

Abstract
CCR5 is the major co-receptor used by HIV-1 and individuals homozygous for a 32bp deletion in CCR5 are profoundly resistant to HIV-1 infection. Using engineered zinc finger nucleases (ZFNs), we were able to disrupt the CCR5 gene in human hematopoietic stem/progenitor cells (HSC) at a mean frequency of 17% of total alleles in a population. This procedure produces both mono and bi-allelically disrupted cells. ZFN-treated HSC retained the ability to engraft NOD/SCID/IL2rγnull mice and gave rise to polyclonal multi-lineage progeny with the CCR5 gene permanently disrupted. Control mice receiving untreated HSC and challenged with CCR5-tropic HIV-1 displayed profound CD4+ T cell loss. In contrast, mice transplanted with ZFN-modified HSC underwent rapid selection for CCR5-negative cells, had significantly lower HIV-1 levels and preserved human cells throughout their tissues. The demonstration that a minority of CCR5-modified HSC can populate an infected animal with HIV-1-resistant, CCR5-negative progeny suggests the use of ZFN-modified autologous HSC as a clinical approach to treating HIV-1.
INTRODUCTION
The entry of HIV-1 into target cells involves the sequential binding of the viral gp120 Env protein to the CD4 receptor and a chemokine co-receptor (1). CCR5 is the major co-receptor used by HIV-1, and is expressed on key T cell subsets that are depleted during HIV-1 infection including memory T cells (2). A genetic mutation that deletes 32 bp from the CCR5 gene (CCR5Δ32) is relatively common in Western European populations and provides profound resistance to HIV-1 infection and AIDS in homozygotes (3,4). The absence of any other significant phenotype associated with a lack of CCR5 (5–7) has spurred the development of therapies aimed at blocking the virus-CCR5 interaction, and CCR5 antagonists have proved to be an effective salvage therapy in patients with drug-resistant strains of HIV-1 (8).
Recently, the ability of CCR5-negative hematopoietic stem/progenitor cells (HSC) to generate HIV-resistant progeny that can suppress HIV-1 replication in vivo was demonstrated in an HIV-infected patient undergoing HSC transplantation from a homozygous CCR5Δ32 donor during treatment for acute myeloid leukemia (9). The donor cells were able to confer long-term control of HIV-1 replication and to restore the patient’s CD4+ T cell levels in the absence of any antiretroviral drugs. This clinical finding provides powerful human data to support the potential of gene or stem cell therapies based on the elimination of CCR5. However, the risks associated with HSC transplantation, as well as the impracticality of obtaining sufficient numbers of matched CCR5Δ32 donors (10), mean that the more widespread use of this approach will require methods that allow the effective and safe generation of CCR5-negative cells in a patient’s own HSC.
Towards this goal, various gene therapy approaches to block CCR5 expression are currently being evaluated, including the expression in hematopoietic cells of CCR5-specific intrabodies, ribozymes and siRNAs (11–14). The targeted cell populations include both mature peripheral T cells or HSC, and the loss of CCR5 in HSC appears to have no adverse effects on hematopoiesis (12,13,15).
An alternative approach that we are pursuing is the use of engineered zinc finger nucleases (ZFNs) to permanently disrupt the CCR5 open-reading frame. ZFNs comprise a series of linked zinc finger peptides that can be engineered to bind to DNA sequences in a highly specific manner, fused to an endonuclease domain (16). The concerted binding of two juxtaposed ZFNs on the DNA, followed by dimerization of the two endonuclease domains, results in a double stranded break (DSB) at the targeted DNA sequence. Such DSBs are rapidly repaired by cellular repair pathways, most notably the mutagenic non-homologous end-joining (NHEJ) pathway, which leads to efficient disruption of the gene due to the addition or deletion of nucleotides at the break site (17,18). A significant advantage of this approach is that permanent gene disruption can occur following only transient expression of the ZFNs, so that a heritable and absolute CCR5-negative phenotype can be achieved with no requirement for the long-term expression of a foreign transgene.
CCR5-targeted ZFNs are currently being evaluated in a clinical trial targeting mature CD4+ T cells (19). However, disruption of CCR5 in HSC is likely to provide a more effective and long-term anti-viral effect, giving rise to CCR5-negative cells in both the lymphoid and myeloid compartments that HIV-1 infects. To evaluate this approach, we optimized the delivery of CCR5-specific ZFNs to human HSC and analyzed the consequences of this modification for the stem cells by transplantation into NOD/SCID/IL2rγnull (NSG) mice, which support both human hematopoiesis (20) and HIV-1 infection (13). Following challenge of the mice with a CCR5-tropic strain of HIV-1, we observed rapid selection for human cells that were CCR5-negative, a significant reduction in viral load and protection of human T cell populations in the key tissues that HIV-1 infects, including the gut mucosa. These findings suggest that ZFN engineering of autologous HSC could enable long-term control of HIV-1 replication in patients.
RESULTS
Efficient disruption of the CCR5 gene in human CD34+ HSC
ZFN activity generates a site-specific DSB in DNA that can be converted to a permanent gene disruption following the action of the cellular NHEJ repair pathway. Since DSB formation requires only transient expression of ZFNs, gene delivery methods that are suitable to express ZFNs include plasmid DNA nucleofection, integrase-defective lentiviral vectors and adenoviral vectors (16,19,21). Although non-viral methods such as nucleofection are attractive, this procedure has previously been associated with relatively high rates of toxicity in human CD34+ HSC and loss of engraftment potential (22), although more recently, less toxic outcomes have been described (23–25). We evaluated different parameters in order to identify nucleofection conditions that allowed the efficient disruption of the CCR5 gene by ZFNs, while limiting the associated toxicity. The extent of CCR5 disruption obtained was quantified using PCR amplification across the CCR5 locus, denaturing and re-annealing of the products, followed by digestion with the Cel 1 nuclease that preferentially cleaves DNA at regions of distorted duplexes caused by mismatches. This assay detects a linear range of CCR5 disruption between 0.69 and 44% of the total alleles in a population, and has an upper limit of sensitivity of approximately 70–80% disruption (19 and data not shown). Cel 1 analysis was used to assess the levels of CCR5 disruption since only a minority of human CD34+ HSC express CCR5 (26), so that it is not possible to monitor the effects of this modification by FACS analysis of the population.
Using CD34+ HSC harvested from umbilical cord blood and optimized nucleofection conditions, we achieved mean disruption rates of 17% +/- 10 (n=21) of the total CCR5 alleles in the HSC population (Fig. 1a). Similar results were also achieved using CD34+ cells isolated from fetal liver (data not shown). Previous studies in both cell lines (16) and primary human T cells (19) have shown that the percentage of bi-allelically modified cells in a population treated with ZFNs is between 30–40% of the total number of disrupted alleles detected by the Cel I assay, so that we estimate that 5–7% of the HSC would be homozygous CCR5-negative, although this was not directly measured.
Figure 1.

ZFN-mediated disruption of CCR5 gene in HSC. (a) Representative gel showing extent of CCR5 disruption in CD34+ HSC, 24 hours after cells were nucleofected with ZFN-expressing plasmids (ZFN) or mock nucleofected (mock). Neg. is untreated HSC. CCR5 disruption was measured by PCR amplification across the ZFN target site, followed by Cel I nuclease digestion and quantitation of products by PAGE. (b) Graph showing mean +/− SD percentage of human CD45+ cells in peripheral blood of mice at 8 weeks post-transplantation with either untreated, mock nucleofected, or ZFN nucleofected HSC, (n=5 each group). (c) FACS profiles of human cells from various organs of one representative mouse transplanted with ZFN-treated HSC. Cells were gated on FSC/SSC to remove debris. Staining for human CD45, a pan leukocyte marker, was used to reveal the level of human cell engraftment in each organ. CD45+ gated populations were further analyzed for subsets, as indicated: CD19 (B cells) in bone marrow, CD14 (monocytes/macrophages) in lung, CD4 and CD8 (T cells) in thymus and spleen, and CD3 (T cells) in the small intestine (lamina propria). The CD45+ population from the small intestine was further analyzed for CD4 and CCR5 expression. Peripheral blood cells from CD45+ and lymphoid gates were analyzed for CD4 and CD8 expression. The percentage of cells in each indicated area is shown. No staining was observed with isotype-matched control antibodies (Supplementary Fig. 1 online), or in animals receiving no human graft (data not shown).
At the same time, we evaluated the toxicity of the process by measuring the induction of apoptosis in the HSC cultures. Although we found that nucleofection increased toxicity 3-fold compared to untreated cells, the inclusion of the ZFN plasmids had no additional effect when compared to mock nucleofected controls (data not shown). Therefore, although nucleofection has some inherent toxicity for human CD34+ cells, the expression of ZFNs did not increase this further. Overall, we consider that any adverse effects on cell viability resulting from the nucleofection procedure may be offset by the high levels of CCR5 disruption achieved, as well as the speed and simplicity of the procedure when compared to viral vector systems (19,21).
ZFN-modified HSC are capable of multi-lineage engraftment in NSG mice
NSG mice can be engrafted with human HSC (20) and thereby provide a rigorous readout of the hematopoietic potential of genetically modified HSC. We evaluated the effects of nucleofection and/or CCR5 disruption on HSC by transplanting both untreated and ZFN-treated human HSC into 1-day old mice, previously irradiated with low-dose (150 cGy) radiation. Under these conditions, we observed rapid and efficient engraftment of human cells, typically resulting in 40% human CD45+ leukocytes in the peripheral blood of mice at 8 weeks post-transplantation. Additionally, the animals did not experience any obvious toxicity or ill health, as has been reported for animals receiving higher radiation doses (27). Using this optimized engraftment procedure, we found that ZFN-treated HSC engrafted NSG mice as efficiently as untreated HSC controls (Fig. 1b), with no statistically significant difference between the two groups (Student’s t test, p-value = 0.26).
Eight to 12 weeks following transplantation of the ZFN-treated HSC, we measured the extent of engraftment of various mouse tissues with human CD45+ leucocytes, as well as with cells from specific hematopoietic lineages (Fig. 1c). The human cells were detected using human-specific antibodies, whose specificity was confirmed using both unengrafted animals, as well as with isotype-matched antibody controls on engrafted animals (Supplementary Figure 1 online). High levels of human CD45+ cells were found in both the peripheral blood and tissues, typically ranging from 5–15% of the intestine, >50% of blood, spleen and bone marrow and >90% of the thymus (Supplementary Table 1). CD4+ and CD8+ T cells were present in multiple organs including the thymus, spleen, and both the intraepithelial and lamina propria regions of the small and large intestines. B cell progenitors were detected in the bone marrow and CD14+ macrophage/monocytes were detected in the lung. Of particular interest was the presence of a large population of human CD4+CCR5+ cells in the intestine, since T cells in the gut-associated lymphoid tissue (GALT) represent a significant population that is targeted by both HIV-1 in humans and SIV in primates (28–34). Overall, the profile of the human cells found in mice receiving ZFN-treated HSC was indistinguishable from mice transplanted with unmodified HSC, both in terms of the percent human cells in each tissue, as well as the frequencies of different subsets (Supplementary Table 1 online), suggesting that ZFN-modified HSC are functionally normal.
ZFN-treated HSC produce CCR5-disrupted progeny that persist through secondary transplantation
To evaluate whether ZFN treatment of the bulk CD34+ population was capable of modifying true SCID-repopulating HSC, we harvested the bone marrow of an animal 18 weeks after engraftment with ZFN-treated HSC. The extent of disruption of the human CCR5 gene in genomic DNA extracted from the bone marrow of this mouse was 11% (Table 1). The bone marrow was used to transplant three 8 week old recipients. At the same time, bone marrow was harvested from a control animal previously engrafted with untreated HSC, and was used to transplant 3 additional animals. Analysis of the peripheral blood of the secondary recipients 8 weeks later revealed that all 6 animals were engrafted and that there was no significant difference in the percentage of human CD45+ leukocytes between the ZFN-treated and control HSC groups. Furthermore, human cells in the blood of mice transplanted with bone marrow from the ZFN-treated donor had levels of CCR5 disruption that slightly exceeded the rate of gene disruption detected in the original donor marrow (range 12 to 20%) (Table 1). These data demonstrate that ZFN activity can lead to permanent disruption of the CCR5 gene in SCID-repopulating HSC, and that such modified cells retain their engraftment and differentiation potential.
Table 1.
Secondary transplantation of ZFN-treated HSC
| donor animalsa | % CD45b blood | % Cel 1c BM | secondary recipients | % CD45b blood | % Cel 1c blood |
|---|---|---|---|---|---|
| ZFN (1) | 41 | 11 | ZFN (3) | 34 +/− 5 | 16 +/− 4 |
| neg. (1) | 47 | 0 | neg. (3) | 37 +/− 7 | 0 +/− 0 |
Bone marrow (BM) was harvested from donor mice engrafted with ZFN-treated HSC (ZFN) or untreated HSC (neg.) and used to transplant three secondary recipients for each BM.
Levels of human CD45+ cells were measured in blood of both donor and recipient mice at 8 weeks post-transplantation.
CCR5 disruption rates, measured by Cel I analysis of donor BM at time of harvest, and in blood of recipient mice at 10 weeks post-transplantation.
Protection of CD4+ T cells in peripheral blood of NSG mice transplanted with ZFN-treated HSC following HIV-1 infection
Engrafted animals at 8–12 weeks post-transplantation, that had received either unmodified (neg.) or ZFN-treated HSC, were challenged with the CCR5-tropic virus HIV-1BAL. This strain of HIV-1 has previously been used in humanized mouse models, where it causes a robust infection and significant CD4+ T cell depletion (35,36). These observations mimic the human infection, where depletion of CD4+ CCR5+ lymphocytes results from a combination of direct infection, systemic immune activation and the upregulation of CCR5 on thymocytes (36,37,38). Following infection, blood samples were collected from the mice every two weeks and analyzed for HIV-1 RNA levels, T cell subsets and the extent of CCR5 disruption. At 8–12 weeks post-infection, animals were euthanized and necropsy analysis performed on multiple tissue samples (Supplementary Fig. 2 online).
Changes in the ratio of CD4+ to CD8+ T cells in the peripheral blood are characteristic of progressive infection in AIDS patients (39,40). We therefore examined the CD4:CD8 ratio in the peripheral blood samples from individual mice, both pre- and post-infection. We found that the mean CD4:CD8 ratio pre-infection was similar for both the untreated and ZFN-treated groups. Following HIV-1 challenge, the ratios became highly skewed in the control group due to the pronounced loss of CD4+ cells, while the ZFN-treated animals maintained normal ratios (Fig. 2a,b).
Figure 2.
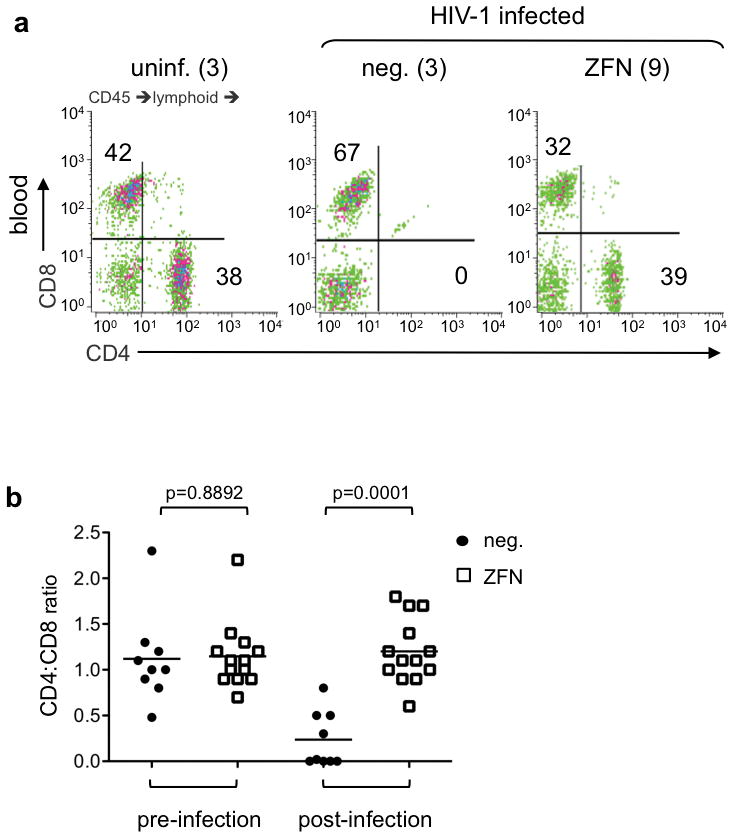
Protection of human CD4+ T cells in peripheral blood of HIV-infected mice transplanted with ZFN-modified HSC (a) FACS plots showing human CD4+ and CD8+ T cells in peripheral blood of representative animals from each of three cohorts: uninfected mice previously engrafted with either untreated or ZFN-treated HSC (uninf.), and HIV-1 infected animals previously engrafted with either untreated HSC (neg.) or ZFN-treated (ZFN) HSC, at 4 weeks post-infection. The total number of animals analyzed in each cohort is indicated. Cells were gated on FSC/SSC to remove debris, on human CD45, and a lymphoid gate applied. Percentage of cells in indicated compartments is shown. (b) Ratio of human CD4+ to CD8+ lymphocytes in peripheral blood of individual mice transplanted with untreated (neg.) or ZFN-modified HSC, measured pre-infection, and at 6–8 weeks post-infection. Statistical analysis comparing neg. and ZFN cohorts at each time-point is shown.
Protection of human cells and selection for CCR5-negative progeny following HIV-1 infection
We next analyzed the human cells present in various mouse tissues, 12 weeks after infection with HIV-1BAL. NSG mice transplanted with untreated HSC displayed a characteristic loss of certain human cell populations, while the ZFN-treated cohort retained normal FACS profiles throughout their tissues, despite HIV-1 challenge (Fig. 3a). In the intestines and spleen, which are the organs harboring the highest percentage of human CD4+CCR5+ cells in this mouse model (Supplementary Fig. 3 online), we observed specific depletion of the CD4+ T cells from the spleen, and the complete loss of all human lymphocytes from the intestines of the untreated HSC animals, while these populations were fully preserved in the ZFN-treated cohort (Fig. 3b). In the bone marrow, which is not a major target organ of HIV-1 infection, as expected, the levels of CD45+ cells were similar in each of the three groups.
Figure 3.

Effects of HIV-1 infection on human cells in transplanted NSG mice. (a) FACS analysis of human cells in tissues of representative NSG mice from three cohorts: uninfected mice previously engrafted with either untreated or ZFN-treated HSC (uninf.), and HIV-1 infected animals previously engrafted with either untreated HSC (neg.) or ZFN-treated (ZFN) HSC. Mice were necropsied at 12 weeks post-infection, or at the equivalent time-point for uninfected animals. The total number of animals analyzed in each cohort is indicated. FACS analysis was performed as described in Figure 1. Small intestine sample is lamina propria, and similar results were obtained when samples from the large intestine were analyzed. Percentage of cells in indicated compartments is shown. (b) Immunohistochemical analysis of human CD3 expression in small intestine (SI), and CD4 expression in spleen of representative NSG mice, transplanted with untreated (neg.) or ZFN-treated (ZFN) HSC, with and without HIV-1 infection. Animals were necropsied at 12 weeks post-infection, or at the same time-point for uninfected animals. Control animals receiving no human HSC (no graft) were also analyzed. The number of animals analyzed in each cohort is shown. Bars represent 50 μM.
Notably, HIV-1 infection resulted in the loss of virtually all human cells from the thymus of mice receiving untreated HSC by 12 weeks post-infection (Fig. 3a). Depletion of both single and double positive thymocytes has been reported to occur as a consequence of the up-regulation of CCR5 on these cells following HIV-1 infection (36,38), and likely contributed to both the observed depletion of the thymus, as well as a reduction in the numbers of mature CD4+ and CD8+ T cells observed in other tissues.
HIV-1 infection rapidly selects for CCR5-negative T cells
We examined whether the survival of human T cells during HIV-1BAL infection in mice receiving ZFN-treated HSC was the result of selection for ZFN-modified CCR5-negative cells, and not just the survival of cells that were phenotypically CCR5-negative, by measuring the percentage of CCR5-disrupted alleles in the blood of mice at sequential time-points following HIV-1 challenge. We used both the Cel 1 heteroduplex mismatch assay and a specific PCR amplification that detects a 5bp duplication at the ZFN target site that typically accounts for 10–30% of total modifications induced by the CCR5 ZFNs (19). Both assays revealed rapid selection for ZFN-disrupted alleles, reaching the upper limit of the Cel 1 assay by 4 weeks post-infection (Fig. 4a).
Figure 4.

HIV-1 infection selects for disrupted CCR5 alleles. (a) Mean +/- SD levels of CCR5 disruption (Cel 1 assay, black bars) in sequential peripheral blood samples taken from mice transplanted with ZFN-treated HSC and infected with HIV-1. Upper limit of linearity of Cel I assay is 44% (19) and is indicated by the dotted line; upper limit of sensitivity of assay is 70–80%. White bars show the frequency of a common 5bp duplication at the ZFN target site that typically comprises 10–30% of total CCR5 mutations (19). Numbers of mice analyzed at each time point, and in each assay, are shown above the appropriate bar. (b) Mean +/− SD levels of CCR5 disruption (Cel I assay) in indicated tissues from mice transplanted with ZFN-treated HSC and necropsied at 12 weeks post-infection (black bars), or at an equivalent time-point for uninfected ZFN-treated animals (white bars). Numbers analyzed in each group are shown above the appropriate bar. One representative Cel I analysis from the large intestine (lamina propria) of uninfected and infected mice is shown. Animals receiving untreated HSC gave no Cel I digestion products at any time-point analyzed (data not shown). Asterisk indicates levels too low to quantify. (c) Contour FACS analyses of human CD4+ cells in the small intestine (lamina propria) and spleen of one representative animal from each indicated cohort are shown. Cells were gated on FSC/SSC to remove debris and gated on human CD45 and CD4. Numbers indicate the percentage of cells that are CCR5+. (d) Mean +/− SD numbers of human CD4+ cells (grey bars) and CD4+CCR5+ cells (white bars) per 5,000 human CD45+ cells analyzed from different sections of the intestine, and from the indicated cohorts. Asterisk indicates levels too low to quantify. Number of animals analyzed in each cohort is indicated. Abbr. S, small intestine; L, large intestine; E, intraepithelial lymphocytes; P lamina propria lymphocytes; BM, bone marrow.
We also examined the levels of CCR5 disruption in multiple tissues from ZFN-treated animals, either uninfected, or 12 weeks after HIV-1BAL challenge. This analysis revealed a dramatic increase in the extent of CCR5 disruption caused by HIV-1 infection (Fig. 4b). In addition, FACS analysis of CD4+ cells from the spleen and both the small and large intestine revealed that in contrast to uninfected animals, where approximately 25% of CD4+ cells were also CCR5+, very little or no CCR5 expression was detected in the CD4+ T cells that persisted in the ZFN-treated animals (Fig. 4c,d). Together, these data suggest that the protection of CD4+ lymphocytes in ZFN-HSC transplanted mice is a consequence of the selection for CCR5-disrupted, HIV-1 resistant cells that are derived from ZFN edited HSC.
Heterogeneity of CCR5 modifications suggests polyclonal origins
ZFN-induced DSBs repaired by the NHEJ pathway result in highly heterogeneous changes at the targeted locus (19). We used this property to investigate whether the CCR5-negative cells that developed in mice transplanted with ZFN-treated HSC were polyclonal in origin. Sequencing of 60 individual CCR5 alleles amplified from the large intestine lamina propria of an HIV-1-infected mouse transplanted with ZFN-treated HSC revealed that 59 out of the 60 alleles analyzed harbored mutations at the ZFN target site (Fig. 5). As previously reported for this ZFN pair (19), a high proportion (13 out of 59) of the mutated loci contained a characteristic 5bp duplication, with the remaining 46 clones comprising 36 unique sequences. In contrast, all alleles sequenced from a mouse receiving untreated HSC contained the wild-type sequence (data not shown). The high degree of diversity observed for these mutations strongly suggests that multiple stem or progenitor cells had been modified by the ZFNs. These findings also predict that the overwhelming majority of cells selected by HIV-1BAL infection would be homozygous for CCR5 gene disruption and consequently phenotypically CCR5-negative, which is in agreement with the data from CCR5 FACS analysis (Fig. 4c).
Figure 5.

ZFN activity produces heterogeneous mutations in CCR5. Sequence analysis was performed on 60 cloned human CCR5 alleles, PCR amplified from intraepithelial cells from the large intestine of an HIV-infected mouse previously transplanted with ZFN-treated HSC, and at 12 weeks post-infection. The number of nucleotides deleted or inserted at the ZFN target site in each clone is indicated on the right of each sequence, together with the number of times the sequence was found. Dashes (-) indicate deleted bases compared to the wildtype sequence, uppercase letters are point mutations, underlined upper case letters are inserted bases. Some specific mutations of CCR5 occurred more frequently, in particular a 5bp duplication at the ZFN target site that was identified 13 times (bottom sequence). No mutations in CCR5 were observed in a similar analysis performed on control samples from a mouse receiving non-modified HSC (data not shown).
Presence of ZFN-modified cells controls HIV-1 replication in vivo
HIV-1 levels were measured in the peripheral blood of infected animals at multiple time-points following infection, and in tissues harvested at necropsy between 8 and 12 weeks post-infection. Quantitative PCR analysis of HIV-1 RNA in peripheral blood revealed that peak viremia occurred at 6 weeks post-infection for animals transplanted with either untreated or ZFN-treated HSC (Fig. 6a), although the levels were significantly lower in the ZFN cohort. By 8 weeks post-infection, viral loads in both cohorts were dropping but there continued to be a statistically significant difference between the two groups (p = 0.001). Measurements of p24 levels in the blood by ELISA corroborated these findings, with a significant difference in antigenemia observed as early as the 6 week time point (data not shown).
Figure 6.
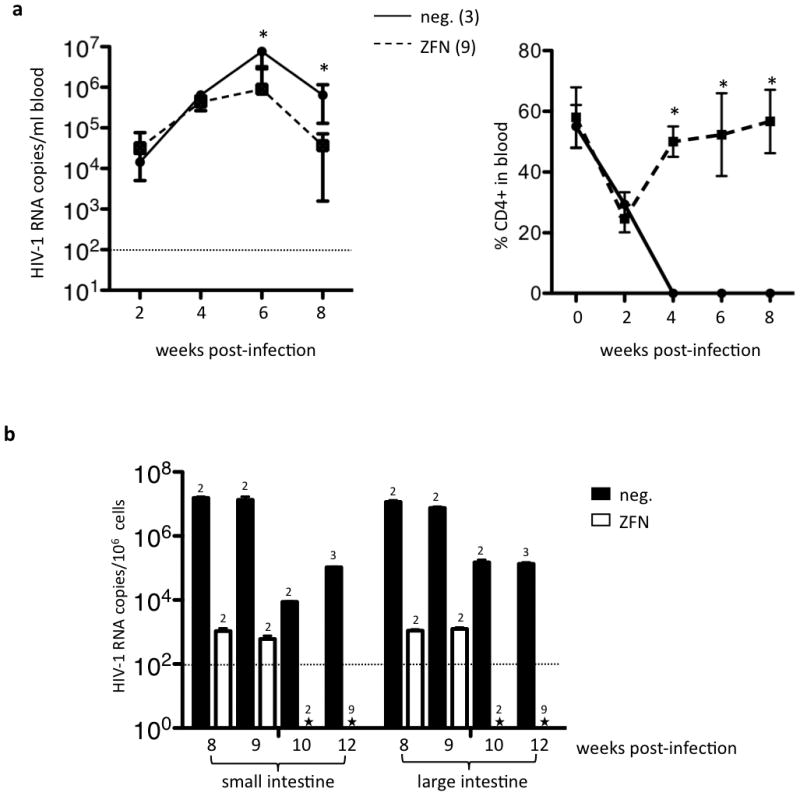
Control of HIV-1 replication in mice receiving ZFN-treated HSC. (a) Mean +/− SD levels of HIV-1 RNA (left) and percent CD4+ human T cells (right) in peripheral blood of mice transplanted with untreated (neg.) or ZFN-treated HSC, at indicated times post-infection. Dashed line is limit of detection of assay. Asterisk indicates a statistically significant difference between two groups (p<0.05). (b) Mean +/− SD HIV-1 RNA levels in small and large intestine lamina propria from neg. or ZFN mice, from animals necropsied between 8 and 12 weeks post-infection. Numbers of mice analyzed at each time point are shown above the appropriate bar. Dashed line indicates limits of detection of assay. Asterisk indicates undetectable levels.
These differences in virus levels between the two cohorts are even more striking when the levels of human CD4+ T cells are also considered (Fig. 6a), since the loss of CD4+ T cells in the untreated mice by 4 weeks post-infection likely contributed to the lowering of overall viral levels seen as the infection progressed. This continued presence of virus in the blood, despite the acute loss of CD4+ cells, is reminiscent of observations in patients, were high viral load measurements in serum are typically observed during progression to AIDS, when T cell death is rapidly occurring (41). In contrast, CD4+ T cell levels rebounded in the ZFN-treated mice after the 2 week nadir, and recovered to normal levels by 4 weeks post-infection. Serum HIV-1 levels began to decline in these animals by 8 weeks post-infection. In contrast to these findings with HIV-1BAL, challenge of ZFN HSC mice with a CXCR4-tropic HIV-1 strain did not result in any decrease in virus levels at 8 weeks, and no preservation of CD4 T cells, confirming that this anti-viral mechanism is CCR5 specific (Supplementary Fig. 4 online).
We also measured HIV-1 loads in intestinal samples. In tissues harvested at 8–9 weeks post-infection, we observed up to 4 orders of magnitude lower levels of virus in the ZFN-treated animals when compared to the untreated HSC controls, and this fell to undetectable levels in the ZFN mice by 10 weeks post-infection (Fig. 6c). This suppression of HIV-1 replication occurred despite the maintenance of normal numbers of human T lymphocytes in the intestines and other tissues for the ZFN cohort (Fig. 3). These observations are consistent with a strong selective pressure for CCR5-negative cells during active HIV-1 replication, leading to the suppression of virus replication in the mice as susceptible, CCR5-expressing cells are replaced by an HIV-resistant, CCR5-negative population.
DISCUSSION
Despite major advances in anti-retroviral therapy (ART), HIV-1 infection remains an epidemic cause of morbidity and mortality. Effective ART often involves costly, multi-drug regimens that are not well tolerated by a significant percentage of patients (42), and even successful adherence to ART does not eradicate the virus, so that a rapid rebound in HIV-1 levels can occur if therapy is discontinued (43). Consequently, an alternate approach to controlling HIV-1 replication that is being considered is the engineering of the body’s immune cells to be resistant to infection by the virus (44). In this regard, the CCR5 co-receptor is a particularly attractive target because of the HIV-resistant phenotype of homozygous CCR5Δ32 individuals (3), who tolerate this genotype with minimal consequences (5–7). In the present study, we identified conditions that allowed the efficient disruption of the CCR5 gene in human CD34+ HSC, and further demonstrated that such modified HSC were able to generate CCR5-negative, HIV-resistant progeny in a mouse model of human hematopoiesis and HIV-1 infection, ultimately leading to the profound suppression of HIV-1 replication in vivo. These findings suggest that modification of a patient’s own HSC by CCR5-specific ZFNs could provide a permanent supply of HIV-resistant progeny, able to replace cells killed by HIV-1, and leading to immune reconstitution and the long-term control of virus replication in the absence of anti-retroviral drugs.
The high levels of CCR5 disruption that we achieved in human HSC were possible because of an efficient gene editing technology based on zinc finger nucleases. ZFNs can be designed to bind to a specific genomic DNA sequence and, by generating a targeted DSB, can affect the efficient, specific and permanent knockout of the targeted gene (19,45–47). In contrast to other genetic approaches to the control of HIV-1, ZFN-mediated gene disruption is a one-time, “hit-and-run” process that requires only transient expression of the ZFNs in HSC during a brief period of ex vivo culture. Long-term expression of a therapeutic transgene is not required and, once established, the genetic mutation is present for the life of the cell and its progeny. In this way a major shortcoming of other gene therapy technologies, the need for continued expression of a foreign transgene, is avoided. Moreover, unlike small molecule, antibody, or RNAi-based treatments (44), ZFN treatment results in disruption of the CCR5 open-reading frame and thus has the potential to completely eliminate this important HIV-1 co-receptor from the surface of cells that are bi-allelically modified. By using an optimized nucleofection procedure, we were able to overcome the technical challenges to ZFN-induced genome editing in HSC that have previously been reported (21) and achieve, on average, disruption at 17% of the loci.
The safety and efficacy of CCR5-targeted ZFNs are currently being evaluated in a Phase I clinical trial using T lymphocytes. Pre-clinical studies included an extensive investigation into the specificity of these ZFNs for the CCR5 locus. Off-target events were only observed at significant levels at the homologous CCR2 locus (19). Mouse studies have not found any deleterious phenotype associated with loss of CCR2 (48), and human genetic studies have even suggested a beneficial phenotype from the loss of this gene in HIV-infected individuals (49). Although not specifically analyzed in our study, it is likely that the use of these same CCR5 ZFN reagents in HSC will result in similar, low levels of off-target events, the consequences of which will need to be evaluated in a larger study.
Although T lymphocytes are the primary target of HIV-1 infection, HSC may represent a more attractive cell for ZFN engineering since their modification could allow the long-term production of CCR5-negative cells in a patient. The scientific rationale for CCR5 modification of HSC is strong and is supported by the recent finding that an AIDS leukemia patient undergoing HSC transplantation from a CCR5-negative donor was effectively cured of his infection, with undetectable levels of HIV-1 and rebounds in CD4 T cell levels despite discontinuing ART (9). As shown by our data, ZFN-modified HSCs retain full functionality and give rise to CCR5-negative cells in lineages relevant to HIV-1 pathogenesis, such as T cells and monocyte/macrophages. ZFNs delivered to purified CD34+ cell populations by nucleofection were clearly capable of modifying true SCID-repopulating stem cells, and the high levels of CCR5 editing that we observed in the progeny of engrafted HSC were also maintained following secondary transplantations. Thus, the technology appears well suited to this application.
The experimental mouse model of HIV-1 infection used in these studies revealed a strong selection for CCR5-negative progeny during acute infection with a CCR5-tropic strain of HIV-1. This suggests that homozygous CCR5-negative stem cells, even if the minority, produce sufficient numbers of CCR5-negative progeny to ultimately support immune reconstitution and inhibit HIV-1 replication. Such selection is consistent with clinical observations from genetic diseases such as ADA-SCID, X-linked SCID and Wiskott-Aldrich syndrome, where normal hematopoietic cells have a selective advantage, so that spontaneous monoclonal reversions that have occurred in a small number of patients can lead to the selective outgrowth of these cells and amelioration of symptoms (50–53).
The observation of almost complete replacement of human T cells in the intestines of infected mice with CCR5-negative cells is consistent with this tissue harboring the majority of the body’s CD4+CCR5+ effector memory cells. A characteristic feature of HIV-1 replication in mucosal tissues is an ongoing cycle of T cell death and the accompanying recruitment of replacement T cells, whose activated state means that they are also highly permissive targets for HIV-1 infection (37). This is especially true in the GALT, which represents a key battleground in HIV-1 infection (54–56). We also noted a strong selective pressure for CCR5-negative cells at the level of the thymus. This suggests that T cells would be selected to be CCR5-negative at both a precursor stage in the thymus, as well as the effector CD4+ stage in the mucosa. Ultimately, the presence of HIV-resistant CCR5-negative cells in mucosal tissues would both protect individual cells from infection, and also help to break the cycle of immune hyperactivation that may underlie much of the pathology of AIDS (57).
Does ZFN modification of HSC present a reasonable treatment option? Although ART is clearly highly effective in many patients, the associated costs and potential for side effects can be considerable when extrapolated over the lifetime of a patient. In contrast, a ZFN-engineered HSC therapy could potentially provide a one-shot treatment that would be most suited to the setting of autologous HSC transplantation. Procedures for the phenotypic isolation and processing of HSC for autologous or allogeneic transplantation are well established. The use of a patient’s own stem cells may remove the requirement for full ablation of the marrow hematopoietic compartment, and the immune suppression that is necessary for allogeneic matched donor bone marrow transplantations. Indeed, the accompanying toxicity of such regimens is one of the reasons why allogeneic stem cell transplantations from CCR5Δ32 individuals will not be a feasible treatment option for HIV patients in the absence of other conditions that would necessitate the transplant.
Of note, certain HIV-infected patient populations, such as AIDS lymphoma patients, already undergo full ablation and autologous HSC rescue as part of their therapy (58), and thereby present an opportunity for HSC-based gene therapies (44). In addition, the experience of autologous HSC transplantation in gene therapy for patients with genetic diseases such as ADA-SCID (59–60), chronic granulomatous disease (61) and X-linked adrenoleukodystrophy (62), is that non-myeloablative conditioning can be used to facilitate engraftment of gene-modified autologous HSC with minimal associated toxicity. It is possible that the use of non-myeloablative regimens, together with the selective advantage conferred on CCR5-negative progeny, could prove an effective combination for HIV-1 patients receiving ZFN-treated autologous HSC.
Targeting the CCR5 gene is not expected to provide protection against viruses that use alternate co-receptors such as CXCR4. However only a handful of cases of HIV-1 infection of CCR5Δ32 homozygotes have been reported (63,64). CXCR4-tropic viruses have been associated with accelerated disease progression (65), so that selection for such strains could be an undesirable consequence of targeting CCR5. However, this outcome is not generally observed in patients treated with CCR5 inhibitors unless pre-existing CXCR4-tropic viruses were present before therapy, and resistance to the drugs can occur, instead, because of viral adaptations to the drug-bound form of the CCR5 co-receptor (66,67). Interestingly, the patient receiving the CCR5Δ32 HSC transplantation harbored CXCR4-utilizing viruses before the procedure, yet he did not experience a tropism switch, and instead, successfully controlled his HIV-1 infection long-term (9,10). For the present, similar to the recommendations for the use of CCR5 inhibitor drugs, it may be prudent to restrict CCR5 ZFN treatment of HSC to individuals with no detectable CXCR4-tropic viruses.
In contrast to the acute HIV-1 infection modeled in this study, HIV-1 patients usually present in a chronic phase of the disease, and will frequently have virus levels that are effectively controlled by ART. The requirement for the selective pressure of active HIV-1 replication in the success of this, or other anti-HIV gene therapies is presently unknown. It has been suggested that low level virus replication continues in certain sanctuary sites, even in well-controlled patients on ART (43,68), and this could provide a low level of selection, although drug intensification trials have not provided evidence of any such ongoing HIV-1 replication (69). It is also possible that the high levels of CCR5 disruption we achieved without selection, if extrapolated to patients, could be sufficient to provide a therapeutic effect even in the absence of a strong selective pressure. Alternatively, ZFN knockout of CCR5 in HSC could be viewed as a backup strategy, expected to play an increasingly important role in controlling HIV-1 if ART fails or is withdrawn. It may also be possible to incorporate ART treatment interruptions into an overall therapeutic strategy, as recently described for HIV-infected individuals receiving autologous HSC engineered with anti-HIV ribozymes, where gene marked progeny were found at higher levels in patients who underwent treatment interruptions (70).
In summary, these data demonstrate for the first time that transient ZFN treatment of human CD34+ HSC can result in efficient levels of gene disruption, while yielding cells that remain competent to engraft and support hematopoiesis. In the presence of CCR5-tropic HIV-1, CCR5-negative progeny rapidly replaced cells depleted by the virus, leading to a polyclonal population that ultimately preserved human immune cells in multiple tissues. Our findings indicate that the modification of only a minority of human HSC was sufficient to provide the same strong anti-viral benefit as was conferred by a complete CCR5Δ32 stem cell transplantation in a patient (9). This suggests that a partially modified autologous transplant, administered under only mildly ablative transplantation regimens, may also be effective, opening up the treatment to many more HIV-infected individuals. Finally, the identification of conditions that allow the efficient use of ZFNs in human HSC suggests the use of this technology in other diseases for which HSC modification may be curative.
METHODS
Hematopoietic stem cell isolation
Human CD34+ HSC were isolated from umbilical cord blood collected from normal deliveries at local hospitals, according to guidelines approved by the Childrens Hospital Los Angeles Committee on Clinical Investigation, or as waste cord blood material from StemCyte Corp. (Covina, CA). Immunomagnetic enrichment for CD34+ cells was performed using the magnetic-activated cell sorting (MACS) system (Miltenyi Biotec, Auburn, CA), per the manufacturer’s instructions, with the modification that the initial purified CD34+ population was put through a second column and washed 3 times with 3 ml of the supplied buffer per wash before the final elution. This additional step gave a greater than 99% pure CD34+ population, as measured by FACS analysis using the anti-CD34 antibody, 8G12 (BD Biosciences, San Jose, CA).
Nucleofection of HSC with ZFN expression plasmids
Freshly isolated CD34+ cells were stimulated for 5–12 hours in X-VIVO 10 media (Lonza, Basel, Switzerland) containing 2nM L-glutamine, 50ng/ml SCF, 50 ng/ml Flt-3 and 50ng/ml TPO (R & D Systems, Minneapolis, MN). One × 106 cells were nucleofected with 2.5 μg each of a plasmid pair expressing ZFNs binding upstream (ZFN-L) or downstream (ZFN-R) of codon Leu55 within TM1 of human CCR5 (19). The CD34+ cell/DNA mix was processed in an × series Amaxa Nucleofector (Lonza) using the U-01 setting and the human CD34+ nucleofector solution, according to the manufacturer’s instructions. Following nucleofection, cells were immediately placed in pre-warmed IMDM media (Lonza) containing 26% fetal bovine serum (Mediatech, Inc. Manassas, VA), 0.35% BSA, 2nM L-glutamine, 0.5% 10−3 Mol/L hydrocortisone (Stem cell technologies, Vancouver, BC), 5 ng/ml IL-3, 10 ng/ml IL-6 and 25 ng/ml SCF (R & D Systems). Cells were allowed to recover in this media for 2–12 hours before injection into mice.
Apoptosis assay
CD34+ HSC were collected at 24 hr. post-nucleofection and analyzed for the percent of viable cells marked for apoptosis using the PE apoptosis detection kit (BD Biosciences) according to the manufacturer’s instructions. Cells were stained with 7-AAD (detects viable cells) and annexin V (detects apoptotic cells) and analyzed using a FACScan flow cytometer (BD Biosciences). This double staining allowed the identification of cells in the early stages of apoptosis.
NSG mouse transplantation
NOD.Cg-Prkdc scid Il2rg tm1Wj/SzJ (NOD/SCID/IL2rγnull, NSG) mice (71) were obtained from Jackson Laboratories (Bar Harbor, ME). Neonatal mice within 48 hours of birth received 150 cGy radiation, then 2–4 hours later 1 × 106 ZFN-modified or mock-treated human CD34+ cells in 50 μl PBS containing 1% heparin were injected via the facial vein. For secondary transplantations, bone marrow was harvested by needle aspiration from the upper and lower limbs of 18 week old animals previously engrafted with human CD34+ cells, filtered through a 70 μm nylon mesh screen (Fisher Scientific, Pittsburg, PA), and washed in PBS. The cells were used to transplant three 8-week old mice that had previously received 350 cGy radiation, using retro-orbital injection of 2 × 107 bone marrow cells per mouse.
Analysis of CCR5 disruption
The percentage of CCR5 alleles disrupted by ZFN treatment was measured by performing PCR across the ZFN target site followed by digestion with the Surveyor (Cel 1) nuclease (Transgenomic, Omaha, NE), which detects heteroduplex formation, as previously described (19). Briefly, genomic DNA was extracted from mouse tissues and subject to nested PCR amplification using human CCR5 specific primers, with the resulting radiolabeled products digested with Cel I nuclease and resolved by PAGE. The ratio of cleaved to uncleaved products was calculated to give a measure of the frequency of gene disruption. The assay is sensitive enough to detect single-nucleotide changes and has a linear detection range between 0.69 and 44% (19).
In addition, a common 5bp (pentamer) duplication that occurs following NHEJ repair of the ZFN-cleaved CCR5 locus (19) was detected by PCR. The first round PCR product generated during Cel I analysis was diluted 1:5000 and 5 μl used in a Taqman qPCR reaction using primers (5′-GGTCATCCTCATCCTGATCTGA-3′ and 5′-GATGATGAAGAAGATTCCAGAGAAGAAG-3′ ) and probe (5′-FAM d(CCTTCTTACTGTCCCCTTCTGGGCTCAC) BHQ-1-3′ (Biosearch Technologies, Novato, CA), and analyzed using a 7900HT real time PCR machine (Applied Biosystems, Foster City, CA). At the same time, 5 μl of a 1:50,000 dilution of the PCR product were used in a Taqman qPCR reaction using primers (5′-CCAAAAAATCAATGTGAAGCAAATC-3′ and 5′-TGCCCACAAAACCAAAGATG -3′) and probe (5′-FAM d(CAGCCCGCCTCCTGCCTCC) BHQ-1-3′ (Biosearch Technologies), in order to detect total copies of the human CCR5 gene. Data was analyzed using software supplied by the manufacturer (Applied Biosystems) and the frequency of pentamer insertions in the CCR5 gene calculated. The assay is sensitive enough to detect a single pentamer insertion event in 100,000 cells (data not shown).
ZFN-induced modifications of the CCR5 gene were analyzed by directly sequencing cloned CCR5 alleles, isolated by PCR amplification as described above, and TOPO-TA cloning (Invitrogen). Plasmid DNA was isolated from 60 individual bacterial colonies for each tissue analyzed.
HIV-1 infection and analysis
A cell-free virus stock of HIV-1BaL and a molecular clone of HIV-1NL4-3 were obtained from the AIDS Research and Reference Reagent Program (ARRRP), Division of AIDS, NIAID, NIH from material deposited by Dr. Suzanne Gartner, Dr. Mikulas Popovic, Dr. Robert Gallo and Dr. Malcolm Martin. HIV-1BaL virus was propagated in PM1 cells, obtained from the ARRRP and deposited by Dr. Marvin Reitz and harvested 10 days post-infection. HIV-1NL4-3 viruses were generated by transient transfection of 293T cells (ATCC, Manassas, VA). Viruses were titrated using the Alliance HIV-1 p24 ELISA kit (PerkinElmer, Waltham, MA) and by TCID50 analysis on U373-MAGI cells (ARRRP, deposited by Dr. Michael Emerman and Dr. Adam Geballe). Mice to be infected with HIV-1 were anesthetized with inhalant 2.5% isoflourane, and injected intraperitoneally with virus stocks containing 200 ng p24, 7 × 104 TCID50 units, in 100 μl total volume.
HIV-1 levels in peripheral blood or tissues harvested at necropsy were determined by extracting RNA from 5 × 105 cells using the master pure complete DNA and RNA purification kit (Epicentre Biotechnologies) and performing Taqman qPCR using a primer and probe set targeting the HIV-1 LTR region, as previously described (72). In addition, p24 levels were measured in blood samples by ELISA.
Mouse blood and tissue collection
Peripheral blood samples were collected every 2 weeks starting at 8 weeks of age, using retro-orbital sampling. Whole blood was blocked in FBS (Mediatech, Inc.) for 30 min., the red blood cells were lysed using Pharmlyse solution (BD Biosciences), and cells were washed with PBS. Tissue samples were collected at necropsy and processed immediately for cell isolation and FACS analysis, or kept in freezing media (IMDM plus 20% DMSO) in liquid nitrogen, for later analysis and DNA extraction. Tissue samples were manually agitated in PBS before filtering through a sterile 70 μm nylon mesh screen (Fisher Scientific) and suspension cell preparations produced as previously described (19). Intestinal samples were processed as previously described (73), with the modification that the mononuclear cell population was isolated following incubation in citrate buffer and collagenase enzyme for 2 hrs, followed by nylon wool filtration (Amersham Biosciences, Sweden) and ficoll-hypaque gradient isolation (GE Healthcare, Uppsala, Sweden).
Analysis of human cells in mouse tissues
FACS analysis of human cells was performed using a FACSCalibur instrument (BD Biosciences) with either BD CellQuest Pro version 5.2 (BD Biosciences) or FlowJo software version 8.8.6 for Macintosh (Treestar, Ashland, OR). The gating strategy performed was an initial forward scatter versus side scatter gate to exclude debris, followed by a human CD45 gate. For analysis of lymphocyte populations in peripheral blood, a further lymphoid gate (low side scatter) was also applied to exclude cells of monocytic origin (74). All antibodies used were fluorochrome conjugated and human specific, and obtained from BD Biosciences: CD45 (clone 2D1), CD19 (clone HIB19), CD14 (clone MϕP9), CD3 (clone SK7), CD4 (clone SK3), CD8 (clone HIT8a), CCR5 (2D7). Gates were set using fluorescence minus one controls, where cells were stained with all antibodies except the one of interest. Specificity was also confirmed using isotype matched non-specific antibodies (BD Biosciences) (Supplementary Figure 1 online) and with tissues from animals that had not been engrafted with human cells.
Immunohistochemical analysis of human CD3 and CD4 expression, respectively, in the small intestine and spleen tissue from HSC engrafted mice was performed on fixed paraffin embedded tissue sections, as previously described (73,75). Controls included isotype matched non-specific antibodies, and unengrafted NSG mice.
Statistical analysis
All statistical analysis was performed using GraphPad Prism version 5.0b for Mac OSX (GraphPad Software, San Diego, California). Unpaired two-tailed t-tests were performed assuming equal variance to calculate p-values. A 95% confidence interval was used to determine significance. A minimum of three data points was used for each analysis.
Supplementary Material
Acknowledgments
We would like to thank, Andrew Cuddihy, Shundi Ge, Roger Hollis and Nancy Smiley for expert technical assistance, Carolyn Lutzko, Victor Garcia, Ramesh Akkina, Bruce Torbett and Mike McCune for advice regarding humanized mice and Mike McCune, Victor Garcia and Ron Veazey for communicating unpublished data. This work was supported by funding from the California HIV/AIDS Research Project (P.M.C.), The Saban Research Institute (V.T.), and the National Heart Lung and Blood Institute P01 HL73104 (G.M.C., D.B.K. and P.M.C.).
Footnotes
AUTHOR CONTRIBUTIONS
N.H. performed most of the experiments; J.W., K.K., G.F. and X.W. developed assays and analyzed samples; V.T. contributed to discussions; N.H., G.M.C., D.B.K., P.D.G., M.C.H. and P.M.C. designed the experiments and analyzed data; N.H and P.M.C. wrote the manuscript.
COMPETING INTERESTS STATEMENT
The following authors are full-time employees of Sangamo BioSciences, Inc.; J.W., K.K., G.F, P.D.G and M.C.H.
References
- 1.Wu L, et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;6605:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 2.deRoda Husman AM, Blaak H, Brouwer M, Schuitemaker H. CC chemokine receptor 5 cell-surface expression in relation to CC chemokine receptor 5 genotype and the clinical course of HIV-1 infection. J Immunol. 1999;163:84597–84603. [PubMed] [Google Scholar]
- 3.Samson M, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;6593:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 4.Novembre J, et al. The geographic spread of the CCR5 Delta32 HIV-resistance allele. PLoS Biol. 2005;3:1954–1962. doi: 10.1371/journal.pbio.0030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glass WG, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantarci OH, et al. CCR5Δ32 polymorphism effects on CCR5 expression, patterns of immunopathology and disease course in multiple sclerosis. J Neuroimmunol. 2005;169:137–143. doi: 10.1016/j.jneuroim.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Rossol M, et al. Negative association of the chemokine receptor CCR5 d32 polymorphism with systemic inflammatory response, extra-articular symptoms and joint erosion in rheumatoid arthritis. Arthritis Res Ther. 2009;11:91–98. doi: 10.1186/ar2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dau B, Holodiny M. Novel targets for antiretroviral therapy: clinical progress to date. Drugs. 2009;69:31–50. doi: 10.2165/00003495-200969010-00003. [DOI] [PubMed] [Google Scholar]
- 9.Hutter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 10.Hutter G, Schneider T, Thiel E. Transplantation of selected or transgenic blood stem cells - a future treatment for HIV/AIDS? J Int AIDS Soc. 2009;12:10–14. doi: 10.1186/1758-2652-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson J, et al. Safety and efficacy of a lentiviral vector containing three anti-HIV genes--CCR5 ribozyme, tat-rev siRNA, and TAR decoy--in SCID-hu mouse-derived T cells. Mol Ther. 2007;15:1182–1188. doi: 10.1038/sj.mt.6300157. [DOI] [PubMed] [Google Scholar]
- 12.Bai J, et al. Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol Ther. 2000;3:244–254. doi: 10.1006/mthe.2000.0038. [DOI] [PubMed] [Google Scholar]
- 13.Kumar P, et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swan CH, et al. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006;20:1480–1492. doi: 10.1038/sj.gt.3302801. [DOI] [PubMed] [Google Scholar]
- 15.Swan CH, Torbett BE. Can gene delivery close the door to HIV-1 entry after escape? J Med Primatol. 2006;35:236–247. doi: 10.1111/j.1600-0684.2006.00172.x. [DOI] [PubMed] [Google Scholar]
- 16.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 17.Jasin M, et al. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- 18.Sonoda E, et al. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5:1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa F, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 22.Hollis RP, et al. Stable gene transfer to human CD34(+) hematopoietic cells using the Sleeping Beauty transposon. Exp Hematol. 2006;10:1333–43. doi: 10.1016/j.exphem.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Sumiyoshi T, et al. Stable transgene expression in primitive human CD34+ hematopoietic stem/progenitor cells, using the Sleeping Beauty transposon system. Hum Gene Ther. 2009;12:1607–1626. doi: 10.1089/hum.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Màtés L, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;7:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- 25.Xue X, et al. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood. 2009;7:1319–1330. doi: 10.1182/blood-2009-03-210005. [DOI] [PubMed] [Google Scholar]
- 26.Basu S, Broxmeyer HE. CCR5 ligands modulate CXCL12-induced chemotaxis, adhesion, and Akt phosphorylation of human cord blood CD34+ cells. J Immunol. 2009;183:7478–7488. doi: 10.4049/jimmunol.0900542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe S, et al. Hematopoietic stem cell-engrafted NOD/SCID/IL2Rgamma null mice develop human lymphoid systems and induce long-lasting HIV-1 infection with specific humoral immune responses. Blood. 2007;109:212–218. doi: 10.1182/blood-2006-04-017681. [DOI] [PubMed] [Google Scholar]
- 28.Brenchley JM, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brenchley JM, et al. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–239. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 30.Guadalupe M, et al. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol. 2003;77:11708–11717. doi: 10.1128/JVI.77.21.11708-11717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–1152. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 32.Mattapallil JJ, et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 33.Talal AH, et al. Effect of HIV-1 infection on lymphocyte proliferation in gut-associated lymphoid tissue. J Acquir Immune Defic Syndr. 2001;26:208–217. doi: 10.1097/00042560-200103010-00002. [DOI] [PubMed] [Google Scholar]
- 34.Veazey RS, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 35.Berges BK, et al. HIV-1 infection and CD4 T cell depletion in the humanized Rag2−/−gamma c−/− (RAG-hu) mouse model. Retrovirology. 2006;3:76–90. doi: 10.1186/1742-4690-3-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoddart CA, et al. IFN-alpha-induced upregulation of CCR5 leads to expanded HIV tropism in vivo. PLoS Pathog. 2010;19:e1000766. doi: 10.1371/journal.ppat.1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appay V, Sauce D. Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol. 2008;214:231–241. doi: 10.1002/path.2276. [DOI] [PubMed] [Google Scholar]
- 38.Choudhary SK, et al. R5 human immunodeficiency virus type 1 infection of fetal thymic organ culture induces cytokine and CCR5 expression. J Virol. 2005;79:458–471. doi: 10.1128/JVI.79.1.458-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahn JO, Walker BD. Acute human immunodeficiency virus type 1 infection. N Engl J Med. 1998;339:33–39. doi: 10.1056/NEJM199807023390107. [DOI] [PubMed] [Google Scholar]
- 40.Margolick JB, et al. Impact of inversion of the CD4/CD8 ratio on the natural history of HIV-1 infection. J Acquir Immune Defic Syndr. 2008;42:620–626. doi: 10.1097/01.qai.0000223028.55080.9d. [DOI] [PubMed] [Google Scholar]
- 41.Henrad DR, et al. Natural History of HIV-1 cell-free viremia. JAMA. 1995;274:554–558. [PubMed] [Google Scholar]
- 42.Chen RY, et al. Distribution of health care expenditures for HIV-infected patients. Clin Infect Dis. 2006;42:1003–1010. doi: 10.1086/500453. [DOI] [PubMed] [Google Scholar]
- 43.Richman DD, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 44.Rossi JJ, June CH, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–1454. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bibikova M, et al. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doyon Y, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santiago Y, et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters W, Dupuis M, Charo IF. A mechanism for the impaired IFN-gamma production in C–C chemokine receptor 2 (CCR2) knockout mice: Role of CCR2 in linking the innate and adaptive immune responses. J Immunol. 2000;165:7072–7077. doi: 10.4049/jimmunol.165.12.7072. [DOI] [PubMed] [Google Scholar]
- 49.Smith MW, et al. CCR2 chemokine receptor and AIDS progression. Nat Med. 1997;3:1052–1053. doi: 10.1038/nm1097-1052c. [DOI] [PubMed] [Google Scholar]
- 50.Davis BR, Candotti F. Revertant somatic mosaicism in the Wiskott-Aldrich syndrome. Immunol Res. 2009;44:27–31. doi: 10.1007/s12026-008-8091-4. [DOI] [PubMed] [Google Scholar]
- 51.Hirschhorn R, et al. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet. 1996;3:2. doi: 10.1038/ng0796-290. [DOI] [PubMed] [Google Scholar]
- 52.Hirschhorn R, et al. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;10:721–728. doi: 10.1136/jmg.40.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephan V, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;21:1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 54.Chun TW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. 2008;197:714–720. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- 55.Lackner AA, et al. The gastrointestinal tract and AIDS pathogenesis. Gastroenterology. 2009;136:1965–1978. doi: 10.1053/j.gastro.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Picker LJ. Immunopathogenesis of acute AIDS virus infection. Curr Opin Immunol. 2006;18:399–405. doi: 10.1016/j.coi.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Veazey RS, Marx PA, Lackner AA. The mucosal immune system: primary target for HIV infection and AIDS. Trends Immunol. 2001;11:626–633. doi: 10.1016/s1471-4906(01)02039-7. [DOI] [PubMed] [Google Scholar]
- 58.Krishnan A, et al. Autologous stem cell transplantation for HIV associated lymphoma. Blood. 2001;13:3857–3859. doi: 10.1182/blood.v98.13.3857. [DOI] [PubMed] [Google Scholar]
- 59.Aiuti A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;5577:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 60.Aiuti A, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;5:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 61.Ott MG, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 62.Cartier N, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 63.Biti R, et al. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med. 1997;3:252–253. doi: 10.1038/nm0397-252. [DOI] [PubMed] [Google Scholar]
- 64.Oh DY, et al. CCR5Delta32 genotypes in a German HIV-1 seroconverter cohort and report of HIV-1 infection in a CCR5Delta32 homozygous individual. PLoS One. 2008;3:2747–2753. doi: 10.1371/journal.pone.0002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiser B, et al. HIV-1 coreceptor usage and CXCR4-specific viral load predict clinical disease progression during combination antiretroviral therapy. AIDS. 2008;22:469–79. doi: 10.1097/QAD.0b013e3282f4196c. [DOI] [PubMed] [Google Scholar]
- 66.Ogert RA, et al. Mapping Resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology. 2008;2:387–399. doi: 10.1016/j.virol.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 67.Soulie C, et al. Primary genotypic resistance of HIV-1 to CCR5 antagonist treatment-naïve patients. AIDS. 2008;16:2212–2214. doi: 10.1097/QAD.0b013e328313bf9c. [DOI] [PubMed] [Google Scholar]
- 68.Palmer S, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A. 2008;10:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dinoso JB, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 2009;23:9403–9408. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitsuyasu RT, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med. 2009;15:285–292. doi: 10.1038/nm.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shultz LD, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hematopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 72.Rouet F, et al. Transfer and evaluation of an automated, low-cost real-time reverse transcription-PCR test for diagnosis and monitoring of human immunodeficiency virus type 1 infection in a West African resource-limited setting. J Clin Microbiol. 2005;43:2709–2717. doi: 10.1128/JCM.43.6.2709-2717.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun Z, et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. 2007;204:705–714. doi: 10.1084/jem.20062411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loken MR, et al. Establishing lymphocyte gates for immunophenotyping by flow cytometry. Cytometry. 1990;4:453–459. doi: 10.1002/cyto.990110402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.