

Abstract
AIMS
This study aimed to test whether a pharmacokinetic simulation model could extrapolate nonclinical drug data to predict human efavirenz exposure after single and continuous dosing as well as the effects of concomitant rifampicin and further to evaluate the weight-based dosage recommendations used to counteract the rifampicin–efavirenz interaction.
METHODS
Efavirenz pharmacokinetics were simulated using a physiologically based pharmacokinetic model implemented in the Simcyp™ population-based simulator. Physicochemical and metabolism data obtained from the literature were used as input for prediction of pharmacokinetic parameters. The model was used to simulate the effects of rifampicin on efavirenz pharmacokinetics in 400 virtual patients, taking into account bodyweight and CYP2B6 phenotype.
RESULTS
Apart from the absorption phase, the simulation model predicted efavirenz concentration–time profiles reasonably well, with close agreement with clinical data. The simulated effects of rifampicin co-administration on efavirenz treatment showed only a minor decrease of 16% (95% confidence interval 13–19) in efavirenz area under the concentration–time curve, of the same magnitude as what has been clinically observed (22%). Efavirenz exposure depended on CYP2B6 phenotype and bodyweight. Increasing the efavirenz dose during concomitant rifampicin was predicted to be most successful in patients over 50 kg regardless of CYP2B6 status.
CONCLUSIONS
Our findings, although based on a simulation approach using limited in vitro data, support the current recommendations for using a 50 kg bodyweight cut-off for efavirenz dose increment when co-treating with rifampicin.
Keywords: drug–drug interaction, efavirenz, HIV/AIDS, in vitro–in vivo extrapolation, rifampicin, Simcyp
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Rifampicin is known to reduce the exposure of efavirenz. Despite lack of adequate clinical support, most guidelines recommend an increased efavirenz dose in patients treated with both drugs, although some propose an increase only in patients weighing over 50 kg.
WHAT THIS STUDY ADDS
An example of utilization of in vitro data to predict in vivo consequences of metabolic drug–drug interactions, suggesting answers to specific questions difficult to study in patients. Here the efavirenz–rifampicin interaction was simulated with the specific question of whether or not to increase the efavirenz dose based on bodyweight.
The results from this simulation suggest that increasing the efavirenz dose may be appropriate only in patients with bodyweights over 50 kg.
Introduction
Efavirenz (EFZ) is frequently recommended as the preferred non-nucleoside reverse transcriptase inhibitor (NNRTI) component in highly active antiretroviral therapy (HAART) [1, 2]. Efavirenz is considered to have a more extensive efficacy and safety documentation than other NNRTIs [2]. Compared with other HAART regimens, one containing efavirenz has proved to be superior or similar with respect to viral load suppression [3, 4].
Efavirenz is metabolized mainly by oxidation to 7- or 8-hydroxy-EFZ and by glucuronidation to N-glucuronide-EFZ [5]. The 8-hydroxy pathway was believed to constitute >90% of the oxidative pathway, with CYP2B6 suggested to be the principal enzyme, with minor contributions from CYP1A2 and CYP3A [5]. However, recent work by Ogburn et al. shows the importance of CYP2A6 to the 7-hydroxy-EFZ pathway, where the authors assigned 23% of the combined 7- and 8-hydroxy pathway to CYP2A6 and the rest to CYP2B6 [6]. The enzyme UGT2B7 is involved in the glucuroconjugation pathway [7]. Polymorphisms in UGT2B7 together with CYP2B6 and CYP2A6 polymorphisms have been shown to influence efavirenz mid-dose concentrations in HIV-infected patients [8]. Efavirenz exhibits profound autoinduction of CYP2B6 and, to a lesser degree, CYP3A4 [9, 10]. Moreover, autoinhibition of CYP3A4 has also been reported [11].
Rifampicin is a known inducer of the P450 enzymes CYP2B6, CYP3A, CYP2C19, CYP2C8, CYP2C9 and CYP1A2 [12, 13]. Despite its well-known interaction potential, rifampicin is commonly used in the treatment of HIV/AIDS patients co-infected with tuberculosis. The combination of efavirenz and rifampicin has been shown to reduce the area the under the concentration–time curve (AUC) of efavirenz by 22% [14]. The proposed mechanism of the rifampicin–efavirenz interaction is induction of CYP3A and CYP2B6. Most guidelines therefore recommend an increase in efavirenz dose from 600 to 800 mg day−1 to counter the effects of rifampicin [1, 2, 15]. Moreover, the US Department of Health and Human Services also recommends taking the patient's bodyweight into consideration by suggesting a 50 kg cut-off limit for the efavirenz dose adjustment [1]. Although no such recommendations are included in the drug label and a limited number of studies are available on the subject, this practice is fairly common, it being more practical to dose adjust based on weight compared with phenotype [14, 16–18]. The Centers for Disease Control and Prevention (CDC) have in their 2007 guide suggested that a 22% decrease of efavirenz exposure is unlikely to be of any clinical meaning [19]. However, in a recent food and drug administration case study, it is argued that suboptimal drug concentrations can lead to virological failure, and further clinical trials or in silico simulations are encouraged [17].
The Simcyp™ population simulator is used for simulation of oral absorption, tissue distribution, metabolism and excretion (ADME) of drugs in healthy subjects or diseased populations. Using routinely generated experimental data in preclinical development and the physiochemical properties of the drug, the Simcyp™ platform allows simulations of population pharmacokinetics as well as assessment of metabolic drug interactions in different populations [20]. In Simcyp™ one can predict hepatic clearance, and its variability within a population, using in vitro metabolism data [21].
The primary aim of this study was to evaluate whether a pharmacokinetic simulation model could predict the efavirenz exposure in humans after single and continuous dosing as well as the effects of concomitant rifampicin, based on nonclinical drug information. Secondary aims were to see whether the predictions could be of any guidance in recommending dosing strategies for efavirenz in the presence of rifampicin.
Methods
Efavirenz pharmacokinetics were simulated using a generic physiologically based pharmacokinetic (PBPK) model implemented in the Simcyp™ ADME simulator (version 8.20, Simcyp™ Ltd, Sheffield, UK) [22]. The PBPK model consisted of 11 tissue compartments. The efavirenz volume of distribution (3.33 l kg−1) was scaled from rat to man by means of allometric scaling and, together with efavirenz logP, was used as input for computation of efavirenz tissue partitioning constants (Kp), as proposed by Jansson et al. [23]. Different absorption models, such as the Advanced Dissolution, Absorption and Metabolism (ADAM) model, the Compartmental Absorption and Transit (CAT) model and the first-order absorption model, were examined [22].
The intrinsic metabolic clearance (CLint) was described by:
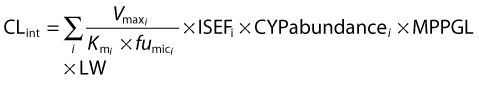 |
(1) |
where i indicates the isoform of cytochrome P450 (CYP450) involved in metabolism, Vmax is the maximal rate of metabolism for an individual enzyme, Km is the Michaelis–Menten constant, fumic is the fraction of unbound drug in microsomal incubations, ISEFi is a scaling factor that compensates for any differences in enzyme activity between different recombinant and hepatic systems [24], CYPabundance is the abundance level for the specific isoform of the CYP450, MPPGL is the amount protein per gram of liver and LW is liver weight. Naturally occurring variability in CYPabundance, MPPGL and LW is provided by default in the software. CYPabundance may explain variability due to functional genetic polymorphism or other ethnicity-related factors as well as developmental changes in neonatal and paediatric populations. The MPPGL is thought to contribute less in explaining the variability in populations than CYPabundance. Liver weight, being a function of body surface area, is accommodating for the variability in bodyweight. An extensive review on the subject has previously been published elsewhere [21].
The relative contributions of different CYP P450 isoforms were calculated as shown in eqn (2):
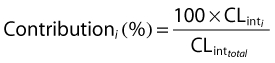 |
(2) |
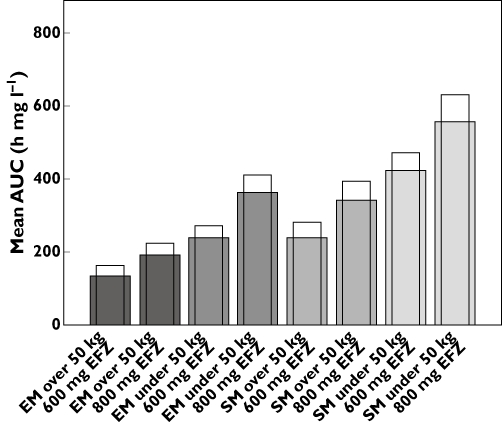
where 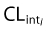 is the intrinsic clearance for a particular isoform and CLint total is the sum of all such intrinsic clearances. Data describing the physicochemical, distribution and metabolic properties of efavirenz were obtained from the literature (Table 1). Microsomal binding of efavirenz (fumic) was computed from lipophilicity and microsomal protein concentration used in the incubations [25]. Hepatic binding (fuhep) for efavirenz and rifampicin measured in vitro was obtained from Shou et al. [26]. Metabolic rate parameters (Vmax and Km) for efavirenz CYP2B6, 1A2 and 3A4 were obtained from recombinantly expressed CYPs [5]. The Vmax values for efavirenz CYP2A6 metabolism in human liver microsomes (HLM) was converted to Vmax (rCYP) for recombinantly expressed systems according to eqn (3)[6, 24]. All CLint values, except for CYP2A6, were corrected for ISEF. The required data for rifampicin, with the exception of data regarding CYP2B6 enzyme induction, was available within the software (Table 1). Induction parameters and coefficients of variations (CV%) not provided by Simcyp™ were estimated from experimental data reported for human hepatocytes incubated with rifampicin or efavirenz. The concentration-dependent degree of induction (E) of CYP2B6 [9, 27] and CYP3A4 [10, 26] was fitted with an Emax model (eqn (4)), WinNonlin version 5.2 (Pharsight Co., Mountain View, CA, USA).
is the intrinsic clearance for a particular isoform and CLint total is the sum of all such intrinsic clearances. Data describing the physicochemical, distribution and metabolic properties of efavirenz were obtained from the literature (Table 1). Microsomal binding of efavirenz (fumic) was computed from lipophilicity and microsomal protein concentration used in the incubations [25]. Hepatic binding (fuhep) for efavirenz and rifampicin measured in vitro was obtained from Shou et al. [26]. Metabolic rate parameters (Vmax and Km) for efavirenz CYP2B6, 1A2 and 3A4 were obtained from recombinantly expressed CYPs [5]. The Vmax values for efavirenz CYP2A6 metabolism in human liver microsomes (HLM) was converted to Vmax (rCYP) for recombinantly expressed systems according to eqn (3)[6, 24]. All CLint values, except for CYP2A6, were corrected for ISEF. The required data for rifampicin, with the exception of data regarding CYP2B6 enzyme induction, was available within the software (Table 1). Induction parameters and coefficients of variations (CV%) not provided by Simcyp™ were estimated from experimental data reported for human hepatocytes incubated with rifampicin or efavirenz. The concentration-dependent degree of induction (E) of CYP2B6 [9, 27] and CYP3A4 [10, 26] was fitted with an Emax model (eqn (4)), WinNonlin version 5.2 (Pharsight Co., Mountain View, CA, USA).
Table 1.
Simcyp™ inputs for efavirenz and rifampicin
| Efavirenz input parameters | Value | Variability (CV%) | Comments and references |
|---|---|---|---|
| MW | 315.67 | – | [11] |
| LogP | 5.4 | – | [38] |
| B : P ratio | 0.74 | – | [39] |
| fuplasma | 0.01125 | – | Predicted by Simcyp™ |
| Caco-2 permeability (10−6 cm s−1) | 8.92 | – | [40] |
| fugut | 1 | – | Assumed |
| Main binding protein | Albumin | – | [11] |
| fumic | 0.3 | – | Predicted [25] |
| fuhep | 0.063 | – | [26] |
| rCYP 3A4 Vmax (pmol min−1 pmol−1 3A4) | 0.16 | – | Baculovirus ISEF [5] |
| rCYP 3A4 Km (µm) | 23.5 | – | Baculovirus ISEF [5] |
| rCYP 3A5 Vmax (pmol min−1 pmol−1 3A5) | 0.6 | – | Baculovirus ISEF [5] |
| rCYP 3A5 Km (µm) | 19.1 | – | Baculovirus ISEF[5] |
| rCYP 1A2 Vmax (pmol min−1 pmol−1 1A2) | 0.6 | – | Baculovirus ISEF [5] |
| rCYP 1A2 Km (µm) | 8.3 | – | Baculovirus ISEF[5] |
| rCYP 2B6 Vmax (pmol min−1 pmol−1 2B6) | 3.5 | – | Baculovirus ISEF[5] |
| rCYP 2B6 Km (µm) | 6.4 | – | Baculovirus ISEF [5] |
| rCYP 2A6 Vmax (pmol min−1 pmol−1 2A6) | 1.08 | – | Converted from Vmax(HLM) [6] as proposed by [24] |
| rCYP 2A6 Km (µm) | 14.7 | – | [6] |
| UGT2B7 Vmax (pmol min−1 mg−1) | 1.5 | – | [7] |
| UGT2B7 Km (µm) | 16.1 | – | [7] |
| CYP 3A4 Indmax | 6.45 | 18.6 | Digitalized data [26] modelled together with[10] |
| CYP3A4 IndC50 (µm) | 3.93 | 52.5 | Digitalized data[26] modelled together with[10] |
| CYP 2B6 Indmax | 5.76 | 13.7 | Modelled from [9] |
| CYP 2B6 IndC50 (µm) | 0.82 | 71.9 | Modelled from [9] |
| Rifampicin input parameters | Value | Variability* (CV%) | Comments and references |
| CYP2B6 Indmax | 8.5† | 30 | [27] |
| CYP2B6 IndC50 (µm) | 1.17 | 30 | [27] |
| fuhep | 0.419 | [26] |
Default Simcyp setting; Indmax, maximal fold induction over vehicle (1 = no induction)
mean value (n = 2)
Vmax, maximal rate of metabolism; fugut, fraction unbound in gut; funep, franction unbound in hepatocytes; fumic, fraction unbound in microsomes; fuplasma, fraction unbound in plasma; Km, Michaelis–Menten constant; ISEF, Inter System Extrapolation Factor; B : P ratio, blood : plasma concentration ratio; MW, molecular weight; Log P, logarithm of octanol : water ratio.
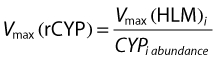 |
(3) |
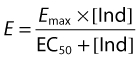 |
(4) |
In eqn (4), Emax is the net maximal fold increase, EC50 is the inducer concentration producing 50% of Emax and [Ind] is the inducer concentration. In cases where the literature did not provide tabulated data points, the data were harvested from graphs by graph digitalizing software.
The pharmacokinetic simulation model was assessed by visually comparing simulated efavirenz plasma concentration–time profiles with concentration–time data from three clinical studies. The three studies comprised a single-dose study in 121 Ugandan healthy subjects (Figure 1A) [28] and two steady-state studies in 74 and 76 HIV-positive patients on HAART performed in Zimbabwe [29] and Norway/Sweden [30], respectively (Figure 1B,C). The frequency of intermediate and slow metabolizers in the Zimbabwean study was reported to be 71%; intermediate and slow metabolizers had a steady-state oral clearance (CL/F) of 7.2 and 4.0 l h−1, respectively, compared with 9.4 l h−1 in the normal population [29]. In Figure 1A,B, the default Simcyp™ frequency of slow CYP2B6 metabolizers (11%) is assumed. In Figure 1C, simulations with slow metabolizer frequencies of 11 and 71% are compared. Weight and sex ratio of the virtual subjects was matched to those reported in the three clinical studies. In addition, model-derived population pharmacokinetic parameters from the single-dose study [28] were compared with simulated estimates. The efavirenz autoinduction was evaluated by comparison of simulated autoinduced CL/F with the clinically observed literature autoinduced CL/F [31]. The extent of the simulated rifampicin–efavirenz drug–drug interaction was compared with clinical results reported by Lopez-Cortes et al. [14], where the degree of efavirenz AUC decrease was used as metric. The possible influence of efavirenz on rifampicin kinetics was not investigated.
Figure 1.
Simulated and observed efavirenz concentration–time profiles after single (A) and repeated dosing of 600 mg (B,C). The continuous lines represent the mean and 95th and 5th percentiles of the observed systemic concentration, while the dashed lines represent the mean and 95th and 5th percentiles of the predicted systemic concentrations in 100 simulated individuals. The circles in A are observed concentrations from a single-dose study in Uganda [28]. The circles (B,C) and diamonds (B) are clinically observed plasma concentrations from Zimbabwe and Sweden/Norway, respectively [29, 30]. In A and B, a defult frequency of slow CYP2B6 metabolizers (11%) is assumed. In C, the bold and narrow dashed lines represent Simcyp predictions with frequencies of 71 and 11%, respectively
The model was used to simulate an efavirenz dose adjustment from 600 to 800 mg in a population of 400 virtual patients. The 400 virtual patients were divided into four groups based on CYP2B6 phenotype and bodyweight (<50 or >50 kg). These four groups where used to simulated eight scenarios with or without concomitant rifampicin, as follows: extensive metabolizers over 50 kg bodyweight receiving 600 or 800 mg efavirenz (scenarios I and II, respectively), extensive metabolizers under 50 kg receiving 600 or 800 mg efavirenz (scenarios III and IV, respectively), slow metabolizers over 50 kg receiving 600 or 800 mg efavirenz (scenarios V and VI, respectively), and slow metabolizers under 50 kg receiving 600 or 800 mg efavirenz (scenarios VII and VIII, respectively). Slow (SM) and extensive metabolizers (EM) were predefined in the Simcyp™ software by enzyme abundance level (SM = 6, EM = 17 pmol (mg protein)−1). Changes in efavirenz steady-state AUC, CL/F, maximum plasma concentration (Cmax) and trough plasma concentration (Ctrough) were used to evaluate the magnitude of the predicted interaction.
Results
The pharmacokinetic simulation model predicted the typical efavirenz concentration–time course and its interindividual variability after single and continuous dosing reasonably well (Figure 1). However, the absorption phase, predicted by a first-order absorption model, was overpredicted. Pharmacokinetic parameters derived from the pharmacokinetic simulation model agreed well with published data after single-dose administration (Table 2). Simulated and clinically derived single-dose AUC, Cmax, time for Cmax (Tmax) and CL/F deviated by 15, 25, 63 and 9.5%, respectively. Upon continuous administration, efavirenz CL/F was predicted to increase from 3.23 to 9.9 l h−1 due to autoinduction, which is comparable to clinical observations (9.4 l h−1) [29]. Efavirenz steady-state oral efavirenz clearance was estimated as 12.6 l h−1 with concomitant rifampicin, which is somewhat lower than clinical data (17.2 l h−1).
Table 2.
Means ± SD for clinical and Simcyp-generated pharmacokinetic parameters for efavirenz after single-dose oral administration (600 mg)
| Clinical data* | Simcyp™ simulation | |
|---|---|---|
| Parameter | Mean (±SD) n = 121 | Mean (±SD) n = 1000 |
| AUC(0.72 h) (mg h l−1) | 112 (34.7) | 90.37 (35.6) |
| Cmax (mg l−1) | 3.67 (1.1) | 4.95 (1.14) |
| Tmax (h) | 4.84 (4.1) | 1.76 (0.33) |
| CL/F (l h−1) | 3.87 (1.36) | 3.23 (1.67) |
[28]. AUC, area under the curve plasma concentration time curve; CL/F, oral clearance; Cmax, maximum plasma concentration; Tmax, time for Cmax.
The simulations of continuous efavirenz administration predicted a mean decrease in efavirenz AUC of 16% [95% confidence interval (CI) 13–19] in the presence of rifampicin. The model predicted mean decreases in Cmax and Ctrough of 14 (95% CI 11–16) and 18% (95% CI 15–21), respectively.
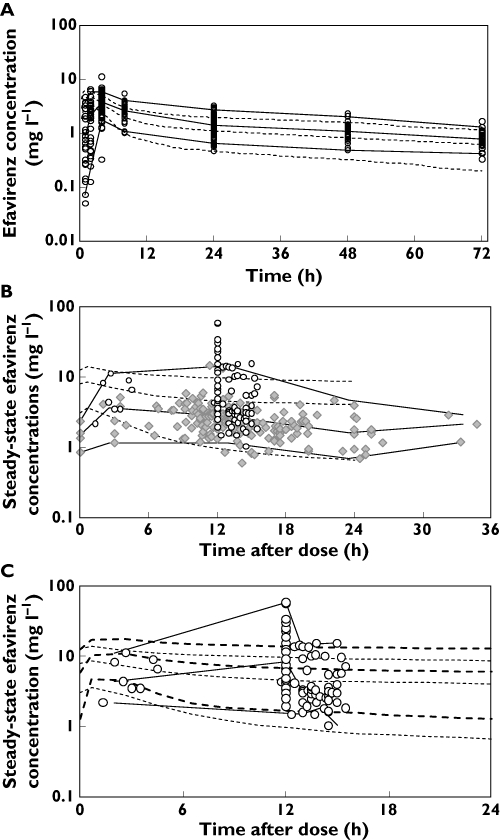
The simulated influence on efavirenz exposure for the eight scenarios is depicted as the ratio of steady-state AUCs with and without concomitant rifampicin (Figure 2). Individuals with a bodyweight exceeding 50 kg receiving either 600 or 800 mg efavirenz were predicted to have the largest decrease in efavirenz AUC due to concomitant rifampicin, whereas individuals with bodyweight less than 50 kg were expected to have the least change in AUC.
Figure 2.
Simulated effects of continuous rifampicin co-administration on efavirenz steady-state area under the concentration–time curve (AUC) in eight scenarios. Abbreviations: EFV, efavirenz; EM, extensive CYP2B6 metabolizers; SM, slow CYP2B6 metabolizers
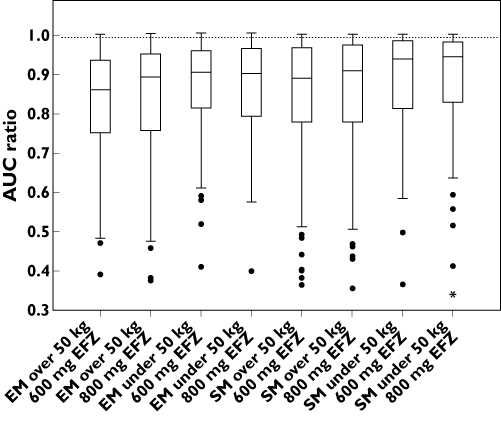
A comparison of the predicted efavirenz (600 mg) and dose-adjusted (800 mg) AUC with and without concurrent rifampicin for the eight simulation scenarios is shown in Figure 3. Males and females were not predicted to differ in efavirenz exposure or the investigated interaction when normalized for bodyweight.
Figure 3.
Simulated median efavirenz steady-state AUC with and without concomitant rifampicin grouped according to bodyweight and CYP2B6 phenotype. The open bars are the median efavirenz steady-state AUC without concomitant rifampicin. The shaded bars are the median efavirenz steady-state AUC when co-administered with rifampicin. Abbreviations: EFV, efavirenz; EM, extensive CYP2B6 metabolizers; SM, slow CYP2B6 metabolizers
The relative contribution of individual isozymes to the overall metabolic elimination of efavirenz was computed in Simcyp™ in the induced and noninduced state for extensive and slow metabolizers separately (Table 3).
Table 3.
Simcyp™-estimated mean contribution of cytochrome P450 and UGT enzymes to single-dosed and steady-state efavirenz (EFZ) CL/F in extensive (EM) and slow (SM) CYP2B6 metabolizers
| Single dose | Steady state | |||
|---|---|---|---|---|
| EFZ (%) | EFZ (%) | |||
| Enzyme | SM | EM | SM | EM |
| CYP1A2 | 45.0 | 31.4 | 21.4 | 10.9 |
| CYP2B6 | 23.0 | 45.9 | 45.9 | 72.6 |
| CYP3A4 | 11.1 | 7.60 | 21.1 | 11.6 |
| CYP3A5 | 6.80 | 4.97 | 3.37 | 1.85 |
| CYP2A6 | 13.8 | 9.79 | 6.7.1 | 3.53 |
| UGT2B7 | 0.34 | 0.23 | 0.14 | 0.07 |
Discussion
The present study is a positive example of when human in vivo pharmacokinetics can be predicted by model simulation based on nonclinical drug data, in this case obtained from the literature. Moreover, the extent of a drug–drug interaction, as exemplified by the inductive effect of concomitant rifampicin on efavirenz pharmacokinetics, was predicted with a fair degree of accuracy.
Following single-dose oral administration, the pharmacokinetic simulation model predicted a median efavirenz CL/F of 3.23 l h−1 (90% CI 2.89–3.56), a value in close agreement with a literature value of 3.87 l h−1 (95% CI 3.45–4.27) [28] (Figure 1A). Furthermore, the model simulations were able to mirror the large interindividual pharmacokinetic variability for efavirenz as evidenced in the general agreement between 90% prediction intervals and scatter of clinical concentration–time data (Figure 1B,C). The frequency of slow metabolizers in the Scandinavian study is unknown, since the subjects may not be ethnically Scandinavian (Figure 1B). The Zimbabwean population had a very high frequency of the CYP2B6 516G→T (*6) variant, where 71% of the subjects were characterized as either intermediate or slow metabolizers [29]. Both frequencies (11 and 71%) are shown in Figure 1C.
Efavirenz Tmax and Cmax after single dose were under- and overpredicted, respectively. Efavirenz has a somewhat atypical absorption profile, which has previously been described as a zero-order input to the dosing compartment followed by sequential first-order absorption [28]. In the present study, a simple first-order absorption model was used in Simcyp™, which may explain some deviation of predicted Tmax and Cmax values. More advanced and mechanistic absorption models, such as the ADAM model or the CAT model, were tested but did not improve the absorption profile and led to considerable longer run times [22]. Caco-2 permeability data provided to Simcyp™ should preferably be calibrated against a high-permeability drug, such as propanolol. No such internal standard was available for efavirenz permeability literature data, which may have contributed to the rather poor prediction of the absorption-rate-dependent parameters, Tmax and Cmax[28]. Efavirenz steady-state concentrations and their interindividual variability were also quite well predicted, albeit with a tendency towards plasma concentrations being overpredicted (Figure 1B).
The efavirenz unbound fraction in microsomal incubations was computed according to Austin et al. [25]. In some cases, this method has proved to underestimate the fraction unbound in the microsomes [32], thus leading to an overprediction of CL/F. An experimentally determined in vitro value for fumic would have been preferable but was not found for efavirenz.
Both CYP2B6 and CYP1A6 have been identified as major contributors to metabolism of efavirenz [5, 6]. Surprisingly, and in contrast to earlier interpretation of in vitro data, the simulation model predicted an 11% contribution of CYP1A2 in extensive metabolizers and as much as 21% in slow metabolizers in the induced state. Although efavirenz has a higher affinity for the CYP2B6 enzyme, the relatively higher CYP1A2 abundance can in part explain why Simcyp™ predicted such a large contribution of CYP1A2 to metabolic clearance. The predicted contributions of the various CYP450 isoforms may, however, change greatly if additional metabolic data become available and should therefore be interpreted with some caution. If predictions of the CYP1A2 contribution should prove to be valid, it may be possible that variation in CYP1A2 abundance, either due to xenobiotic inducers or to genetic polymorphism, may contribute significantly to the variability of efavirenz intrinsic clearance in slow CYP2B6 metabolizers.
The simulations of continuous efavirenz administration under the influence of rifampicin predicted a mean decrease in efavirenz AUC, Cmax and Ctrough of 16, 14 and 18%, respectively. These decreases are somewhat underpredicted compared with clinical observations, where rifampicin has in the literature been reported to decrease efavirenz AUC, Cmax and Ctrough by 22, 24 and 25%, respectively [14]. Efavirenz steady-state CL/F was estimated as 12.6 l h−1 with concomitant rifampicin, which is somewhat lower compared with clinical data (17.2 l h−1) [33]. This underprediction could possibly be attributed to lack of data concerning induction of CYP1A6 and CYP2A6 by rifampicin. Rifampicin has been shown to be a weak inducer of CYP1A6 in vivo and CYP2A6 in vitro[12, 34]. However, the nature of these data is not yet compatible for use with the Simcyp™ software. The consequences of underpredicting rifampicin-induced steady-state efavirenz CL/F are not expected to have a major influence on the relative differences in the level of rifampicin induction between slow and extensive metabolizers nor subjects over and under 50 kg bodyweight.
There were considerable differences in the level of rifampicin effect on efavirenz exposure in the eight scenarios (Figure 3). As anticipated, both weight and genetic polymorphism influence efavirenz exposure substantially, with the combination of low bodyweight (<50 kg) and low metabolism resulting in the highest efavirenz exposure. This combination of traits could be common in emaciated late-stage African AIDS patients, due to the relative high frequency of slow metabolizers in some African populations [35–37]. By excluding patients with low bodyweight (<50 kg) when adjusting the efavirenz dose due to concomitant rifampicin, one could easily eliminate the worst-case scenario where the slow metabolizers weighing under 50 kg are unnecessarily exposed to high efavirenz concentrations due to the dose adjustment. In further support of the >50 kg cut-off, individuals with >50 kg bodyweight were predicted to have an efavirenz AUC, after the dose adjustment during concurrent rifampicin, most similar compared with the standard efavirenz dose given without concurrent rifampicin. Although the influence of genetic polymorphism on efavirenz exposure is suggested to have considerable impact and dosage recommendations based on phenotype continue to gain support, there are increasing concerns about the practical value of genotyping in resource-limited settings, such as parts of the African continent. In this light, a straightforward weight-based dosing recommendation could be of more practical value.
This in vitro–in vivo simulation, although not prospective, lends some support to the recommendation of an increased dose in patients on concurrent rifampicin and efavirenz therapy with bodyweight >50 kg until such time as clinical results become available.
In conclusion, the pharmacokinetic simulation model was able to predict efavirenz pharmacokinetics and its interindividual variability after single and repeated dosing as well as the impact of rifampicin treatment on efavirenz exposure. This suggests the usefulness of in vitro to in vivo extrapolations when assessing the clinical risk of drug–drug interactions. The results show that indiscriminately increasing the efavirenz dose during rifampicin treatment may lead to high plasma concentrations, possibly increasing the risk of adverse drug reactions. Our findings, although based on a simulation approach using limited in vitro data, support the current recommendations for using a 50 kg bodyweight cut-off for efavirenz dose increment when co-treating with rifampicin.
Acknowledgments
Results from the efavirenz Simcyp pharmacokinetic simulation model were compared with clinical data kindly provided by Magnus Gisslén and Collen Masimirembwa, respectively, with colleagues.
Daniel Röshammar is currently an employee of AstraZeneca. However, this work was not funded or supported by any pharmaceutical company.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Washington DC: Department of Health and Human Services; 2009. pp. 1–161. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf (last accessed December 2009) [Google Scholar]
- 2.Josephson F, Albert J, Flamholc L, Gisslen M, Karlstrom O, Lindgren SR, Naver L, Sandstrom E, Svedhem-Johansson V, Svennerholm B, Sonnerborg A. Antiretroviral treatment of HIV infection: Swedish recommendations 2007. Scand J Infect Dis. 2007;39:486–507. doi: 10.1080/00365540701383154. [DOI] [PubMed] [Google Scholar]
- 3.Keiser P, Nassar N, White C, Koen G, Moreno S. Comparison of nevirapine- and efavirenz-containing antiretroviral regimens in antiretroviral-naive patients: a cohort study. HIV Clin Trials. 2002;3:296–303. doi: 10.1310/M47B-R51C-X0MC-K3GW. [DOI] [PubMed] [Google Scholar]
- 4.Lucas GM, Chaisson RE, Moore RD. Comparison of initial combination antiretroviral therapy with a single protease inhibitor, ritonavir and saquinavir, or efavirenz. AIDS. 2001;15:1679–86. doi: 10.1097/00002030-200109070-00011. [DOI] [PubMed] [Google Scholar]
- 5.Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther. 2003;306:287–300. doi: 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]
- 6.Ogburn E, Jones D, Masters A, Xu C, Guo Y, Desta Z. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos. 2010;38:1218. doi: 10.1124/dmd.109.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug Metab Dispos. 2009;37:1793–6. doi: 10.1124/dmd.109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwara A, Lartey M, Sagoe KW, Kenu E, Court MH. CYP2B6, CYP2A6 and UGT2B7 genetic polymorphisms are predictors of efavirenz mid-dose concentration in HIV-infected patients. AIDS. 2009;23:2101–6. doi: 10.1097/QAD.0b013e3283319908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hariparsad N, Nallani SC, Sane RS, Buckley DJ, Buckley AR, Desai PB. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J Clin Pharmacol. 2004;44:1273–81. doi: 10.1177/0091270004269142. [DOI] [PubMed] [Google Scholar]
- 11.Approval documentation for Sustiva™ (Efavirenz) NDA no 020972. 2009. Avalable at http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020972s033,021360s02lbl.pdf (last accessed December 2009)
- 12.Backman JT, Granfors MT, Neuvonen PJ. Rifampicin is only a weak inducer of CYP1A2-mediated presystemic and systemic metabolism: studies with tizanidine and caffeine. Eur J Clin Pharmacol. 2006;62:451–61. doi: 10.1007/s00228-006-0127-x. [DOI] [PubMed] [Google Scholar]
- 13.Huang SM, Temple R, Throckmorton DC, Lesko LJ. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther. 2007;81:298–304. doi: 10.1038/sj.clpt.6100054. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Cortes LF, Ruiz-Valderas R, Viciana P, Alarcon-Gonzalez A, Gomez-Mateos J, Leon-Jimenez E, Sarasanacenta M, Lopez-Pua Y, Pachon J. Pharmacokinetic interactions between efavirenz and rifampicin in HIV-infected patients with tuberculosis. Clin Pharmacokinet. 2002;41:681–90. doi: 10.2165/00003088-200241090-00004. [DOI] [PubMed] [Google Scholar]
- 15.Pozniak AL, Miller RF, Lipman MC, Freedman AR, Ormerod LP, Johnson MA, Collins S, Lucas SB. BHIVA treatment guidelines for tuberculosis (TB)/HIV infection 2005. HIV Med. 2005;6(Suppl 2):62–83. doi: 10.1111/j.1468-1293.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 16.Brennan-Benson P, Lyus R, Harrison T, Pakianathan M, Macallan D. Pharmacokinetic interactions between efavirenz and rifampicin in the treatment of HIV and tuberculosis: one size does not fit all. AIDS. 2005;19:1541–3. doi: 10.1097/01.aids.0000183519.45137.a6. [DOI] [PubMed] [Google Scholar]
- 17.DiGiacinto JL, Chan-Tack KM, Robertson SM, Reynolds KS, Struble KA. Are literature references sufficient for dose recommendations? An FDA case study of efavirenz and rifampin. J Clin Pharmacol. 2008;48:518–23. doi: 10.1177/0091270008315308. [DOI] [PubMed] [Google Scholar]
- 18.Manosuthi W, Sungkanuparph S, Thakkinstian A, Vibhagool A, Kiertiburanakul S, Rattanasiri S, Prasithsirikul W, Sankote J, Mahanontharit A, Ruxrungtham K. Efavirenz levels and 24-week efficacy in HIV-infected patients with tuberculosis receiving highly active antiretroviral therapy and rifampicin. AIDS. 2005;19:1481–6. doi: 10.1097/01.aids.0000183630.27665.30. [DOI] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention. Managing Drug Interactions in the Treatment of HIV-Related Tuberculosis [online] 2007. Available at http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm (last accessed December 2009)
- 20.Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5:211–23. doi: 10.1517/17425250802691074. [DOI] [PubMed] [Google Scholar]
- 21.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140–8. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 22.Turner D. SimCYP User Manual Version 8. Sheffield: SimCYP Inc; 2008. [Google Scholar]
- 23.Jansson R, Bredberg U, Ashton M. Prediction of drug tissue to plasma concentration ratios using a measured volume of distribution in combination with lipophilicity. J Pharm Sci. 2008;97:2324–39. doi: 10.1002/jps.21130. [DOI] [PubMed] [Google Scholar]
- 24.Proctor N, Tucker G, Rostami-Hodjegan A. Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica. 2004;34:151–78. doi: 10.1080/00498250310001646353. [DOI] [PubMed] [Google Scholar]
- 25.Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab Dispos. 2002;30:1497–503. doi: 10.1124/dmd.30.12.1497. [DOI] [PubMed] [Google Scholar]
- 26.Shou M, Hayashi M, Pan Y, Xu Y, Morrissey K, Xu L, Skiles G. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab Dispos. 2008;36:2355. doi: 10.1124/dmd.108.020602. [DOI] [PubMed] [Google Scholar]
- 27.Faucette SR, Wang H, Hamilton GA, Jolley SL, Gilbert D, Lindley C, Yan B, Negishi M, LeCluyse EL. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32:348–58. doi: 10.1124/dmd.32.3.348. [DOI] [PubMed] [Google Scholar]
- 28.Mukonzo JK, Roshammar D, Waako P, Andersson M, Fukasawa T, Milani L, Svensson JO, Ogwal-Okeng J, Gustafsson LL, Aklillu E. A novel polymorphism in ABCB1 gene, CYP2B6*6 and sex predict single-dose efavirenz population pharmacokinetics in Ugandans. Br J Clin Pharmacol. 2009;68:690–9. doi: 10.1111/j.1365-2125.2009.03516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyakutira C, Roshammar D, Chigutsa E, Chonzi P, Ashton M, Nhachi C, Masimirembwa C. High prevalence of the CYP2B6 516G – >T(*6) variant and effect on the population pharmacokinetics of efavirenz in HIV/AIDS outpatients in Zimbabwe. Eur J Clin Pharmacol. 2008;64:357–65. doi: 10.1007/s00228-007-0412-3. [DOI] [PubMed] [Google Scholar]
- 30.Josephson F, Andersson MC, Flamholc L, Gisslen M, Hagberg L, Ormaasen V, Sonnerborg A, Vesterbacka J, Bottiger Y. The relation between treatment outcome and efavirenz, atazanavir or lopinavir exposure in the NORTHIV trial of treatment-naive HIV-1 infected patients. Eur J Clin Pharmacol. 2009;66:349–57. doi: 10.1007/s00228-009-0763-z. [DOI] [PubMed] [Google Scholar]
- 31.Csajka C, Marzolini C, Fattinger K, Decosterd LA, Fellay J, Telenti A, Biollaz J, Buclin T. Population pharmacokinetics and effects of efavirenz in patients with human immunodeficiency virus infection. Clin Pharmacol Ther. 2003;73:20–30. doi: 10.1067/mcp.2003.22. [DOI] [PubMed] [Google Scholar]
- 32.Turner DB, Rowland-Yeo K, Tucker GT, Rostami-Hodjegan A. Prediction of non-specific hepatic microsomal binding from readily available physicochemical properties. Drug Metab Rev. 2006;38:162. [Google Scholar]
- 33.Matteelli A, Regazzi M, Villani P, De Iaco G, Cusato M, Carvalho A, Caligaris S, Tomasoni L, Manfrin M, Capone S. Multiple-dose pharmacokinetics of efavirenz with and without the use of rifampicin in HIV-positive patients. Curr HIV Res. 2007;5:349–53. doi: 10.2174/157016207780636588. [DOI] [PubMed] [Google Scholar]
- 34.Itoh M, Nakajima M, Higashi E, Yoshida R, Nagata K, Yamazoe Y, Yokoi T. Induction of human CYP2A6 is mediated by the pregnane X receptor with peroxisome proliferator-activated receptor- coactivator 1. J Pharmacol Exp Ther. 2006;319:693. doi: 10.1124/jpet.106.107573. [DOI] [PubMed] [Google Scholar]
- 35.Barrett JS, Joshi AS, Chai M, Ludden TM, Fiske WD, Pieniaszek HJ., Jr Population pharmacokinetic meta-analysis with efavirenz. Int J Clin Pharmacol Ther. 2002;40:507–19. doi: 10.5414/cpp40507. [DOI] [PubMed] [Google Scholar]
- 36.Haas DW, Ribaudo HJ, Kim RB, Tierney C, Wilkinson GR, Gulick RM, Clifford DB, Hulgan T, Marzolini C, Acosta EP. Pharmacogenetics of efavirenz and central nervous system side effects: an Adult AIDS Clinical Trials Group study. AIDS. 2004;18:2391–400. [PubMed] [Google Scholar]
- 37.Haas DW, Smeaton LM, Shafer RW, Robbins GK, Morse GD, Labbe L, Wilkinson GR, Clifford DB, D'Aquila RT, De Gruttola V, Pollard RB, Merigan TC, Hirsch MS, George AL, Jr, Donahue JP, Kim RB. Pharmacogenetics of long-term responses to antiretroviral regimens containing Efavirenz and/or Nelfinavir: an Adult Aids Clinical Trials Group Study. J Infect Dis. 2005;192:1931–42. doi: 10.1086/497610. [DOI] [PubMed] [Google Scholar]
- 38.Rowe SM, Fontalbert L, Rabel SR, Maurin MB. Physical chemical properties of efavirenz. AAPS annual meeting and exposition. AAPS PharmSci Supplement. 1999 [Google Scholar]
- 39.Balani SK, Kauffman LR, deLuna FA, Lin JH. Nonlinear pharmacokinetics of efavirenz (DMP-266), a potent HIV-1 reverse transcriptase inhibitor, in rats and monkeys. Drug Metab Dispos. 1999;27:41–5. [PubMed] [Google Scholar]
- 40.Takano R, Sugano K, Higashida A, Hayashi Y, Machida M, Aso Y, Yamashita S. Oral absorption of poorly water-soluble drugs: computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm Res. 2006;23:1144–56. doi: 10.1007/s11095-006-0162-4. [DOI] [PubMed] [Google Scholar]