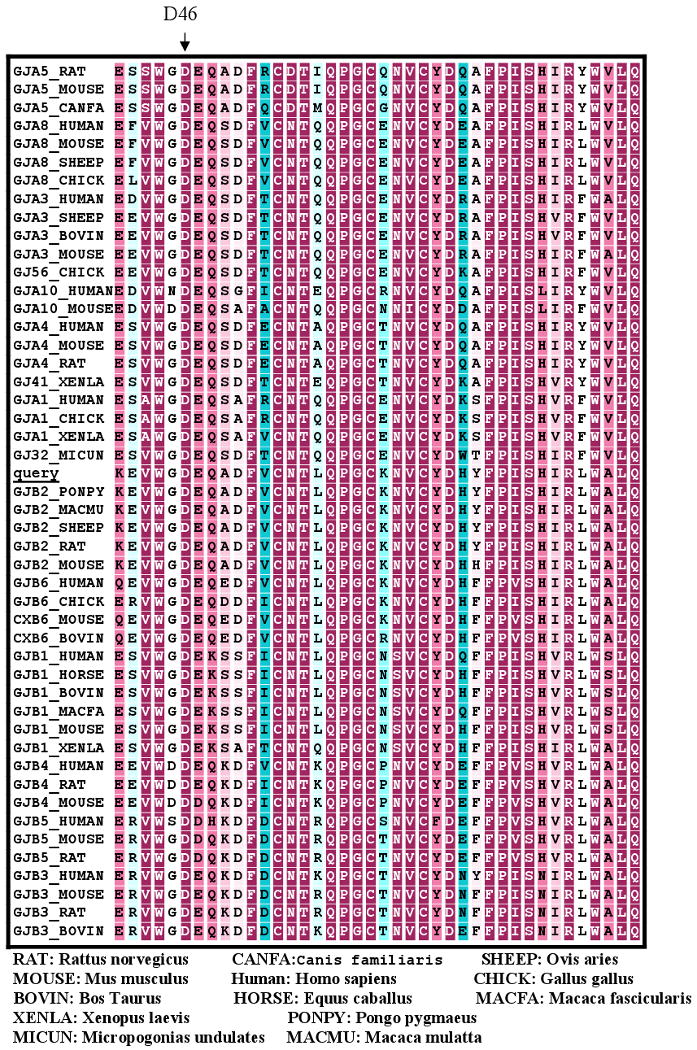
Multiple sequence alignment of the first extracellular loop (E1) of connexin 26 shows high cross-species conservation (D46 indicated by arrow).
Discussion
In this report we present two families with ADNSHL caused by a novel amino acid change – D46N – in GJB2. To date, nine mutations in GJB2 have been linked to ADNSHL whilst 21 mutations cause syndromic hearing loss associated with a variety of skin diseases. Two mutations, R75W and R75Q, cause both syndromic and nonsyndromic hearing loss.
Most of the known dominant mutations, whether syndromic and nonsyndromic, occur in the highly conserved first extracellular loop (E1) of Cx26, which is crucial for voltage gating and connexon-connexon docking (Fig 6) (Richard et al, 2002; Martin and Evans, 2004). Prominent among these mutations are G45E, D50N and D50Y, which cause Keratitis-Ichthyosis-Deafness syndrome [OMIM 148210, Janecke et al, 2005; Richard et al, 2002; Sonoda et al, 2004], and W44C and W44S, which cause ADNSHL [OMIM, 601544; Denoyelle et al, 1998; Tekin et al, 2001; Marziano et al, 2003]. The D46N mutation affects the same region of E1 (Fig 6). The recently resolved crystal structure of a connexin 26 gap junction channel has shown that this segment of E1 forms a 310 helix structure which lines the channel pore [Maeda et al, 2009].
Figure 6.
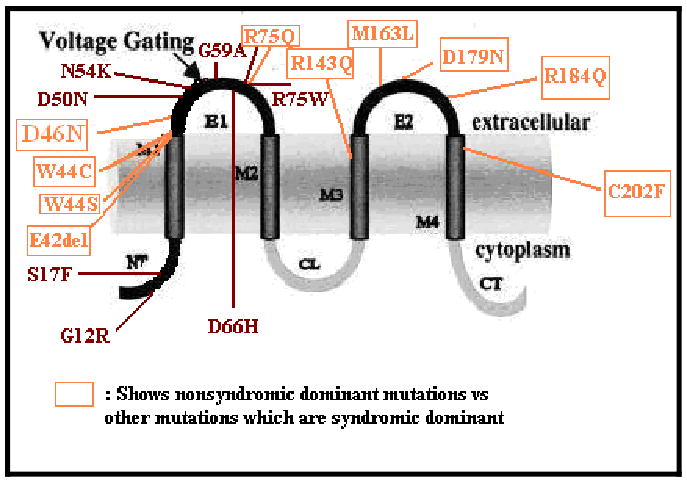
Syndromic and nonsyndromic mutations in GJB2 involved in autosomal dominant hearing loss.
Many mutations in GJB2 that cause ARNSHL cause a simple loss-of-channel activity, however it is less clear how dominant mutations affect gap junction function [Bruzzone et al, 2001]. The broad phenotypic heterogeneity seen with autosomal dominant mutations suggests variability in the molecular defects caused by individual mutations. For example, some dominant GJB2 mutations, like R75W, are known to inhibit formation of functional channels. Expression studies in Xenopus oocytes have shown that R75W, which occurs in a highly conserved region involved in voltage gating, inhibits normal gap junction function of co-expressed wild-type Cx26 in a dominant-negative manner [Richard et al, 1998]. Dye transfer studies using the gap junction permeant tracer Cascade Blue have also demonstrated a disruption of intercellular coupling with W44S, R75W, G59A and D66H. For G59A and D66H mutants, this change correlates with impaired intracellular trafficking and targeting to the plasma membrane [Marziano et al, 2003]. Functional studies also indicate that the mutation T55N produces a protein that is deeply impaired in its intracellular trafficking and fails to reach the plasma membrane [Melchionda et al, 2005].
Although functional studies of D46N are needed to determine the effect of this mutation on channel activity, useful information can be gleaned by studying the structure of wild-type Cx26 gap junction channels. Based on the structural analysis by Maeda and colleagues, D46 is one of the key residues involved in interactions between adjacent connexins. Additionally, D46 and D50 line the gap junction pore at the level of the cell membrane and face the interior of the channel, creating a negatively charged path between adjacent cells [Maeda et al, 2009]. This region is hypothesized to contribute to charge selectivity and size restriction of the gap junction. While the size of the side chain would not be significantly altered, the D46N mutation would reduce the negative charge, potentially altering the permselectivity of the channel.
The strong evolutionary conservation of D46 between species and among connexins, its location lining the pore of the Cx26 gap junction, the predicted importance of its negative charge, and its absence in 200 Iranian control subjects are convincing evidence for the disease-causing effect of this mutation. The identification of this mutation in two families from the same small village in northern Iran is consistent with a founder effect, which we confirmed by genotyping additional microsatellite markers in the DFNB1 region. On this basis, we recommend screening for this mutation in deaf persons from the Northern provinces in Iran.
Acknowledgments
We would like to thank our patients and their families for their collaboration in this research. This project was sponsored by the Iranian National Science Foundation grant numbers 85073/23 and 85033/10. R. Smith is the Sterba Hearing Research Professor, University of Iowa College of Medicine, who supported the project with National Institutes of Health (NIH)-NIDCD grants RO1 DCOO2842 and RO1 DCO3544.
References
- Akiyama M, Sakai K, Arita K, Nomura Y, Ito K, Kodama K, McMillan JR, Kobayashi K, Sawamura D, Shimizu H. A novel GJB2 mutation p.Asn54His in a patient with palmoplantar keratoderma, sensorineural hearing loss and knuckle pads. J Invest Dermatol. 2007;127:1540–1543. doi: 10.1038/sj.jid.5700711. [DOI] [PubMed] [Google Scholar]
- Alexandrino F, Sartorato EL, Marques-de-Faria AP, Steiner CE. G59S mutation in the GJB2 (connexin 26) gene in a patient with Bart-Pumphrey syndrome. Am J Med Genet Part A. 2005;136A:282–284. doi: 10.1002/ajmg.a.30822. [DOI] [PubMed] [Google Scholar]
- Arita K, Aiyama M, Aizawa T, Umetsu Y, Segawa I, Goto M, Sawamura D, Demura M, Kawano K, Shimizu H. A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26. Am J Pathol. 2006;169:416–423. doi: 10.2353/ajpath.2006.051242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CW, Levy ML, Flaitz CM, Reid BS, Manolidis S, Hebert AA, Bender MM, Heilstedt HA, Plunkett KS, Fang P, Roa BB, Chung P, Tang HY, Richard G, Alford RL. A novel GJB2 (connexin 26 mutation, F142L, in a patient with unusual mucocutaneous findings and deafness. J Invest Dermatol. 2003;121:1221–1223. doi: 10.1046/j.1523-1747.2003.12550_4.x. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Gomes D, Denoyelle E, Duval N, Perea J, Veronesi V, Weil D, Petit C, Gabellec MM, D'Andrea P, White TW. Functional analysis of a dominant mutation of human connexin26 associated with nonsyndromic deafness. Cell Commun Adhes. 2001;8:425–431. doi: 10.3109/15419060109080765. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaïb H, Lévi-Acobas F, Weil D, Petit C. Connexin 26 gene linked to a dominant deafness. Nature. 1998;393(6683):319–320. doi: 10.1038/30639. [DOI] [PubMed] [Google Scholar]
- de Zwart-Storm EA, van Geel M, van Neer PA, Steijlen PM, Martin PE, van Steensel MA. A novel missense mutation in the second extracellular domain of GJB2, p.Ser183Phe, causes a syndrome of focal palmoplantar keratoderma with deafness. Am J Pathol. 2008;173:1113–1119. doi: 10.2353/ajpath.2008.080049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zwart-Storm EA, Hamm H, Stoevesandt J, Steijlen PM, Martin PE, van Geel M, van Steensel MA. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet. 2008:45161–166. doi: 10.1136/jmg.2007.052332. [DOI] [PubMed] [Google Scholar]
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma I. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998;351:394–398. doi: 10.1016/S0140-6736(97)11124-2. [DOI] [PubMed] [Google Scholar]
- Green GE, Scott DA, McDonald JM, Woodworth GG, Sheffield VC, Smith RJH. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211–2216. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- Hamelmann C, Amedofu GK, Albrecht K, Muntau B, Gelhaus A, Brobby GW, Horstmann RD. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum Mutat. 2001;18:84–85. doi: 10.1002/humu.1156. [DOI] [PubMed] [Google Scholar]
- Heathcote K, Syrris P, Carter ND, Patton MA. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis. J Med Genet. 2000;37:50–51. doi: 10.1136/jmg.37.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://conseq.tau.ac.il/
- http://davinci.crg.es/deafness/index.php?seccion=mut_db&db=nonsynd&nonsynd=cx26mut
- Janecke AR, Nekahm D, Löffler J, Hirst-Stadlmann A, Müller T, Utermann G. De novo mutation of the connexin 26 gene associated with dominant non-syndromic sensorineural hearing loss. Hum Genet. 2001;108:269–270. doi: 10.1007/s004390100484. [DOI] [PubMed] [Google Scholar]
- Janecke AR, Hennies HC, Günther B, Gansl G, Smolle J, Messmer EM, Utermann G, Rittinger O. GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am J Med Genet Part A. 2005;133A:128–131. doi: 10.1002/ajmg.a.30515. [DOI] [PubMed] [Google Scholar]
- Jonard L, Feldmann D, Parsy C, Freitag S, Sinico M, Koval C, Grati M, Couderc R, Denoyelle F, Bodemer C, Marlin S, Hadj-Rabia S. A famililial case of Keratitis-Ichthyosis-Deafness (KID) syndrome with the GJB2 mutation G45. E Eur J Med Genet. 2008;51:35–43. doi: 10.1016/j.ejmg.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998;62:792–799. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastr uctural analysis. Anal Embryol. 1995;191:101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- Leonard NJ, Krol AL, Bleoo S, Somerville MJ. Sensorineural hearing loss, striate palmoplantar hyperkeratosis, and knuckle pads in a patient with a novel connexin 26 (GJB2) mutation. J Med Genet. 2005;42(1):e2. doi: 10.1136/jmg.2003.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler J, Nekahm D, Hirst-Stadlmann A, Günther B, Menzel HJ, Utermann G, Janecke AR. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur J Hum Genet. 2001;9:226–230. doi: 10.1038/sj.ejhg.5200607. [DOI] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature. 2009;458(7238):597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Maestrini E, Korge BP, Ocaña-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, Munro CS. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- Mani RS, Ganapathy A, Jalvi R, Srikumari Srisailapathy CR, Malhotra V, Chadha S, Agarwal A, Ramesh A, Rangasayee RR, Anand A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Hum Genet. 2008 doi: 10.1038/ejhg.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–812. doi: 10.1093/hmg/ddg076. [DOI] [PubMed] [Google Scholar]
- Martin PEM, Evans HW. Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res. 2004;62:378–387. doi: 10.1016/j.cardiores.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Matos T, Caria H, Galhardo I, Dias O, Andrea M, Correia C, Fialho G. A novel M163L mutation in GJB2 gene associated with autosomal dominant isolated hearing loss. Hear Res. 2008;240:87–92. doi: 10.1016/j.heares.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Melchionda s, Bicego M, Marciano E, Franze A, Morgutti M, Bortone G, Zelante L, Carella M, D' Andrea P. Functional characterization of novel CX26 (T55N) mutation associated to non-syndromic hearing loss. Biochem Biophys Res Commun. 2005;337:799–805. doi: 10.1016/j.bbrc.2005.09.116. [DOI] [PubMed] [Google Scholar]
- Meşe G, Richard G, White TW. Gap junctions: basic structure and function. J Invest Dermatol. 2007;127:2516–2524. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JR, White TW, Martin BL, Turner ML, Holland SM. A novel connexin 26 mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Dermatol. 2004;51:377–382. doi: 10.1016/j.jaad.2003.12.042. [DOI] [PubMed] [Google Scholar]
- Morlé L, Bozon M, Alloisio N, Latour P, Vandenberghe A, Plauchu H, Collet L, Edery P, Godet J, Lina-Granade G. A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss. J Med Genet. 2000;37:368–370. doi: 10.1136/jmg.37.5.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMIM 124500; Vohwinkel syndrome
- OMIM 148210; KID syndrome
- OMIM 148350; PPK
- OMIM 149200; Bart-Pumphrey syndrome
- OMIM, 601544; DFNA3
- Richard G, Brown N, Smith LE, Terrinoni A, Melino G, Mackie RM, Bale SJ, Uitto J. The spectrum of mutations in erythrokeratodermias--novel and de novo mutations in GJB3. Hum Genet. 2000;106:321–329. doi: 10.1007/s004390051045. [DOI] [PubMed] [Google Scholar]
- Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynänen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, Munro CS, Uitto J, Hodgins MB, Richard G. Trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci. 2001;114(Pt 11):2105–2113. doi: 10.1242/jcs.114.11.2105. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Verselis VK, Bennett MV, Bargiello TA. Molecular analysis of voltage dependence of heterotypic gap junctions formed by connexins 26 and 32. Biophys J. 1992;62:183–193. doi: 10.1016/S0006-3495(92)81804-0. discussion 193-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Bale JF, Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- Snoeckx RL, Hassan DM, Kamal NM, Van Den Bogaert K, Van Camp G. Mutation analysis of the GJB2 (connexin 26) gene in Egypt. Hum Mutat. 2005;26:60–61. doi: 10.1002/humu.9350. [DOI] [PubMed] [Google Scholar]
- Sonoda S, Uchino E, Sonoda KH, Yotsumoto S, Uchio E, Isashiki Y, Sakamoto T. Two patients with severe corneal disease in KID syndrome. Am J Ophthalmol. 2004;137:181–183. doi: 10.1016/s0002-9394(03)00739-6. [DOI] [PubMed] [Google Scholar]
- Tekin M, Arnos KS, Xia XJ, Oelrich MK, Liu XZ, Nance WE, Pandya A. W44C mutation in the connexin 26 gene associated with dominant non-syndromic deafness. Clin Genet. 2001;59:269–273. doi: 10.1034/j.1399-0004.2001.590409.x. [DOI] [PubMed] [Google Scholar]
- Uyguner O, Tukel T, Baykal C, Eris H, Emiroglu M, Hafiz G, Ghanbari A, Baserer N, Yuksel-Apak M, Wollnik B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin Genet. 2002;62:306–309. doi: 10.1034/j.1399-0004.2002.620409.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Derm. 2002;118:724–727. doi: 10.1046/j.1523-1747.2002.01735.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Steijlen PM, Bladergroen RS, Hoefsloot EH, van Ravenswaaij-Arts CM, van Geel M. A phenotype resembling the Clouston syndrome with deafness is associated with a novel missense GJB2 mutation. J Invest Dermatol. 2004;123:291–293. doi: 10.1111/j.0022-202X.2004.23204.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004;131C:12–19. doi: 10.1002/ajmg.c.30030. [DOI] [PubMed] [Google Scholar]
- White TW, Paul DL, Goodenough DA, Bruzzone R. Functional analysis of selective interactions among rodent connexins. Mol Biol Cell. 1995;6:459–470. doi: 10.1091/mbc.6.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsumoto S, Hashiguchi T, Chen X, Ohtake N, Tomitaka A, Akamatsu H, Matsunaga K, Shiraishi S, Miura H, Adachi J, Kanzaki T. Novel mutations in GJB2 encoding connexin-26 in Japanese patients with keratitis-ichthyosis-deafness syndrome. Br J Dermatol. 2003;148:649–653. doi: 10.1046/j.1365-2133.2003.05245.x. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Huang D, Yu F, Zhu X, Kang D, Yuan H, Han D, Dai P. A de novo GJB2 (connexin 26) mutation, R75W, in a Chinese pedigree with hearing loss and palmoplantar keratoderma. Am J Med Genet A. 2008 doi: 10.1002/ajmg.a.32461. Epub ahead of print. [DOI] [PubMed] [Google Scholar]