

Abstract
Genomic instability is a feature of the human Xp22.31 region wherein deletions are associated with X-linked ichthyosis, mental retardation and attention deficit hyperactivity disorder. A putative homologous recombination hotspot motif is enriched in low copy repeats that mediate recurrent deletion at this locus. To date, few efforts have focused on copy number gain at Xp22.31. However, clinical testing revealed a high incidence of duplication of Xp22.31 in subjects ascertained and referred with neurobehavioral phenotypes. We systematically studied 61 unrelated subjects with rearrangements revealing gain in copy number, using multiple molecular assays. We detected not only the anticipated recurrent and simple nonrecurrent duplications, but also unexpectedly identified recurrent triplications and other complex rearrangements. Breakpoint analyses enabled us to surmise the mechanisms for many of these rearrangements. The clinical significance of the recurrent duplications and triplications were assessed using different approaches. We cannot find any evidence to support pathogenicity of the Xp22.31 duplication. However, our data suggest that the Xp22.31 duplication may serve as a risk factor for abnormal phenotypes. Our findings highlight the need for more robust Xp22.31 triplication detection in that such further gain may be more penetrant than the duplications. Our findings reveal the distribution of different mechanisms for genomic duplication rearrangements at a given locus, and provide insights into aspects of strand exchange events between paralogous sequences in the human genome.
INTRODUCTION
The distal portion of the short arm of the human X chromosome (Xp22.3) is a region that undergoes frequent genomic rearrangements. In the pseudoautosomal region PAR1, at the tip of the X chromosome, an obligatory recombination occurs in every male meiosis to maintain the homology between chromosomes X and Y PAR1 regions. Proximal to the PAR1 boundary of the X chromosome, a series of historical duplication and inversion events occurred. Several gene families, such as the sulfatase gene family, VCX/Y gene family and the CD99 gene family, may have arisen from evolutionary genomic segmental duplications. These rearrangements occurring both within this region and between the homologous regions on the X–Y chromosomes, shaped the intricate genomic structure therein during primate genome evolution (1–3). Consistent with genomic instability, rearrangements causing disease in Xp22.3 have been frequently observed. Deletions in males (females in a few cases) are associated with contiguous gene syndromes (4). Unbalanced translocations between X and Y homologous regions were also reported in different patients (5,6).
Xp22.31 is one of the most extensively studied genomic intervals on the short arm of the X chromosome; deletion of the steroid sulfatase gene [STS (MIM 300747)] accounts for 90% of X-linked ichthyosis [XLI (MIM 30870)] (4,7,8). Complex traits, including X-linked nonspecific mental retardation [MRX (MIM 309530)] and attention deficit hyperactivity disorder [ADHD (MIM 143465)], have also been observed in addition to XLI (9–11). Interspersed around these deletions are S232 low copy repeats (LCRs). In the reference human haploid genome, there are six paralogous copies of S232 LCRs, four at Xp22.31 and two at Yq11.22 (Table 1). Each of them contains two variable number of tandem repeat (VNTR) elements, termed repeating unit 1 and 2 (RU1 and RU2) (12). The RU2 element consists of a variable-sized monomeric unit of ∼26–37 bp, with an embedded polymorphic tetranucleotide repeat. It consists of purine-rich highly asymmetric sequences without cytosines on one strand (12). Yen et al. (13) showed that unequal recombination involving two of the S232 elements flanking the STS region frequently produces the 1.6 Mb recurrent deletion. Fine mapping of recombination sites in four patients carrying the common deletion narrowed the breakpoint region into the RU2 element, implicating nonallelic homologous recombination (NAHR) (14) as the mechanism for these deletions (10).
Table 1.
Sizes and percent identities of S232 LCRs
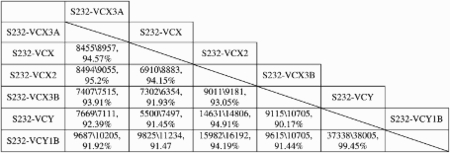 |
Each pair of S232 LCRs are aligned with each other and the numbers in the table represent: size of the LCR in the row (bp)\size of LCR in the column (bp), fraction matching. Data are obtained from the Segmental Duplications track in UCSC genome browser (http://genome.ucsc.edu/).
Recently, a 13-mer, cis-acting, homologous recombination (HR) stimulating motif (5′-CCNCCNTNNCCNC-3′) has been identified from population-based studies of historical recombinants and shown to be associated with 40% of the allelic homologous recombination (AHR) hotspots identified from the HapMap Phase II data (15). In addition, the same motif was found to bind a protein, PR-domain containing 9 (PRDM9), thought to be involved in hotspot specification. This protein possesses histone H3K4 trimethylase activity and contains multiple zinc finger motifs (16–18). Empirical studies suggest that HR crossovers, or Holliday structure resolution, occur within a 400 bp range either upstream or downstream from this motif (19). Interestingly, almost every repeat unit within the RU2 element contains one copy of this motif. Based on the sequence of the haploid reference genome, the RU2 elements in the six paralogous S232 LCRs contain 12–28 copies of this motif. If calculated on a genome-wide megabase scale, the concentration of the 13-mer motifs is highest in the Xp22.31 region (15). However, the exact position at which crossover occurs with respect to the associated hotspot motif remains to be elucidated.
To date, efforts to understand rearrangements in the Xp22.31 region have focused on deletions and translocations, but rarely duplications. This may partly be due to the fact that the duplication is present in some ‘phenotypically normal’ individuals; therefore, it has been considered as a benign copy number variant (CNV). Recently, Li et al. (20) summarized clinical phenotypes in 23 patients with Xp22.31 duplications together with 12 other patients with similar duplications reported in the literature. However, the clinical significance of Xp22.31 duplications is still debated and further investigation is necessary.
We have identified frequent duplication events at Xp22.31 (0.46%) in clinical samples referred for chromosome microarray analysis (CMA) in the Medical Genetics Laboratories (MGL) at Baylor College of Medicine (BCM). We systematically investigated 61 unrelated subjects with CMA-detected copy gains involving the Xp22.31 region to determine the size, extent and genomic content of these duplications. We confirm NAHR as a major mechanism in the presence of large highly homologous directly oriented LCRs and extend our mechanistic observations to the strand exchange level. Among these NAHR-mediated events, breakpoint-sequencing data reveal enrichment of breakpoints in proximity to the HR hotspot motif, and suggest that multiple hotspot motifs in tandem may have an additive effect on stimulating HR. Surprisingly, both recurrent triplications and complex rearrangements were observed in different subjects. To investigate the potential clinical significance of the Xp22.31 duplication and triplication, we studied the phenotypes and prevalence of these two types of rearrangements. In addition, we compared the phenotypes of duplications and triplications to investigate whether dosage increments correlate with either penetrance or severity of the phenotype.
RESULTS
Fine mapping of Xp22.31 duplications
Samples from 69 subjects (61 unrelated) who were ascertained in the MGL clinical diagnostic laboratory as having Xp22.31 gains were anonymized and analyzed by array comparative genomic hybridization (aCGH) using region-specific custom arrays (Fig. 1 and Supplementary Material, Fig. S1). Forty-four (72%) unrelated cases were found to have a 1.6 Mb common recurrent duplication flanked by two S232 LCRs, which is the apparent reciprocal rearrangement to the previously reported recurrent deletion (13). Nine cases carry apparently simple nonrecurrent duplications ranging in size from ∼350 kb to ∼1.9 Mb. Three (BAB2861, BAB3084 and BAB3089) of them have one breakpoint located within the S232 LCR. Surprisingly, we found that the region is apparently triplicated in three unrelated cases as evidenced by the log2 ratio of the aCGH result (∼1.58 for male; 1 for female). These triplications have a similar size and extent and thus genomic content as the common recurrent duplications. The recurrent duplications occur ∼14-fold more frequently than the recurrent triplications (Fig. 1B). In addition, apparently complex rearrangements were also detected by aCGH in five cases. In BAB 2833, a 45 kb segment is triplicated at the distal portion of the recurrent duplication and a 92 kb segment is duplicated proximal to the recurrent duplication. In the other four cases, complex rearrangements with a duplication–normal–duplication pattern were observed. Of note, in these cases with apparent complex rearrangements, almost half (10 of 21) of all breakpoints are located within one of the S232 LCRs.
Figure 1.

Summary of aCGH results. (A) A schematic representation (top) of Xp22.31 genomic region based on the reference genome (hg18). Individual genes are shown as black rectangles. The vertical yellow shadow areas represent S232 LCRs, with the relative orientations of each of the LCR indicated. Below, horizontal bars depict the involved genomic intervals for each subject from the interpretations of aCGH results. Red bars represent duplications and blue bars represent triplications; white normal copy. Colors of subject numbers indicate genders, with black depicting male and red female individuals; 44 subjects (27 males, 17 females) carry recurrent duplications as shown in the top row. (B) Distribution of different rearrangement types. Note that BAB2861 is classified as carrying a complex rearrangement here because breakpoint analysis described later revealed complex rearrangements in this subject.
Recurrent triplication can occur at Xp22.31
Our aCGH data suggested that three cases might carry recurrent triplications in Xp22.31. The triplications were inherited from mothers in two male patients, BAB2817 and BAB2822, but de novo in a female patient, BAB2828. Three independent experimental molecular approaches, fluorescent in situ hybridization (FISH), multiplex ligation-dependent amplification (MLPA) and quantitative PCR (qPCR), were used to verify the copy number gains. In each case, all three additional approaches provided CNV information, showing triplication consistent with the findings initially revealed by aCGH (Fig. 2, Supplementary Material, Figs S2 and S3).
Figure 2.

Ascertainment of triplications in patients and carrier parents. Shown above is the genome structure (horizontal black line) and below the genomic location of the probes for qPCR (green), MLPA (blue) and FISH (black), with the positions shown as vertical tics. Copy number calls of the three different approaches are compared in representative male (left) and female (right) subjects. The x-axis represents the relative genomic locations of each probe. Results in subject BAB2817 and mother BAB2818 are shown in this figure. Additional subjects/parents carrying triplication can be found in Supplementary Material, Figures S2 and S3.
Breakpoints located within the LCRs
Previously, Van Esch et al. (10) showed that the breakpoint of Xp22.31 deletion is located within the RU2 element in the S232 LCRs in four male subjects. In our cohort, aCGH results suggest that S232 LCRs are involved in breakpoints of the anticipated reciprocal recurrent duplications and triplications, as well as other rearrangement types including complex rearrangements and nonrecurrent simple duplications. We hypothesized that the RU2 element, where the AHR hotspot motif is enriched (15), also acts as a ‘hotspot’ for NAHR-mediated duplications and rearrangements occurring by other mechanisms.
To test our hypothesis, we designed PCR assays to amplify the breakpoint junctions for fine mapping of the crossover region. Because of the high copy number (8/10 copies in a male/female duplication versus four copies in an XLI-affected male with deletion) and the unusual structure of the S232 LCR, breakpoint junctions have been challenging to map and sequence. We sequenced the breakpoints of 41 subjects, including 34 subjects with recurrent duplications (BAB 2814, BAB2815, BAB2827, BAB2829, BAB2830, BAB2835, BAB2831, BAB2837, BAB2840, BAB2841, BAB2842, BAB2843, BAB2844, BAB2845, BAB2848, BAB2849, BAB2850, BAB2851, BAB2853, BAB2854, BAB2856, BAB2858, BAB2859, BAB2862, BAB2863, BAB2864, BAB2938, BAB3078, BAB3081, BAB3082, BAB3083, BAB3085, BAB3086 and BAB3092), all three subjects with recurrent triplications (BAB2817, BAB2822 and BAB2828), one subject with a complex rearrangement (BAB3088) and three subjects with nonrecurrent duplications (BAB2861, BAB3084 and BAB3089). Consistent with our hypothesis, all of these cases have either one or two breakpoints located within the RU2 element (Fig. 3). There is one exception in the BAB2822 triplication subject. In this subject, one crossover resides within the RU2 element, whereas the other occurs within a 98 bp interval (chrX:8 095 471–8 095 568 or chrX:6 459 040–6 459 137) that is ∼1.7 kb distal to the RU2 elements, but still within an S232 paralogue (Fig. 3). Of note, PCR amplification of all recombinant RU2 elements resulted in fragments of 600–700 bp based on agarose gel electrophoresis migration and comparison with size standards, suggesting one to four copies of the repeat unit, which is significantly shorter than the length of repeat unit arrays in RU2 elements as listed in the reference sequence (12–28 copies). Sanger sequencing could not resolve the RU2 element copy number as the sequence read terminated upon entry into the repeats, potentially due to its unusual structure and high GC content. When we used a bacterial artificial chromosome (BAC) clone, RP11–527B14 that contains the S232–VCX2 repeat, as the PCR template, the size estimate of the amplified product suggested ∼4 copies of the repeat unit in its RU2 element, whereas the sequence of this BAC (GeneBank accession no. AC097626) suggested 25 copies.
Figure 3.

Breakpoints within S232 LCR. (A) Schematic representation of the structure of S232 LCR and location of breakpoints. One member of the S232 LCR family, S232-VCX2, is chosen arbitrarily to show the content of the S232 LCR. The orange bar depicts the RU2 element, which has variable length among different individuals. The black bar depicts the VCX2 gene. The red arrow on the left indicates a 98 bp crossover region (chrX:8 095 471–8 095 568) of one recombination event in BAB2822. It is located ∼1.7 kb distal to the RU2 element. The three dashed red vertical arrows pointing to RU2 indicate the approximate region of crossover in 41 rearrangements. The exact crossover region in the hypervariable RU2 element could not be further refined. (B) The repeat unit in RU2. The first line shows the representative sequence of a repeat unit from an RU2 element summarized from the sequence from the reference genome. Each unit contains two to five copies of TCCC. The underlined sequence is variable among different repeat units and the sequence shown here (CCTCTTCC) is the most commonly seen one. The RU2 element is composed of a tandem array of such repeat units. The second line shows the same repeat unit sequence but with the homologous recombination hotspot motif highlighted in red. The third and fourth lines show the same repeat unit sequence but with the homopurine–homopyrimidine mirror repeats highlighted in green. These green sequences have the potential to adopt the H-DNA conformation. (C) The 2 bp breakpoint interval for the rearrangement in BAB3084 maps within the hotspot motif. The 4 bp breakpoint interval for BAB2861 and BAB3089 maps adjacent to the hotspot motif.
To gain more insight into the role of the hotspot motifs in potentially stimulating the rearrangements, it is important to know the precise region of crossover or strand exchange within the hotspot motifs in RU2. Unique specification of breakpoints has been a challenge for the recurrent duplication/deletion cases, wherein both sides of the breakpoint map within the RU2 elements, whose sequence exhibits considerable interindividual variation. However, our aCGH analysis detected a special configuration of rearrangement that solves this problem. In this configuration, only one side of the breakpoint maps to the hypervariable RU2 element, whereas the other side of the breakpoint maps to a nonvariable sequence. The exact intervals of crossovers were defined in three such cases (Fig. 3C). In BAB3084, the crossover occurred in a 2bp region within the hotspot motif. In BAB2861 and BAB3089, the breakpoint mapped to the same 4bp interval adjacent to the hotspot motif. Microhomologies were observed at the breakpoints in all three cases.
Unexpectedly, bioinformatic analysis of sequences from subject BAB2828 indicates that the breakpoints do not map to the directly oriented LCR pairs that usually mediate recurrent rearrangements. Instead, they map to inverted LCRs S232-VCX and S232-VCX2, potentially suggesting that this subject carries an inversion haplotype (Fig. 4), which will be further elaborated upon in the Discussion section.
Figure 4.

Alternative interpretations of the inversions in subjects BAB2833, BAB3094 and BAB2828. (A) aCGH interpretation based on the reference haplotype. The small blue bars below array interpretation of BAB2828 indicate the mapping position of the sequence from one breakpoint junction in this subject. (B) aCGH interpretation based on a hypothesized inversion haplotype. Inversion haplotype 1 is predicted to be mediated by the LCRs S232-VCX3A and S232-VCX3B. Inversion haplotype 2 could be mediated by the LCRs S232-VCX3A and S232-VCX. Compared with (A), interpretations in(B) simplify the mechanistic processes needed to produce the rearrangements. Note that BAB3094 may not represent a complex rearrangement (Fig. 1) given this interpretation. Similar interpretive challenges posed by a haploid reference human genome that does not incorporate structural variation information have been reported recently (33,41).
Breakpoints that do not involve S232 LCRs
To investigate the underlying molecular mechanisms, we sequenced the breakpoints of six subjects with simple nonrecurrent duplications. The exact coordinates of the tandem duplications and microhomologies found at the breakpoints are summarized in Table 2. Two- or three-base pair microhomologies were detected in five of six subjects, implicating either a fork stalling and template switching (FoSTeS)/microhomology-mediated break induced replication (MMBIR) (21,22) with a single template switch or a non-homologous end joining (NHEJ) mechanism. Long or short interspersed nuclear elements (LINEs or SINEs) are found at the breakpoints of three nonrecurrent duplications. In subjects BAB2824, BAB3090 and BAB3093, the proximal breakpoint is located within an AluJ element in the first patient, and within an L1-LINE element in the other patients.
Table 2.
Summary of nonrecurrent duplications
| Subject no. | Coordinate of tandem duplication | Microhomology at breakpoint | Possible mechanism |
|---|---|---|---|
| BAB2824 | chrX:6 558 299–7 371 465 | AA | FoSTeS/MMBIR×1 or NHEJ |
| BAB2852 | chrX:6 764 860–7 131 598 | AC | FoSTeS/MMBIR×1 or NHEJ |
| BAB3087 | chrX:7 480 688–8 077 529 | None | NHEJ |
| BAB3090 | chrX:7 049 533–7 400 035 | CAC | FoSTeS/MMBIR×1 or NHEJ |
| BAB3093 | chrX:6 558 299–7 371 465 | AT | FoSTeS/MMBIR×1 or NHEJ |
| BAB3096 | chrX:5 865 411–6 592 588 | GAA | FoSTeS/MMBIR×1 or NHEJ |
In the subjects in which aCGH results suggested complex rearrangements, the sequences of all breakpoints located outside of LCRs were characterized. Results are summarized in Figure 5. Microhomologies of 1–4bp were observed at eight of nine breakpoints, supporting a replication-based rearrangement mechanism for formation. Subject BAB3088 lacks microhomology at the breakpoints, favoring NHEJ as the underlying mechanism. In subjects BAB2833 and BAB3095, we detected short tandem duplications at one breakpoint. These locally duplicated short sequence segments are consistent with fingerprints of serial replication slippage (SRS) (23). Within two subjects, BAB2833 and BAB3094, the proximal and the distal sequences of one breakpoint map to opposite strands. These findings are consistent with the subject's rearrangements having occurred on a chromosomal inversion haplotype (Fig. 4).
Figure 5.

Breakpoint sequences of complex rearrangements. (A) The mapping of distal and proximal breakpoint sequences. For each subject, the structure at the breakpoint suggested by the breakpoint sequence is shown above the copy number interpretation from aCGH results. The ends of each breakpoint are mapped to the reference genome, represented by green, pink or orange bars connected by dashed lines. (B) Sequence of breakpoints aligned to distal and proximal reference sequences. The color code is in accordance with colors in (A). The strand of reference sequences are indicated by ‘+’ or ‘−’. The red boxes outline microhomologies identified at the breakpoint junctions. The underlined sequences are the segments proposed to be involved with rearrangements by the SRS mechanism.
Breakpoint analyses also uncovered complexity that was not initially revealed by aCGH. The breakpoint of subject BAB2861 contains a 40 bp microduplication that was not detected by aCGH (Fig. 5). Complex rearrangements at this locus were probably mediated by FoSTeS/MMBIR with multiple template switches, reflecting a low processivity DNA polymerase, utilizing the microhomologies at both ends of this 40 bp segment.
Clinical consequences of the recurrent Xp22.31 duplication and triplication
The Xp22.31 gains are among the most frequent findings in the clinical cytogenetic laboratories (20,24). Nevertheless, it has been debated whether CNV gain of this genomic interval is benign or disease causing. In order to address this conundrum, we focused on the two types of recurrent gains, the recurrent duplication and triplication, and studied their clinical features, patterns of inheritance, X-chromosome inactivation (XCI) status and population frequencies.
Following informed consent, we obtained detailed clinical information for 14 subjects with recurrent duplications and all three subjects with recurrent triplications (ages from 14 months to 10 years) (Tables 3 and Supplementary Material, Table S1). Patients with Xp22.31 recurrent duplications generally presented with a neurocognitive and behavioral phenotype, including developmental delay, which was the primary reason for referral to neurology or genetics. Seven of 11 males (64%) and 2 of 3 females (67%) presented with moderate to severe delay involving motor and/or language areas. Our findings are consistent with recent observations of developmental problems in 69% of the patients with Xp22.31 duplications (20). Additionally, 7 of the 11 males (64%) with duplications had social interaction deficits or behavioral abnormalities, including stereotypic features such as hand flapping and avoidance of eye contact, that are consistent with features seen in autistic spectrum disorder.
Table 3.
Summary of clinical phenotypes for the subjects with the recurrent Xp22.31 duplication or triplication and the comparison with the summarized data from the literature
| Recurrent duplications (n = 12) |
Recurrent Triplications (n = 3) |
All duplications from the literature (n = 35)20 | |||
|---|---|---|---|---|---|
| Male (n = 11) | Female (n = 3) | Male (n = 2) | Female (n = 1) | ||
| Gastro-esophageal reflux | 3 (27%) | – | 1 (50%) | – | 3 (9%) |
| Delay | |||||
| Gross motor | 6 (55%) | 2 (67%) | 2 (100%) | 1 | 24 (69%) |
| Speech | 7 (64%) | 2 (67%) | 2 (100%) | 1 | |
| Developmental regression | 1 (9%) | 1 (33%) | – | – | – |
| Seizures | 1 (9%) | 1 (33%) | – | – | 4 (11%) |
| Hypotonia | 3 (27%) | 1 (33%) | 1 (50%) | 1 | 7 (20%) |
| Behavior problems | |||||
| Autism spectrum | 7 (64%) | – | – | – | 13 (37%) |
| ADHD | – | – | 2 (100%) | – | |
| Macrocephaly >95% | 4 (36%) | – | – | 1 | – |
| Microcephaly <5% | – | 1 (33%) | – | – | 4 (11%) |
| Short stature | 1 (9%) | 1 (33%) | 1 (50%) | – | 2 (6%) |
| MRI/CT brain abnormalities | 2 (18%) | 1 (33%) | – | – | – |
| EEG abnormalities | 2 (18%) | 1 (33%) | – | – | – |
‘–’ indicates the feature is not present in the corresponding category or this information is not available. In the subjects with duplications, there are two siblings and one set of twins. Note that the data from the last column are phenotypes collected from different types of Xp22.31 gains (not restricted to recurrent duplications).
For the Xp22.31 triplications, all three subjects (100%) presented with developmental delay and both males (100%) presented with aggressive behavior with features of ADHD. Despite the small number of subjects with triplications available for analysis, the triplication seems to be potentially more penetrant than the duplication with respect to a possible association with abnormal phenotypes. In the family of the male subject BAB2817, the triplication carrier mother had short stature and learning difficulties. The mother had two girls (carrier status unknown due to unavailability of blood samples) with another partner. Both girls had short stature and learning difficulties; one girl had developmental delay. The carrier mother of the other subject BAB2822 had microcephaly.
Among all the anonymized subjects with recurrent duplications, 17 subjects had parental studies performed, and the duplication was inherited in all cases. Eleven male subjects apparently inherited the duplication maternally; in six female subjects, the duplications were paternal in two and maternal in four. We performed XCI studies in all the affected females and healthy mothers to test whether skewed XCI is associated with manifestation of abnormal phenotypes. However, the majority of the subjects showed random or noninformative patterns of XCI in their blood DNA, implicating no direct association of XCI with disease manifestation (Supplementary Material, Table S2).
Given the presence of both affected and healthy carriers, we sought to compare the frequencies of this rearrangement in the clinically ascertained population and that in the general population. In our MGL sample cohort, the prevalence of the recurrent duplication is 0.289% (58 of 20 095). When the data are parsed by gender, the male or female prevalence is 0.226 or 0.382%, respectively. Our control cohort consists of 5088 individuals from five different dbGaP cohorts. We identified a total of 21 control individuals with the recurrent duplication, for a prevalence of 0.41% (male or female prevalence equals 0.182 or 0.523%, respectively). Therefore, the overall prevalence of the common recurrent duplication is not significantly different between cases and controls (Pearson's χ2 test, P= 0.1573). When comparing the male or female prevalence separately, the male prevalence is higher in the affected cohort than in the healthy cohort whereas the reverse is true for the female prevalence. However, neither of these differences is significant (Fisher's exact test, P= 1 for male, P= 0.28 for female). Haplotype analysis in duplication carriers in the control cohort indicates that duplications occurred on different haplotype backgrounds, consistent with these duplications being recurrent as opposed to being inherited from a common ancestor. The recurrent triplication is not found in the control cohort (data not shown). Of note, 10 of the 58 (17.2%) subjects with the recurrent duplication and 1 of the 3 subjects with the recurrent triplication carry additional chromosomal CNV that potentially contribute to their clinical phenotypes (Supplementary Material, Table S3).
DISCUSSION
Using NAHR events to study features at the HR hotspots
Previously, investigations into the molecular signature at HR hotspots have relied on sites of AHR surmised from population genetic variation among single nucleotide polymorphisms (SNPs) including multisite variants (25) or SNP data from the HapMap project (15). The latter approach led to the identification of the 13-mer HR hotspot-associated motif. In our work, we studied recombination products that occurred by the NAHR mechanism, instead of AHR, to further examine the features at the HR hotspots and capitalize on paralogous sequence variants (PSVs), which is much more frequent than the previously used SNPs, as markers to refine crossovers. Because of our clinical testing screen, we have assembled a large collection of subjects to increase the potential diversity of rearrangement types identified. Our findings have demonstrated the power of this approach.
Recombination hotspots for all rearrangement types are enriched in S232 LCRs
Our results suggest that the recombination breakpoints for all rearrangement types cluster within the RU2 elements. There are three dynamic features of the RU2 repeat that may potentially cause genomic instability and predispose to rearrangements: (i) it includes a tandem array of a 26–37 bp repeating unit and thus represents a minisatellite (26) or VNTR (27); furthermore, each repeat monomer harbors a tetranucleotide microsatellite-like structure; (ii) embedded within the repeating unit is the recombination hotspot-associated motif (5′-CCNCCNTNNCCNC-3′); and (iii) the RU2 element contains a homopurine–homopyrimidine mirror repeat (H palindrome), which has been proposed to facilitate formation of the H-form DNA conformation (Fig. 3B) (28,29). Any single or combination of these features may account for the frequent involvement of S232 LCRs in recombinations.
The number of the HR hotspot motifs in the RU2 element may reflect their recombinogenic potential. In Xp22.31, there are two pairs of directly oriented LCRs, S232-VCX3A/S232-VCX2 and S232-VCX/S232-VCX3B. However, NAHR causing recurrent deletion/duplication occurs only between the former pairs. We propose that this phenomenon may be explained by the increased number of tandem motifs in the reference genome in the former (24 and 25, respectively) than in the latter (12 and 28, respectively) LCR pairs. Additionally, the LCRs of the former pair share greater sequence identity than the latter pair (95.2 versus 91.93%). Nevertheless, it must be recognized that the absence of any phenotype associated with duplications involving the latter LCRs may have biased our ascertainment. Furthermore, both structure (e.g. minisatellite) and conformation (potential H-form) of DNA, rather than primary DNA sequence motifs, could potentially contribute to regional genomic instability.
The vast majority (∼72%) of our cases carry recurrent duplications, indicating that NAHR is the major rearrangement mechanism at this locus, probably due to the enrichment of both directly oriented LCRs and the HR hotspot motifs within the LCRs. Approximately 90% (55 of 61) of the patients studied herein have breakpoints, ranging from one to four, located in S232 LCRs. S232 LCRs were overrepresented in rearrangements mediated by NAHR as well as other mechanisms, indicating that the HR hotspot motif may potentially act as a cis-acting element for facilitating rearrangements occurring by diverse recombinational mechanisms. This motif might (i) stimulate DNA lesions in the nearby region, perhaps by PRDM9-facilitated entry of factors inducing a DNA break or (ii) facilitate template switching or strand invasions given the reiterative microhomology found in the breakpoint sequences.
Our data from PCR analyses and size fractionation by gel electrophoresis suggest ∼1–4 copies of the repeat unit in RU2 after recombination. One interpretation for this observation is that the RU2 element is shortened by the recombination, perhaps by replication slippage during recombination, which may reduce their recombinogenic potential, therefore maintaining the relative genome stability in this region. This interpretation potentially suggests that the recurrent duplication may arise by some replication-based mechanism in addition to, or instead of, the widely accepted NAHR mechanism. However, it should also be noted that the apparent shortening of the RU2 element repeat copy number relative to the human reference genome may reflect that our PCR assay is more efficient in amplifying short RU2 repeats or the inability of the polymerase utilized in PCR to extend through this complex repeat. Further investigations of strand exchanges using a multitude of techniques may be required to understand the features and underlying mechanisms of rearrangements stimulated by either tandem arrays of the hotspot motif or potential unusual DNA conformations.
Recurrent triplication at Xp22.31
Here, we report for the first time that recurrent triplications can occur at Xp22.31. Due to the limitations of the dynamic range of aCGH with increasing copy number, it has been challenging to differentiate triplications from duplications, particularly on autosomal chromosomes. The identification of triplication was facilitated by using high-density aCGH. Previously, triplications have been reported in other genomic loci (30–35). In these cases, the triplications are usually embedded in complex rearrangements and their mechanism for formation is proposed to be FoSTeS/MMBIR. From the four cases with triplications in our study, FoSTeS/MMBIR could be the underlying mechanism for one case: BAB2833, particularly with inversion. With two breakpoints obtained in LCRs, the triplication in BAB2822 seems to be generated by two NAHR events. It is unclear whether these two events are concomitant with each other or not. With our knowledge about genomic disorders growing, the need to have diagnostic arrays that are robust enough to differentiate triplications from duplications is becoming more evident.
Nonrecurrent, including complex, duplications at Xp22.31 are generated by different mechanisms
The breakpoint sequences enabled us to surmise potential substrates and attempt to understand the molecular mechanisms that produced such rearrangements. Microhomology is the most prevailing feature observed at the breakpoints. It is found in 13 of 15 (87%) nonrecurrent breakpoints. Interestingly, two recent studies identified microhomology as a prevalent feature at the breakpoints of either pathogenic (30 of 38; 79%) (36) or apparently benign (219 of 315; 70%) (37) CNVs. Although NHEJ can possibly account for the mechanism for these rearrangements, more and more evidence that links formation of microhomology with replication-based mechanism has accumulated (21,32,33,38). Notably, in subjects BAB2833 and BAB3095, both carrying complex rearrangements, the breakpoint sequences show concurrent rearrangements between distantly located and closely (within the same replication fork) located segments, strongly suggesting a replicative mechanism (both FoSTeS/MMBIR and SRS). In addition, the breakpoint sequence of BAB3095 suggests replication template switching between positive and negative DNA strands, further supporting replicative rearrangement mechanism.
Two subjects with complex rearrangements, BAB3088 and BAB3091, seem to carry a combination of a recurrent rearrangement (duplication in BAB3088 and deletion in BAB3091, respectively) and a nonrecurrent rearrangement (deletion in BAB3088 and duplication in BAB3091, respectively). These proposed structures are strongly supported by the breakpoint sequences of nonrecurrent rearrangements in both subjects (Fig. 5) and of a recurrent duplication in subject BAB3088. The two rearrangements in each subject are likely to be caused by different mechanisms, with recurrent rearrangements apparently by the NAHR mechanism and nonrecurrent rearrangements by NHEJ or FoSTeS/MMBIR×1 mechanisms depending on whether microhomology is present. We cannot conclude whether the two rearrangements occurred in the same meiosis without tracing the de novo event that produced them.
Structural variation may exist at Xp22.31
NAHR between LCRs arranged in an inverted orientation can cause inversion (39). Such inversions may convey a phenotype by disrupting genes or regulatory regions, or altering chromatin structures and potentially causing position effects. We have not directly experimentally demonstrated the presence of an inversion chromosome in the parent of origin; nevertheless, the pattern of complex rearrangements seen in subjects BAB2833, BAB3094 and BAB2828 suggests that these individuals may carry an inversion polymorphism in their personal genome with respect to the haploid reference human genome sequence (Fig. 4). Such an inversion haplotype can simplify mechanistic processes that produced this rearrangement, and more parsimoniously explains the aCGH observed complexity in these three subjects. In support of our prediction, the segment between S232-VCX and S232-VCX2 in the chimpanzee genome is inverted with respect to the human genome (40). If inversion polymorphism exists in our patient cohort, this may explain why we cannot readily determine the breakpoint junctions in some of our cases.
Inversions may also occur in a nonrecurrent fashion, i.e. by rearrangements not involving LCR. These inversions may be misinterpreted as being overly complex by aCGH in comparison to the haploid reference genome (33,41). However, unlike the LCR-mediated inversions proposed above, such inversions are unpredictable based on our current knowledge of genomic structure and the aCGH technique. Therefore, the LCR-rich Xp22.31 region presents a terrific opportunity to further investigate the impact of structural variations on our haploid–reference–genome-based interpretation of aCGH data.
The Xp22.31 duplication: a benign CNV or disease-causing variant
It has been controversial whether the Xp22.31 duplication is disease causing or merely a benign CNV. Although the clinical features for subjects with Xp22.31 duplications are variable, our detailed clinical analyses showed that these patients generally presented with neurocognitive and behavioral phenotypes, which argues for the pathogenic potential of the duplication. In support of a causal relationship between CNV gain and observed clinical phenotypes, one of the genes duplicated, VCX3A, was recently found to be expressed in human brain, and modulates the stability and translation of mRNAs involved in neuronal differentiation and arborization (42). Either deficiency or SNPs of the STS gene has been associated with cognitive impairment, ADHD, autism (AUTSX2 [MIM 300495]) and disorders of social communication (11,43).
With the duplication almost always inherited, most of the time from mothers and in a few cases from fathers, the interpretation of potential phenotypic association is particularly challenging to discern (44). We proposed that incomplete penetrance may account for the absence of abnormal phenotypes in some carriers and performed a case–control study to test this hypothesis. However, we detected an unexpectedly high prevalence of the Xp22.31 recurrent duplication in the control cohort, which argues against potential pathogenicity. Nevertheless, it should be noted that the subtle clinical features and behavioral phenotypes may obfuscate the definition of ‘normal' phenotype and result in misdiagnosis in the control cohort; prevalence difference between ethnic groups may also potentially add to the difficulty for interpretation. A familial approach would greatly help understand the issue. However, since most of our subjects are anonymized, we do not have parental clinical information for them.
A genomic dosage model has been proposed to explain manifestation of some disease traits, in which a combination of two or more genetic alterations is needed to present a clinical phenotype that is otherwise not as severe or not as penetrant (45). Examples of two genetic changes acting additively or synergistically include a patient with Potocki-Lupski syndrome (PTLS [MIM 610883]) duplication as well as hereditary neuropathy with liability to pressure palsy (HNPP [MIM 162500]) deletion (46), microdeletions in Thrombocytopenia-Absent Radius syndrome (47) and duplication/deletion of the 15q24 region (48). Recently, the two-hit model was statistically tested for the 16p12.1 deletion syndrome in which a 520 kb CNV within 16p12.1 occurs in combination with another genomic CNV (49). In line with these findings, we have shown at least 17.2% (this estimation is conservative since a number of our patients were tested on a relatively lower-coverage BAC array) of the patients with recurrent Xp22.31 duplication carry additional large genomic changes. We assessed whether the additional CNVs by themselves are sufficient to cause the abnormal phenotypes by literature review. In 81.8% (9 of 11) of the cases, the secondary CNVs alone are not unambiguously pathogenic (Supplementary Material, Table S3), further supporting the second-hit model. Consistent with the idea that the Xp22.31 duplication does not cause a strong enough genomic burden to convey a disease phenotype, our data suggest that the recurrent triplications may be more penetrant than the duplications. Therefore, we suggest that the recurrent Xp22.31 duplication may predispose an individual to disease; but manifestation of the disease phenotype requires additional genetic changes, including modifiers in the genomic background, additional changes elsewhere in the genome and additional changes at the Xp22.31 locus (e.g. triplications and other complex rearrangements).
With these considerations, it remains uncertain whether the recurrent Xp22.31 duplications alone are associated with abnormal phenotypes. Further clinical study is warranted for more individuals with the Xp22.31 recurrent or simple nonrecurrent duplications, triplications and other complex rearrangements in order to reach conclusions whether these changes are pathogenic or benign CNVs.
MATERIALS AND METHODS
Subjects
The MGL has performed CMA testing with aCGH assay on 20 095 samples that were referred for clinical diagnosis from 20 February 2004 to 1 July 2009. A total of 92 (0.46%) unrelated subjects (49 females and 43 males) were found to have Xp22.31 duplications by the clinical CMA array. Among the 92 subjects, 69 anonymized subjects (59 unrelated individuals, 1 set of twins, 2 siblings and 6 parents) were selected randomly for further rearrangement analyses. All studies were approved by the Institutional Review Board (IRB) of BCM.
Targeted clinical aCGH and custom high-density aCGH analysis
The samples were initially analyzed in the MGL on consecutive versions of CMA arrays (50–55). The criteria for Xp22.31 duplication case identification by aCGH was based on copy number gain of all or either one or two of the BAC clones RP11-483M24, GS1-227L7 and RP11-143E20 or by oligonucleotides emulating the genomic interval chrX:6 455 604–8 109 387 (hg18).
To fine map the duplications identified by the clinical arrays, we designed two versions of Agilent customized HD-CGH microarrays interrogating specifically the Xp22.31 region. The two array designs were in either the Agilent 8 × 15K (#G4427A) or the 8 × 60K (#G4126A) format. Probes (14 261 in the 8 × 15K format and 24 358 in the 8 × 60K format) were selected from the Agilent eArray system (https://earray.chem.agilent.com/earray/), with an average spacing of 400/250 bp, spanning 4 Mb at Xp22.31 (chrX:5 000 000–9 000 000) and 1.6 Mb at Yq11.22 (chrY:14 310 000–16 000 000). The 8 × 60K array contains probes that represent LCR sequences whereas the 8 × 15K array utilized only unique sequence oligonucleotides of interrogating probes. Labeling, hybridization and microarray analyses were performed as previously described (56).
FISH analysis
FISH was used to assess the copy numbers in subjects/parents who have Xp22.31 triplication suggested by aCGH data. Confirmatory FISH analyses were performed with BAC clones using standard procedures. The BAC clones RP11-483M24 at Xp22.31 and RP11-46A23 at Xp21.2 were used as test and control probes, respectively. Terrific broth media with 20 µg/ml chloramphenicol was used to grow the BAC clones of interest. DNA was extracted from BAC clones (Eppendorf Plasmid Mini Prep kit, Hamburg, Germany) and directly labeled with SpectrumOrangeTM/SpectrumGreenTM (test/control) dUTP by nick-translation (Vysis, Downer Grove, IL). A Power Macintosh G3 System using MacProbe software version 4.4 (Applied Imaging, San Jose, CA, USA) was used to capture the FISH images.
MLPA analysis
MLPA was used to assess the copy numbers in subjects/parents who have Xp22.31 triplication suggested by aCGH data. Probes (Supplementary Material, Table S4) were designed using a web-based program H-MAPD (http://genomics01.arcan.stonybrook.edu/mlpa/cgi-bin/mlpa.cgi). SALSA MLPA reagents were commercially available from MRC-Holland (Amsterdam, The Netherlands). The analysis was carried out following the manufacturer's instructions. Ligation products were PCR amplified and resolved on a 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). For quantitative analysis, peak heights of the patient and normal gender-matched control were analyzed using GeneMarker v1.5 software (Softgenetics, State College, PA, USA).
quantitative PCR
TaqMan® copy number assays (Applied Biosystems) were used to assess the copy numbers in subjects/parents who have Xp22.31 triplication suggested by the aCGH data. Three predesigned primer–probe sets were used (ID: Hs04508520_cn, Hs00930823_cn and Hs00091141_cn). TaqMan copy number reference assay-RNase P from Applied Biosystems was used as reference. Experiments were carried out according to the manufacturer's protocol. Four technical replicates were used for each genomic DNA sample. Reactions were run on the ABI 7900HT fast system. Results were analyzed by the CopyCallerTM software v1.0 (Applied Biosystems).
PCR analyses for breakpoint sequences
Breakpoint junctions of nonrecurrent rearrangements were obtained by long-range PCR with the TAKARA LA TaqTM kit (RR002M for regular buffer or RR02AG for GC buffer I) (TAKARA Bio Inc.) as previously described (33). For breakpoints of recurrent duplications, we used LCR-specific primers to amplify the hypothesized crossover interval. A two-step mismatch PCR strategy was employed to ensure the specificity and efficiency of amplification (57). Detailed primer sequences and particular PCR strategies are available in Supplementary Material, Table S5.
XCI studies
XCI studies were performed based on the protocol described by Allen et al. (58) with modification as described previously (59).
Algorithms for identifying the recurrent Xp22.31 duplications in the control population
The primary controls for the study were Illumina genotypes of 6809 subjects obtained from the Database of Genotypes and Phenotypes (dbGaP, http://www.ncbi.nlm.nih.gov/gap). Our analysis was confined to unrelated adult individuals of European descent from five datasets (accessions phs000092.v1.p1, phs000004.v1.p1, phs000093.v2.p2, phs000001.v2.p1 and phs000142.v1.p1). After allele detection and genotype calling were performed with Genome Studio software (Illumina, Inc., San Diego, CA, USA), B allele frequencies (BAFs) and log R ratios were exported as text files for PennCNV analysis. CNVPartition was run as a plug-in within the Genome Studio browser with settings: confidence threshold 50, minimum number of probes 5. Sample-level quality control analysis was performed using PennCNV software. Samples were excluded from further analysis if any of the following criteria were met: standard deviation of log R ratios >0.35, BAF drift >0.1, waviness factor >0.05 or number of CNVs identified >2 standard deviations above the mean of each dataset. CNVs in pericentromeric and immunoglobulin regions were also excluded. A total of 5088 individuals met these criteria and were included in our analysis. CNV regions called by both PennCNV and CNVPartition were identified using the overlap function for rare CNVs in PLINK.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by the National Institute of Neurological Disorders and Stroke (National Institutes of Health, grant R01NS058529) to J.R.L., Texas Children's Hospital General Clinical Research Center (grant M01RR00188) and Intellectual and Developmental Disabilities Research Centers (grant P30HD024064). A.E. is supported by NIH 5K08DK081735. P.S. is supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the participating subjects and families for their time and effort to collaborate. We also thank Drs Andrea Ballabio and Feng Zhang for their critical review and Marjorie A. Withers for her outstanding technical support. The control datasets used for the analyses described in the manuscript were used with permission and derived from dbGaP through accession numbers phs000092.v1.p1, phs000004.v1.p1, phs000093.v2.p2, phs000001.v2.p1 and phs000142.v1.p1. The BAC clone RP11-527B14 was kindly provided by Dr Steven Scherer from the Human Genome Sequencing Center at Baylor College of Medicine.
Conflict of Interest statement. J.R.L. is a consultant for Athena Diagnostics and Ion Torrent Systems, and is a coinventor on multiple United States and European patents for DNA diagnostics. Furthermore, the Department of Molecular and Human Genetics at BCM derives revenue from molecular diagnostic testing (MGL).
REFERENCES
- 1.Ross M.T., Grafham D.V., Coffey A.J., Scherer S., McLay K., Muzny D., Platzer M., Howell G.R., Burrows C., Bird C.P., et al. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. doi:10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meroni G., Franco B., Archidiacono N., Messali S., Andolfi G., Rocchi M., Ballabio A. Characterization of a cluster of sulfatase genes on Xp22.3 suggests gene duplications in an ancestral pseudoautosomal region. Hum. Mol. Genet. 1996;5:423–431. doi: 10.1093/hmg/5.4.423. doi:10.1093/hmg/5.4.423. [DOI] [PubMed] [Google Scholar]
- 3.Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. doi:10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 4.Ballabio A., Bardoni B., Carrozzo R., Andria G., Bick D., Campbell L., Hamel B., Ferguson-Smith M.A., Gimelli G., Fraccaro M., et al. Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proc. Natl Acad. Sci. USA. 1989;86:10001–10005. doi: 10.1073/pnas.86.24.10001. doi:10.1073/pnas.86.24.10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guioli S., Incerti B., Zanaria E., Bardoni B., Franco B., Taylor K., Ballabio A., Camerino G. Kallmann syndrome due to a translocation resulting in an X/Y fusion gene. Nat. Genet. 1992;1:337–340. doi: 10.1038/ng0892-337. doi:10.1038/ng0892-337. [DOI] [PubMed] [Google Scholar]
- 6.Yen P.H., Tsai S.P., Wenger S.L., Steele M.W., Mohandas T.K., Shapiro L.J. X/Y translocations resulting from recombination between homologous sequences on Xp and Yq. Proc. Natl Acad. Sci. USA. 1991;88:8944–8948. doi: 10.1073/pnas.88.20.8944. doi:10.1073/pnas.88.20.8944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballabio A., Sebastio G., Carrozzo R., Parenti G., Piccirillo A., Persico M.G., Andria G. Deletions of the steroid sulphatase gene in ‘classical’ X-linked ichthyosis and in X-linked ichthyosis associated with Kallmann syndrome. Hum. Genet. 1987;77:338–341. doi: 10.1007/BF00291422. doi:10.1007/BF00291422. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Martin A., Gonzalez-Sarmiento R., De Unamuno P. X-linked ichthyosis: an update. Br. J. Dermatol. 1999;141:617–627. doi: 10.1046/j.1365-2133.1999.03098.x. doi:10.1046/j.1365-2133.1999.03098.x. [DOI] [PubMed] [Google Scholar]
- 9.Fukami M., Kirsch S., Schiller S., Richter A., Benes V., Franco B., Muroya K., Rao E., Merker S., Niesler B., et al. A member of a gene family on Xp22.3, VCX-A, is deleted in patients with X-linked nonspecific mental retardation. Am. J. Hum. Genet. 2000;67:563–573. doi: 10.1086/303047. doi:10.1086/303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Esch H., Hollanders K., Badisco L., Melotte C., Van Hummelen P., Vermeesch J.R., Devriendt K., Fryns J.P., Marynen P., Froyen G. Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum. Mol. Genet. 2005;14:1795–1803. doi: 10.1093/hmg/ddi186. doi:10.1093/hmg/ddi186. [DOI] [PubMed] [Google Scholar]
- 11.Brookes K.J., Hawi Z., Kirley A., Barry E., Gill M., Kent L. Association of the steroid sulfatase (STS) gene with attention deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:1531–1535. doi: 10.1002/ajmg.b.30873. doi:10.1002/ajmg.b.30873. [DOI] [PubMed] [Google Scholar]
- 12.Li X.M., Yen P.H., Shapiro L.J. Characterization of a low copy repetitive element S232 involved in the generation of frequent deletions of the distal short arm of the human X chromosome. Nucleic Acids Res. 1992;20:1117–1122. doi: 10.1093/nar/20.5.1117. doi:10.1093/nar/20.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yen P.H., Li X.M., Tsai S.P., Johnson C., Mohandas T., Shapiro L.J. Frequent deletions of the human X chromosome distal short arm result from recombination between low copy repetitive elements. Cell. 1990;61:603–610. doi: 10.1016/0092-8674(90)90472-q. doi:10.1016/0092-8674(90)90472-Q. [DOI] [PubMed] [Google Scholar]
- 14.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. doi:10.1016/S0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 15.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. doi:10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 16.Myers S., Bowden R., Tumian A., Bontrop R.E., Freeman C., MacFie T.S., McVean G., Donnelly P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. doi:10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parvanov E.D., Petkov P.M., Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. doi:10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. doi:10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F., Potocki L., Sampson J.B., Liu P., Sanchez-Valle A., Robbins-Furman P., Navarro A.D., Wheeler P.G., Spence J.E., Brasington C.K., et al. Identification of uncommon recurrent Potocki-Lupski syndrome-associated duplications and the distribution of rearrangement types and mechanisms in PTLS. Am. J. Hum. Genet. 2010;86:462–470. doi: 10.1016/j.ajhg.2010.02.001. doi:10.1016/j.ajhg.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F., Shen Y., Kohler U., Sharkey F.H., Menon D., Coulleaux L., Malan V., Rio M., McMullan D.J., Cox H., et al. Interstitial microduplication of Xp22.31: causative of intellectual disability or benign copy number variant? Eur. J. Med. Genet. 2010;53:93–99. doi: 10.1016/j.ejmg.2010.01.004. doi:10.1016/j.ejmg.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. doi:10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 22.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. doi:10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J.M., Chuzhanova N., Stenson P.D., Ferec C., Cooper D.N. Meta-analysis of gross insertions causing human genetic disease: novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. doi:10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- 24.Shaffer L.G., Bejjani B.A., Torchia B., Kirkpatrick S., Coppinger J., Ballif B.C. The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am. J. Med. Genet. C Semin. Med. Genet. 2007;145C:335–345. doi: 10.1002/ajmg.c.30152. doi:10.1002/ajmg.c.30152. [DOI] [PubMed] [Google Scholar]
- 25.Lindsay S.J., Khajavi M., Lupski J.R., Hurles M.E. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. doi:10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeffreys A.J., Wilson V., Thein S.L. Hypervariable ‘minisatellite’ regions in human DNA. Nature. 1985;314:67–73. doi: 10.1038/314067a0. doi:10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura Y., Leppert M., O'Connell P., Wolff R., Holm T., Culver M., Martin C., Fujimoto E., Hoff M., Kumlin E., et al. Variable number of tandem repeat (VNTR) markers for human gene mapping. Science. 1987;235:1616–1622. doi: 10.1126/science.3029872. doi:10.1126/science.3029872. [DOI] [PubMed] [Google Scholar]
- 28.Vojtiskova M., Mirkin S., Lyamichev V., Voloshin O., Frank-Kamenetskii M., Palecek E. Chemical probing of the homopurine–homopyrimidine tract in supercoiled DNA at single-nucleotide resolution. FEBS Lett. 1988;234:295–299. doi: 10.1016/0014-5793(88)80102-9. doi:10.1016/0014-5793(88)80102-9. [DOI] [PubMed] [Google Scholar]
- 29.Glover J.N., Pulleyblank D.E. Protonated polypurine/polypyrimidine DNA tracts that appear to lack the single-stranded pyrimidine loop predicted by the ‘H’ model. J. Mol. Biol. 1990;215:653–663. doi: 10.1016/S0022-2836(05)80175-5. doi:10.1016/S0022-2836(05)80175-5. [DOI] [PubMed] [Google Scholar]
- 30.Wolf N.I., Sistermans E.A., Cundall M., Hobson G.M., Davis-Williams A.P., Palmer R., Stubbs P., Davies S., Endziniene M., Wu Y., et al. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus–Merzbacher disease. Brain. 2005;128:743–751. doi: 10.1093/brain/awh409. doi:10.1093/brain/awh409. [DOI] [PubMed] [Google Scholar]
- 31.Bi W., Sapir T., Shchelochkov O.A., Zhang F., Withers M.A., Hunter J.V., Levy T., Shinder V., Peiffer D.A., Gunderson K.L., et al. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. doi:10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho C.M., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J., et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. doi:10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. doi:10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beunders G., van de Kamp J.M., Veenhoven R.H., van Hagen J.M., Nieuwint A.W., Sistermans E.A. A triplication of the Williams-Beuren syndrome region in a patient with mental retardation, a severe expressive language delay, behavioural problems and dysmorphisms. J. Med. Genet. 2010;47:271–275. doi: 10.1136/jmg.2009.070490. doi:10.1136/jmg.2009.070490. [DOI] [PubMed] [Google Scholar]
- 35.Ungaro P., Christian S.L., Fantes J.A., Mutirangura A., Black S., Reynolds J., Malcolm S., Dobyns W.B., Ledbetter D.H. Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J. Med. Genet. 2001;38:26–34. doi: 10.1136/jmg.38.1.26. doi:10.1136/jmg.38.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A., et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. doi:10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 37.Conrad D.F., Bird C., Blackburne B., Lindsay S., Mamanova L., Lee C., Turner D.J., Hurles M.E. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet. 2010;42:385–391. doi: 10.1038/ng.564. doi:10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F., Carvalho C.M., Lupski J.R. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. doi:10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. doi:10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 40.Newman T.L., Tuzun E., Morrison V.A., Hayden K.E., Ventura M., McGrath S.D., Rocchi M., Eichler E.E. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005;15:1344–1356. doi: 10.1101/gr.4338005. doi:10.1101/gr.4338005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F., Seeman P., Liu P., Weterman M.A., Gonzaga-Jauregui C., Towne C.F., Batish S.D., De Vriendt E., De Jonghe P., Rautenstrauss B., et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. doi:10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiao X., Chen H., Chen J., Herrup K., Firestein B.L., Kiledjian M. Modulation of neuritogenesis by a protein implicated in X-linked mental retardation. J. Neurosci. 2009;29:12419–12427. doi: 10.1523/JNEUROSCI.5954-08.2009. doi:10.1523/JNEUROSCI.5954-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kent L., Emerton J., Bhadravathi V., Weisblatt E., Pasco G., Willatt L.R., McMahon R., Yates J.R. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J. Med. Genet. 2008;45:519–524. doi: 10.1136/jmg.2008.057729. doi:10.1136/jmg.2008.057729. [DOI] [PubMed] [Google Scholar]
- 44.Stankiewicz P., Pursley A.N., Cheung S.W. Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am. J. Med. Genet. A. 2010;152A:1089–1100. doi: 10.1002/ajmg.a.33216. doi:10.1002/ajmg.a.33216. [DOI] [PubMed] [Google Scholar]
- 45.Lupski J.R. Structural variation in the human genome. N. Engl. J. Med. 2007;356:1169–1171. doi: 10.1056/NEJMcibr067658. doi:10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
- 46.Potocki L., Chen K.S., Koeuth T., Killian J., Iannaccone S.T., Shapira S.K., Kashork C.D., Spikes A.S., Shaffer L.G., Lupski J.R. DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am. J. Hum. Genet. 1999;64:471–478. doi: 10.1086/302240. doi:10.1086/302240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klopocki E., Schulze H., Strauss G., Ott C.E., Hall J., Trotier F., Fleischhauer S., Greenhalgh L., Newbury-Ecob R.A., Neumann L.M., et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am. J. Hum. Genet. 2007;80:232–240. doi: 10.1086/510919. doi:10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Hattab A.W., Zhang F., Maxim R., Christensen K.M., Ward J.C., Hines-Dowell S., Scaglia F., Lupski J.R., Cheung S.W. Deletion and duplication of 15q24: molecular mechanisms and potential modification by additional copy number variants. Genet. Med. 2010;12:573–586. doi: 10.1097/GIM.0b013e3181eb9b4a. doi:10.1097/GIM.0b013e3181eb9b4a. [DOI] [PubMed] [Google Scholar]
- 49.Girirajan S., Rosenfeld J.A., Cooper G.M., Antonacci F., Siswara P., Itsara A., Vives L., Walsh T., McCarthy S.E., Baker C., et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010;42:203–209. doi: 10.1038/ng.534. doi:10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheung S.W., Shaw C.A., Yu W., Li J., Ou Z., Patel A., Yatsenko S.A., Cooper M.L., Furman P., Stankiewicz P., et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet. Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. doi:10.1097/01.GIM.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 51.Lu X., Shaw C.A., Patel A., Li J., Cooper M.L., Wells W.R., Sullivan C.M., Sahoo T., Yatsenko S.A., Bacino C.A., et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS One. 2007;2:e327. doi: 10.1371/journal.pone.0000327. doi:10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu X.Y., Phung M.T., Shaw C.A., Pham K., Neil S.E., Patel A., Sahoo T., Bacino C.A., Stankiewicz P., Kang S.H., et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. doi:10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ou Z., Kang S.H., Shaw C.A., Carmack C.E., White L.D., Patel A., Beaudet A.L., Cheung S.W., Chinault A.C. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet. Med. 2008;10:278–289. doi: 10.1097/GIM.0b013e31816b4420. doi:10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao L., Shaw C.A., Lu X.Y., Sahoo T., Bacino C.A., Lalani S.R., Stankiewicz P., Yatsenko S.A., Li Y., Neill S., et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: a study of 5,380 cases. Am. J. Med. Genet. A. 2008;146A:2242–2251. doi: 10.1002/ajmg.a.32399. doi:10.1002/ajmg.a.32399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boone P.M., Bacino C.A., Shaw C.A., Eng P.A., Hixson P.M., Pursley A.N., Kang S.H., Yang Y., Wiszniewska J., Nowakowska B.A., et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. doi:10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shinawi M., Liu P., Kang S.H., Shen J., Belmont J.W., Scott D.A., Probst F.J., Craigen W.J., Graham B., Pursley A., et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioral problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2010;47:322–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. doi:10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 59.Ramocki M.B., Peters S.U., Tavyev Y.J., Zhang F., Carvalho C.M., Schaaf C.P., Richman R., Fang P., Glaze D.G., Lupski J.R., et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann. Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. doi:10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.