

Abstract
Eukaryotic chromatin can be highly dynamic and can continuously exchange between an open transcriptionally active conformation and a compacted silenced one. Post-translational modifications of histones have a pivotal role in regulating chromatin states, thus influencing all chromatin dependent processes. Methylation is currently one of the best characterized histone modification and occurs on arginine and lysine residues. Histone methylation can regulate other modifications (e.g. acetylation, phosphorylation and ubiquitination) in order to define a precise functional chromatin environment. In this review we focus on histone methylation and demethylation, as well as on the enzymes responsible for setting these marks. In particular we are describing novel concepts on the interdependence of histone modifications marks and discussing the molecular mechanisms governing this cross-talks.
Keywords: histone modifications, histone methylation, cross-talk, epigenetic, chromatin
INTRODUCTION
In mammals the genomic information is organized into chromatin. The structural and functional unit of chromatin is the nucleosome, that consists of an octamer of the core histones H2A, H2B, H3 and H4 around which 147 bp of DNA are wrapped [1]. In addition, the linker histone H1 binds the DNA entering and exiting the nucleosome and protects the linker DNA, further compacting chromatin. Chromatin is not a static structure, but in order to allow vital cellular processes to occur, it needs to be dynamically modulated. Three main mechanisms have been proposed to regulate chromatin compaction and decompaction. First, chromatin remodeling complexes use the energy liberated from ATP hydrolysis to actively move and reposition nucleosomes along the DNA [2]. Second, histone variants are incorporated at specific locations where they define a precise chromatin state [3] and third, covalent modifications of histones or DNA can be key to regulation of chromatin structure and all DNA dependent processes [4, 5].
So far the best studied histone modifications are located within the flexible N-terminal tail of the core histones. With the recent improvement of the sensitivity of mass-spectrometrical techniques, new, previously uncharacterized modifications have been identified in vivo both in the tails and in the core domain of histones [6–10]. However, for many histone modifications their functional role is not yet fully understood.
HOW DO HISTONE MODIFICATIONS WORK?
There are two main mechanisms explaining the impact of histone modifications on chromatin functions.
The first is the disruption of contacts between adjacent nucleosomes or between histones and DNA e.g. by charge changes. The best example for this is histone lysine acetylation. Due to its capacity to neutralize the positive charge of lysines, histone acetylation can weaken the affinity between histone and DNA, thus creating a more accessible and open chromatin state [11]. In line with these findings, the development of a strategy to produce recombinant nucleosomes fully modified at a specific site showed that histone H4 lysine 16 (H4K16) acetylation inhibits the formation of the condensed 30 nm fiber and the establishment of higher order of chromatin structure [12].
The second mechanism to regulate chromatin dynamics is the recruitment of specific binding proteins by histone marks. According to the so called ‘histone code’ hypothesis [13, 14], single histone modifications or combinations of modifications can be recognized by effector proteins or protein complexes that read these marks, convert them into specific functional chromatin states and regulate downstream responses. It is now well established that chromo-like domains (chromo, MBT, Tudor) and non-related plant homeo domains (PHD) specifically bind methylated lysines, whereas acetylation is specifically recognized by bromodomains and phosphorylation by 14-3-3 proteins [15–17].
Interestingly, the modification specific binding modules and the catalytic domains setting these marks can often be found within the same protein or protein complex, suggesting that pattern of histone modifications can be ‘read and written’ by the same interacting factor(s). For example the histone methyltransferases G9a and its interaction partner Glp1 bind H3K9me1/me2 via their ankyrin repeats and methylate neighboring histones on H3K9 via a distinct catalytic domain [18]. This product-binding capacity of G9a/Glp1 illustrates a general ‘feed forward loop’ mechanism how cells can maintain and propagate histone modifications and functionally defined chromatin states [19].
While modifications of the histone tails might regulate nucleosome function by affecting the binding of effector proteins, modifications within the histone fold domain are more probable to directly regulate nucleosome structure [20, 21].
LYSINE METHYLATION AND DEMETHYLATION
Methylation can occur at different aminoacid residues such as lysine, arginine and histidine. Methylation of lysines and arginines has been extensively studied and implicated in multiple cellular processes [4]. Histone methylation is so far the most complex modification, since its function depends on the precise methylation site and the degree of modification. Lysine residues can be mono-, di- or tri-methylated, whereas arginines can be mono- or di-methylated. In addition arginines can be symmetrically or asymmetrically di-methyleted. Therefore we will focus here on histone methylation and in particular on its complex cross-talk with other modifications.
Site and state-specific lysine methylation of histones is catalyzed by a group of lysine methyltransferases (KMT) containing the evolutionarily conserved SET domain [Su(var), enhancer of zeste, Tritorax] (Table 1). They have been sub-grouped into seven main families, named according to their founding member: SUV39, SET1, SET2, EZ, RIZ, SMYD and SUV4-20 [22]. In addition few orphan members have been identified: SET7/9 and SET8 (also known as PRSET7). Proteins within the same family share high similarity within the SET domain as well as in the surrounding sequences [22]. To date the only identified non-SET domain-containing lysine KMTase is DOT1, specific for H3K79 methylation in the core region of H3 [23, 24].
Table 1:
Histone methylase and demethylase, their target sites and their chromatin function
| KMT | Human nomenclature | Modification sites | Implicated in | |
|---|---|---|---|---|
| SET domain containing histone lysine methylases | SUV39 family | |||
| SUV39H1 | KMT1A | H3K9me2/me3 |
|
|
| SUV39H2 | KMT1B | H3K9me2/me3 |
|
|
| G9a | KMT1C | H3K9me1/me2 | Transcriptional silencing | |
| GLP1 (euHMT) | KMT1D | H3K9me1/me2 | Transcriptional silencing | |
| ESET (SETDB1) | KMT1E | H3K9me2/me3 | Transcriptional silencing of euchromatic genes | |
| CLLD8 (SETDB2) | KMT1F | H3K9me2/me3 |
|
|
| SET1 family | ||||
| MLL1 (HRX, ALL1) | KMT2A | H3K4me1/me2 | Transcriptional activation | |
| MLL2 (ARL) | KMT2B | H3K4me1/me2/me3 | Transcriptional activation | |
| MLL3 | KMT2C | H3K4me1/me2/me3 | Transcriptional activation | |
| MLL4 (HRX2) | KMT2D | H3K4me1/me2/me3 | Transcriptional activation | |
| MLL5 | KMT2E | H3K4me3 | Transcriptional activation | |
| SET1 (ASH2) | KMT2 | H3K4me1/me2/me3 | Transcriptional activation | |
| SET2 family | ||||
| WHSC1 (NSD2) |
|
|
||
| WHSCL1 (NSD3) |
|
Transcriptional repression | ||
| NSD1 | KMT3B |
|
|
|
| SET2 (HYPB) | KMT3A | H3K36me3 |
|
|
| ASH1 | KMT2H | H3K4me3 | Transcriptional activation | |
| RIZ family | ||||
| RIZ (PRDM2) | KMT8 | H3K9 specificity not known | Transcriptional regulation of ER genes during neuronal differentiation | |
| BLIMP1 (PRDM1) | No enzymatic activity | Transcriptional repression | ||
| SMYD family | ||||
| SMYD1 | KMT3D | H3K4me1/me2/me3 | Transcriptional silencing | |
| SMYD2 | KMT3C | H3K36me1/me2/me3 | Transcriptional regulation | |
| SMYD3 | KMT3E | H3K4me2/me3 | Transcriptional activation | |
| EZ family | ||||
| EZH1 | KMT6B | H3K27me1/me2/me3 | HOX genes silencing | |
| EZH2 | KMT6A | H3K27me1/me2/me3 | HOX gene silencing | |
| SUV4-20 family | ||||
| SUV4-20H1 | KMT5B | H4K20me2/me3 | Transcriptional silencing, DNA repair | |
| SUV4-20H2 | KMT5C | H4K20me2/me3 | Transcriptional silencing, DNA repair | |
| Others | ||||
| SET7/9 | KMT7 | H3K4me1/me2 | Transcriptional activation | |
| SET8 (PR-SET7) | KMT5A | H4K20me1 | Transcriptional activation | |
| NON SET domain-containing KMTs | DOT family | |||
| DOT1 | KMT4 | H3K79me1/me2/me3 | Transcriptional activation | |
| Histone lysine demethylases | LSD family | |||
| LSD1 | KDM1 | H3K4me1/me2 H3K9me1/me2 |
|
|
| LSD2 | KDM2 | H3K4me1/me2 | Transcriptional repression | |
| JMJC family | ||||
| JHDM1a/b | KDM2A/B | H3K4me1/me2/me3 H3K36me2/me3 | Suppression of spontaneous mutation (C. elegans) heterochromatin maintenance at the mating type locus (S. cerevisiae) | |
| JHDM2 | KDM3A | H3K9me1/me2 | Transcriptional activation AR-dependent genes | |
| JHDM3/JMJD2 | KDM4 |
|
Transcriptional activation and repression | |
| JMJD2a | KDM4A | H3K9me1/me2/me3 H3K36me2/me3 | Transcriptional regulation | |
| JMJD2b/c | KDM4B/C | H3K9me1/me2/me3 | AR-dependent gene activation | |
| JMJD3 | H3K27me2/me3 | |||
| JARIDa/b/c/d | KDM5A/B/C/D | H3K4me1/me2/me3 |
|
|
| PHF2 | JHDM1E | H3K9me1 | rDNA genes activation | |
| PHF8 | JHDM1F | H3K9me1/me2 | rDNA genes activation | |
| UTX/UTY (KDM6A) | KDM6A | H3K27me2/me3 | HOX gene activation | |
| Histone arginine methylases | PRMT family | |||
| PRMT1 | HRMT1L2 | H4R3 | Transcriptional activation | |
| PRMT2 | HRMT1L1 | No enzymatic activity | Co-activator in ER-dependent transcription | |
| PRMT3 | HRMT1L3 | Ribosomal proteins | Ribosome biosynthesis | |
| PRMT4 (CARM1) | HRMT1L4 | H3R17, H3R26 |
|
|
| PRMT5 | HRMT1L5 |
|
|
|
| PRMT6 | HRMT1L6 | H3R2 H2AR3 | Transcriptional repression | |
| PRMT7 | HRMT1L7 | H2AR3 H4R3 | Transcriptional regulation of imprinted genes | |
| PRMT8 | HRMT1L8 | H4 (specificity not known) | Unknown | |
| PRMT9 (isoform 4) | HRMT1L9 | H2AR3, H4R3 | Unknown | |
| PRMT10, PRMT11 |
|
Not determined | Unknown | |
| Histone arginine demethylases | PADI family | |||
| PADI4 | H3R2me2 H3R8me3 H3R17me2 H3R26me2 H4R3me2 | Transcriptional repression and activation | ||
| JMJD6 |
|
Transcriptional regulation during differentiation |
So far methylatiopn of five residues within the N-terminal tail (H3K4, H3K9, H3K27, H3K36 and H4K20) of histones H3 and H4, and of two residues in the globular domain (H3K64 and H3K79) of histone H3 have been functionally characterized. In addition, the linker histone H1 can also be methylated at H1.4K26. In general, H3K9, H3K27 H3K64 H4K20 and H1.4K26 methylation have been implicated in transcriptional silencing [25] whereas, H3K4, H3K36 and H3K79 methylation are associated with transcriptionally active regions [25]. However, depending on the methylation states and the genomic location the same modification might have different functional outcomes.
H3K9 methylation is involved in euchromatic gene silencing as well as in heterocromatin formation [26, 27]. H3K27 methylation has an important role in the repression of HOX genes during development and in X chromosome inactivation and imprinting [28–30]. More recently H3K64me3 has been shown by our lab to be enriched at pericentric heterochromatin and to be associated with repeat sequences and transcriptionally inactive genomic regions [31].
In the case of H4K20 each methylation state is implicated in different biological processes. H4K20me1 peaks in M phase and is involved in cell-cycle progression and chromosome condensation [32–34]. Outside of mitosis H4K20me1 is a mark for active transcription [35]. H4K20me2 has a role in DNA repair [36] and H4K20me3 is enriched in heterochromatin and is implicated in heterochromatin maintenance and telomere stability [37, 38].
H3K4 methylation occurs in mammals in several distinct genomic distributions. Strong enrichments of H3K4me3 are found at transcription start sites (TSS) of active genes whereas H3K4me2 is present across the body of genes, where they contribute to transcriptional initiation and mRNA processing respectively [10, 35, 39]. H3K4me1 peaks instead at the 3′ end of active genes both in yeast and mammals [40, 41]. Targeting of H3K4 methylation to these sites can occur via the interaction of H3K4 specific KMTs with the active, phosphorylated form of RNA Pol II, providing a direct link with transcription [42]. Interestingly large domains of H3K4 methylation covering both genic and intergenic regions are evident at specific locations such as the HOX genes cluster. A deeper analysis of the HOXA and HOXB loci identified in these regions multiple promoters generating non-coding RNA [43]. These intergenic transcripts have been shown to enhance gene accessibility [44, 45] and in the case of the HOX cluster they might contribute to its tightly controlled pattern of expression during differentiation.
H3K36 methylation is coupled to the process of active transcriptional elongation and it is enriched towards the 3′ end of target genes [46]. However, when present within protein-coding regions it prevents inappropriate transcriptional initiation of intragenic sequences [47].
The function of H3K79 methylation in transcription is still somehow contradictory. In flies and in mammals H3K79 methylation has been suggested to act as an active mark [48, 49]. However, in a parallel study, Barski et al. [50] showed by using the Solexa sequencing that H3K79me1 is enriched at active genes, whereas H3K79me3 is present at transcriptionally repressed genes in human cells.
Modifications of histones H2A and H2B have been less studied [51]. However, deletion of the N terminal tail of H2A or H2B in yeast showed that they affect the expression of a large number of genes and that have then an important role in gene regulation. Recently by Solexa sequencing of the human genome, H2BK5 methylation has been mapped to active genes, suggesting a possible link with transcription [50].
In addition to the core histones, the linker histone H1 is methylated at H1.4K26. This mark is implicated in heterochromatin formation and transcriptional repression by Ezh2 [52, 53].
Although lysine methylation has been considered for a long time as an irreversible modification, it is now well established that like acetylation and phosphorylation also histone methylation can be a dynamic modification. To date two groups of histone lysine demethylases (KDM) have been identified. The first group of amine oxidase-domain containing enzymes is represented by LSD1 (also known as AOF2) and LSD2 (also known as AOF1). LSD1 demethylates H3K4me1/me2 in vitro and in vivo [54] and its activity on nucleosomes substrates requires the transcriptional co-repressor CoREST [55]. LSD1 is also recruited to androgen receptor (AR)-regulated genes where it acts as activator. It has been shown that interaction of LSD1 activity with AR redirects LSD1 enzymatic activity towards H3K9me1/me2 [56]. Moreover, LSD is also stimulated by HDAC1 (histone deacetylase 1) revealing a functional interconnection between histone demethylation and deacetylation [57]. LSD2 has been recently identified and shown to be specific for H3K4me1/me2 [58]. As opposed to LSD1, LSD2 does not form a biochemically stable complex with the co-repressor protein CoREST. Furthermore, LSD2 contains a CW-type zinc finger motif with potential zinc-binding sites that are not present in LSD1 [58].
The second group of KMTs is represented by the Jumonji domain-containing proteins (jmjC), the members of this group are Fe(II) and 2-oxoglutarate (2OG) dependent oxygenases [59]. Unlike LSD1 that can remove only mono and dimethyl groups from lysine residues, jmjC enzymes are able to revert all three histone lysine methylation states [60, 61]. Based on the presence of additional domains beside the jmjC domain, JmjC histone demethylases (JHDM) enzymes have been classified into seven evolutionary conserved subgroups (JHDM1, PHF2/PHF8, JARID, JHDM3/JMJD2, UTX/UTY, JHDM2 and JmjC domain only). Although for many of these proteins the enzymatic activity as histone demethylases still needs to be proven, generally the substrate specificity for the known JHDMs rely on both the JmjC domain and additional domains within each enzyme. For the most histone methylation marks demethylase have been identified, however so far no demethylase enzymes have been found for H3K64 and H3K79 methylation [60].
ARGININE METHYLATION AND DEMETHYLATION
Whereas lysine methylation has been intensively globally mapped in multiple organisms, we currently know much less about the distribution and functions of arginine methylation. Arginine methylation is catalyzed by a family of enzymes called protein arginine methyltransferases (PRMTs). All members are able to monomethylate arginine, but according to the type of dimethylation they are classified into two classes: type I (PRMT1, 3, 4, 6, 8) asymmetrically dimethylate arginine; and type II (PRMT5, 7, 9) symmetrically dimethylate arginine [62]. The PRMT family members are ubiquitously expressed and evolutionary conserved from yeast to human. To date, 11 mammalian PRMTs have been identified. They share a conserved catalytic domain, but differ in their cellular localization as well as in their substrate specificity. PRMT2, PRMT10 and PRMT11 enzymatic activity has not yet been demonstrated [63]. Interaction of PRMTs with different cofactors has also been proposed to modulate their enzymatic activities and specificity at defined loci [63].
H3R2, H3R8 H3R17 H3R26, H4R3 H2AR3 are known PRMT targets in vivo [63] (Table 1). Like different lysine methylation states, symmetric or asymmetric methylation of the same arginine can mark distinct chromatin regions. However, in contrast to lysine methylation the functional role of arginine methylation in chromatin structure and transcription has been so far underexplored. Moreover, general levels of arginine methylation seemed to be lower compared to lysine methylation, indicative of a more restricted function in gene regulation rather than a general structural role in chromatin formation.
When asymmetrically methylated by PRMT1, H4R3me2 acts as a transcription activating mark of several ER regulated genes [64, 65] and is essential in vivo for the establishment and maintenance of open chromatin domains marked by H3 and H4 acetylation [66]. However, when symmetrically dimethylated by PRMT5, H4R3me2 is instead involved in transcriptional silencing, as expected since PRMT5 is also a subunit of the repressive MB2/NURD histone deacetylating complex [67]. In the case of the beta-globin locus transcriptional silencing by PRMT5 is achieved via the recruitment of DNMT3A and subsequent DNA methylation of the promoter region [68]. In agreement with a potential connection between arginine methylation and DNA methylation, H3R8me2 and H4R3me2 have been shown to regulate rDNA promoter activity in a DNA methylation-dependent manner [69]. Unlike other symmetrically methylated arginines, H3R8me2 catalyzed by PRMT5 can also act as an activating mark by cooperating with the SWI/SNF chromatin-remodeling complex in the regulation of genes involved in myogenic differentiation [70]. Moreover, the level of H2A/H4R3me2, another PRMT5 site, are dynamically modulated during epigenetic reprogramming of primordial germ cells (PGCs), pointing at an important function for this modification in the mouse germ cell lineage [71].
H3R17me2 and H3R26me2 are methylated by PRMT4 (also known as CARM1) and are implicated in nuclear receptor (NR)-mediated activation of transcription and the regulation of pluripotent genes during the early mouse development [72, 73].
H3R2me2 is mainly catalyzed by PRMT6. This modification is enriched at heterochromatin and silent euchromatin and depleted at the TSS of active promoters, thus acting as a repressive mark [74, 75]. H3R2me2 can also be targeted by CARM1 although to a lesser extent and it cooperates with histone acetylation in the activation of NR regulated genes [72].
Like other modifications involved in chromatin-regulated processes also arginine methylation needs to be reverted in order to maintain a functional equilibrium within the cells. However, very little is known about arginine demethylases. To date two proteins have been implicated in this mechanism: PADI4 and JMJD6. PADI4 is the only member of the protein arginine deiminase I (PADI) family with nuclear localization where it catalyses deimination of monomethylarginines to citrullines. PADI4 recognizes arginine residues surrounded by unstructured amino acid sequences and has therefore a broad range of substrate specificity. Monomethylarginines on both H3 (R2, R8, R17 and R26) and H4 (R3) are PADI4 targets [76]. However, this enzyme fails to meet the requirements for a true arginine demethylase since it converts the arginine to a citrulline.
One recent publication showed that JMJD6, a member of the JMJ lysine demethylases, can specifically demethylate H3R2 and H4R3 me1/me2 both in vitro and in vivo [77], however there were so far no follow ups to this discovery.
HISTONE MODIFICATIONS CROSS-TALK
It is now well established that there is an intense cross-talk between histone modifications to drive distinct downstream functions. Cross regulation can occur in different flavors: on the one hand, one modification can promote/block the addition of another modification. On the other hand, one modification can stimulate/block the removal of another modification. Moreover, the cross-talk can occur on the same histone (cross-talk in cis; Figure 1A), between histones within the same nucleosome (cross-talk in trans; Figure 1B) or across nucleosomes (nucleosome cross-talk). An increasing number of histone modifying complexes are found to contain more than one distinct enzymatic activities. These enzymes can act in concert to determine the functional status of chromatin by coordinating multiple histone modifications (Figure 1C). For each of these scenarios multiple examples have been described and we will discuss some of the most exiting ones below. Cross-talks also have major implications for our understanding and interpretation of genome-wide mapping data of histone modifications. These approaches usually profile the average presence of histone modifications within a cell population at certain genomic regions. However, they do not provide any information about which marks co-occur at a given nucleosome and about the molecular mechanisms leading to the deposition of these marks on chromatin.
Figure 1:

Mechanisms of cross-talk between histone modifications. (A) Cross-talk in cis; acetylation of H3K18 and H3K23 by CBP can promote the methylation of H3R17 by the methyltransferase CARM1, resulting in activation of estrogene-responsive genes [90]. (B) Cross-talk in trans; H2BK120 ubiquitination by RAD6 is recognized by the WDR82 subunit of the SET1A/B COMPASS complex and it is a prerequisite for efficient H3K4 methylation by SET1A/B and transcriptional activation of target genes in mammals. (C) Multifunctional histone modifications complexes, simplified model of PRC complex function; in Drosophila the Polycomb repressive complexes PRC1, PRC2 and PhoRC are recruited to chromatin in a hierarchical manner and they coordinate distinct histone modifications. The Pho subunit of PhoRC complex binds specific PRE (Polycomb responsive element) elements in the DNA. The PRC2 complex is recruited to this PRE via interactions between the Pho protein and E(Z) (drosophila homolog of human EZH2), the methyltarnsferase subunit of the PRC2 complex. E(Z), methylates H3K27 forming a binding site for PC, a subunit of the PRC1 complex. dRING, the E3 ligase within the PRC2 complex can mediate ubiquitination of H2AK119. dRING is also a subunit of the dRAF (dRING associated factors), an additonal Polycomb complex in Drosophila. dRAF contains the histone demethylase KDM2, which coordinates removal of H3K36me3 with stimulation of H2AK119 ubiquitination by dRING. dRAF cooperates with PRC1 in gene silecing by Polycomb complexes. Additionally non-coding RNAs had recently been implicated in the trageting of Polycomb complexes (not shown).
Only combining our knowledge of how histone modifications influence each other and cross-talk with the high-resolution maps obtained by high throughput sequencing will allow us to answer fundamental biological questions and to make further progresses in deciphering the multiple roles of histone modifications in DNA-dependent processes.
THE METHYL-PHOSPHO SWITCH
One of the first examples for cross-regulation of histone modifications in cis is between H3K9 methylation and the neighboring H3S10 phosphorylation [78]. H3S10 phosphorylation is required for chromosome condensation and segregation during mitosis [79]. H3K9me3 can be specifically bound by the chromodomain of heterochromatin protein 1 (HP1) and has a pivotal role in heterochromatin formation and propagation of pericentric heterochromatin [80, 81]. However, in mitosis HP1 is released from condensed chromatin despite the persistence of its recruiting mark H3K9me3 [82, 83]. To explain this apparent contradiction the methyl-phospho switch model has been proposed. It suggests that H3S10 phosphorylation displaces HP1 from chromatin by inhibiting its binding to the adjacent H3K9me3. Moreover, the loss of HP1 from chromatin during mitosis occurs in concomitance with an increase in H3S10 phosphorylation levels and prior to the loading of condensins to chromatin [78]. Interestingly, low levels of H3S10 phosphorylation have been detected also in interphase where H3S10 phosphorylation is found at the promoter of immediate early (IE) responsive genes. Like in mitosis also in interphase removal of HP1 from chromatin depends on H3S10 phosphorylation and is a prerequisite for transcriptional activation [84]. In support of an interdependence between these two marks, H3S10 phosphorylation levels are significantly increased in Suv39h double null MEFs cells [85].
This methyl-phospho switch model is not limited to directly neighboring residues. Recently two non-adjacent sites within H3 have been found to modulate each other, providing an additional intriguing example of methyl-phospho cross-talk (Figure 2). H3T6 phosphorylation by PKCβI is a novel mark for transcriptional activation of several AR (androgen-receptor)-regulated genes. It can block H3K4 demethylation by the demethylases LSD1 (specific for H3K4me1/me2) and JARID1B (specific for H3K4me2/me3) and it redirects their enzymatic activity towards H3K9 methylation [86]. In support of this model, PKCβI co-localizes with AR and LSD1 at target gene promoters and phosphorylates H3T6 upon androgen receptor activation. Depletion of PKCβI by RNAi abrogates H3T6 phosphorylation, enhances H3K4 demethylation by LSD1 and as a consequence AR dependent transcription is inhibited. While H3T6 phosphorylation blocks LSD1 and JARID1B activities, H3T11 phosphorylation by PRK1 acts in a different way by increasing the activity of LSD1 (H3K9me1/me2) and JMJD2C (H3K9me2/me3) for H3K9 methylation [87] (Figure 2). In agreement with a role for H3T11 phosphorylation in transcriptional activation of AR dependent genes it has been shown that their expression is reduced upon PRK1 depletion, due to impairments in H3K9me1/me2 demethylation and in the establishment of H3K9 and K14 acetylation. Moreover, DNA damage induction in cells lacking H3T11 phosphorylation correlates with reduced binding of the histone acetyltransferase GCN5 and reduced H3K9 acetylation at cyclin B1 and cdk1 promoters [88], further supporting an active transcriptional role for this mark.
Figure 2:
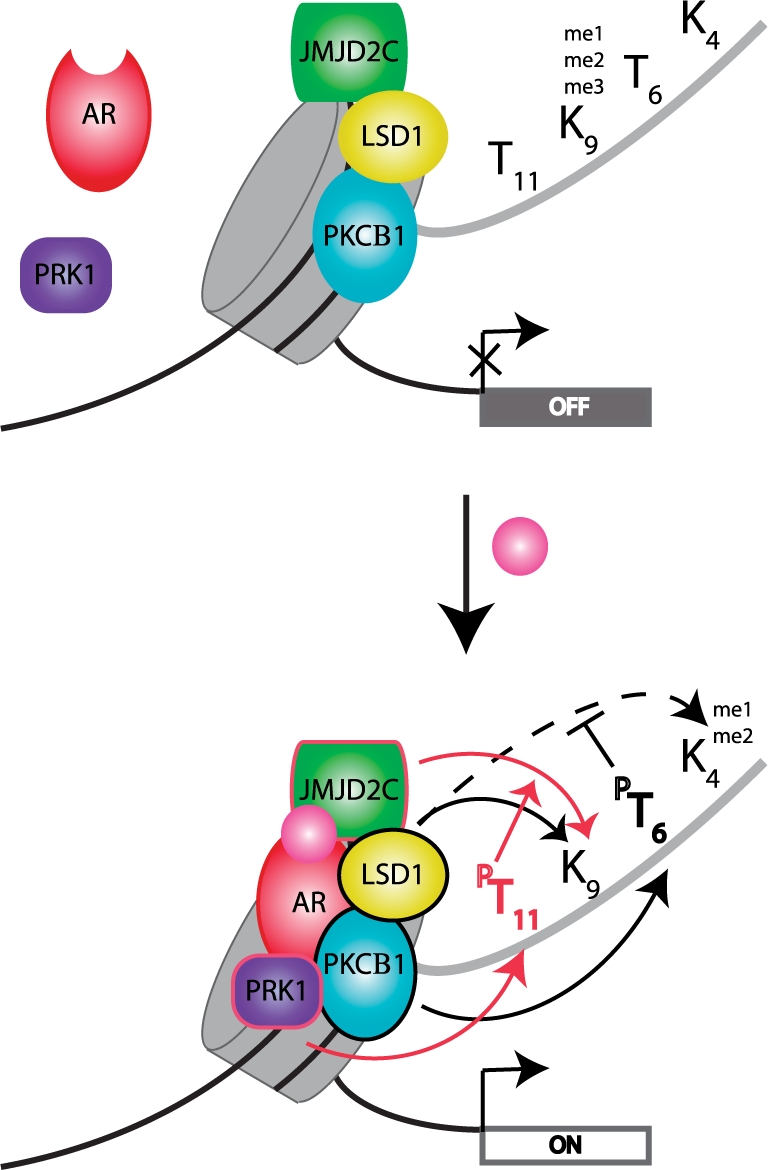
Cross-talk between H3 methylation and phosphorylation in AR-dependent transcriptional regulation. Top panel: in the absence of ligand, the androgen-receptor (AR) is present in the cytoplasma and AR-regulated genes are silenced by the presence of H3K9 methylation. PKCβ1, LSD1 and JMJD2C are already present on chromatin. Bottom panel: association of ligand-activated AR with PRK1 leads to activation of PKCβI. Phosphorylation of H3T6 by activated PKCβI prevents LSD1 from demethylating H3K4me2/me1 (dashed black arrow) but not H3K9me2/me1 and AR-dependent genes get activated. In addition, PRK1 phosphorylates H3T11. This mark enhances JMJD2C demethylating activity for H3K9me3 further contributing to transcriptional activation. The PKCβ1 mediated pathway is indicated by black arrows. The PRK1 mediated pathway is indicated by red arrows. A colour version of this figure is available at http://bfg.oxfordjournals.org/.
The discovery of novel methylation sites in close proximity to phosphorylated residues suggests that the methyl-phospho switch mechanism can modulate the binding of regulator proteins to multiple histone methylation marks. One additional example is the interaction of HP1 with H1.4K26me, blocked by the phosphorylation of the adjacent S27 [52]. Additionally, H3K27 is followed by S28 and H3K4 is preceded by T3. All these residues are modified in vivo by either methylation (H3K27 and H3K4) or phosphorylation (H3S27 and H3T3). In light of the numerous data pointing at a cross-talk between phosphorylation and methylation, it is tempting to speculate that also the recruitment of binding complexes to H3K27me3 and to H3K4me3 might undergo methyl-phospho switch regulation.
CROSS-TALK BETWEEN HISTONE METHYLATION AND ACETYLATION/DEACETYLATION
Although a methyl-phospho switch for arginine methylation has not been described so far, several examples of cross-talk between arginine methylation and histone acetylation have been documented. H4R3me2 by PRMT1 has been associated with transcriptional activation [89]. However, very little is known about the molecular mechanisms employed. One intriguing possibility is that H4R3me2 defines chromatin domains poised for transcription by regulating histone acetylation at specific target promoters. In agreement with this, H4R3me2 by PRMT1 activates transcription of the MMTV promoter by facilitating H4 acetylation by p300 [90]. RNAi of PRMT1 in erythroid cells, results in the loss of H4R3me2 as well as of H3 and H4 acetylation at the beta-globin locus, further supporting the previous in vitro data [66].
Like PRMT1, CARM1 has also been implicated as co-activator in the transcription of nuclear receptor (NR)-regulated genes [91]. CARM1 methylates H4R17 in vivo after CBP recruitment and sequential acetylation of H3K18 and H3K23 at the estrogen stimulated pS2 promoter. Interestingly H3K14 acetylation, another target site of p300/CBP does not induce CARM1 activity, providing a support for the specificity of this cross-talk [92].
While PRMT1 and CARM1 cooperate with histone acetylation to activate transcription, PRMT5 has been shown to act as a transcriptional co-repressor via modulating histone deacetylation levels at specific loci [69]. One example of such a repressive cross-talk is the silencing of ST7 and NM23 tumor suppressor genes by PRMT5. Expression of these genes is reduced in cell line overexpressing PRMT5 and their silencing correlates with an increase in H3R8me2 levels and concomitant loss of H3K9 acetylation at their promoters [93].
HETEROCHROMATIN FORMATION: A CONSERVED MECHANISM
Transcriptionally repressed chromatin is required at telomeric and subtelomeric regions to stabilize chromosome ends and to ensure correct chromosome segregation in mitosis [94]. Due to its vital function, the molecular basis leading to the formation of heterochromatin have been conserved throughout evolution together with the employment of even more specialized mechanisms according to the genome complexity.
In mammals heterochromatin formation requires several sequential steps and a precise cross-talk between histone deacetylation and methylation. First, SIRT1 specifically deacetylates H4K16ac and H3K9ac at the promoter region. Second, it interacts and deacetylates H1.4K26ac establishing a repressive chromatin environment. Finally, chromatin compaction is achieved by further reduction of active marks both at the promoter and at the coding region and by the increase of repressive marks as H3K9me3 and H4K20me3 [38, 95, 96].
Similar players are involved in heterochromatin gene silencing both in worms and yeast. In Caenorhabditis elegans an interplay between SIR-2.1, histone H1 (HIS24) and MES-2 (ortholog of enhancer of zeste methyltransferase) has been recently shown to occur at subtelomeric regions in order to maintain constant level of H3K27 methylation [97]. In Saccharomyces cerevisiae, H3K79me3 is involved in telomeric silencing [98].
CROSS-TALK BETWEEN METHYLATED RESIDUES
PHD fingers are known to specifically recognize methylated lysines [99] and to regulate transcription via interacting with co-activators and co-repressors or by recruiting additional enzymatic activities. Recently, a new family of PHD finger proteins has been found to contain an additional jmjC demethylation module and to be directly responsible for both histone methylation and demethylation, providing a new example of cross-talk among methylation marks [100].
A member of this family is PHD finger 8 (PHF8) protein. PHF8 demethylates H3K9me1/me2 both in vitro and in vivo. Its PHD finger domain recognizes specifically H3K4me3. This interaction is fundamental to stimulate PHF8 demethylase activity and to induce the expression of rDNA genes [101]. Interestingly, a point mutation within the jmjC domain of PFH8 that abolishes its demethylase activity and transcriptional activation has been associated to X-linked mental retardation, linking histone modification cross-regulation and rDNA transcription to neuronal disease [102, 103]. Since many jmjC proteins contain PHD or Tudor domains, it seems that modulation or recruitment of demethylation activities by a pre-existing modified residue might be a common mechanism for this new class of enzymes to propagate histone modification states [104]. In support of this idea the mechanism of action described for PHF8 is conserved by another member of the same family, the jmjC containing protein PHF2 [105].
An additional example of cross-talk between histone methylation marks is the one between H3R2me2 and H3K4me3 [74, 75, 106]. By profiling the presence of several histone marks at the promoters of 151 human genes, Guccione et al. [74] found a counter-correlation between H3R2me2 and H3K4me3. The presence of H3K4me3 at gene promoters is positively correlating with messenger RNA levels, whereas H3R2me2 is not. However, H3R2me2 can be enriched within the body of genes regardless of their transcriptional status. Further analysis allowed to get insights into the molecular mechanisms that governs this negative cross-talk and showed that H3R2 methylation prevents the recruitment of WDR5, one of the subunits of the ASH2/MLL complex, responsible in human for H3K4 methylation. In line with this, genomic regions containing H3K4me3 cannot be methylated at H3R2 by PRMT6. The discovery that WDR5 is absent from regions enriched in H3R2me2 and that PRMT6 depletion affects both H3R2 and H3K4 methylation levels provide additional evidences for cross-talk in vivo between these two methylation sites. This negative cross-talk is highly specific for the H3R2me2, as it has shown that H3R2me1 has distinct functional characteristics and it correlates with active transcription [106]. However, it is still not understood how PRMT6 is recruited to chromatin and how H3R2me2 clearance occurs when a gene is activated. The involvement of additional proteins, the action of a not yet identified arginine demethylase as well as histone replacement are all valid possibilities.
Although the molecular basis of many cross-talks between methylation sites have not been identified yet, recent findings support a model in which the coordinated removal of repressive marks and the deposition of activating marks are important for the stringent regulation of transcription during cellular differentiation [61].
CROSS-TALK BETWEEN LYSINE METHYLATION AND UBIQUITINATION
Although less studied, ubiquitination of lysines can occur within histones and regulate other modifications in different chromatin dependent processes.
So far a lot of attention has been given to the cross-talk between H2B ubiquitination and/or H3K4 and H3K79 methylation, and the molecular mechanisms governing this process have been identified and found to be conserved from yeast to human. H2BK120 monoubiquitination is catalyzed by the mammalian RAD6/BREI complex [107] and is a mark linked to transcriptional elongation [108, 109]. Most importantly, it is required for efficient methylation of H3K4me3 and H3K79me2/me3 both in yeast and mammals [110, 111]. It has been shown that Swd2, one of the subunits of the Set1C/COMPASS complex, responsible in yeast for H3K4 methylation, plays a crucial role in translating the H2B ubiquitination signal into H3 methylation [112]. In mammals, several Set1C/COMPASS-like complexes exist and WDR82, the human homolog of Swd2 has been recently identified as a specific subunit of the SET1A/SET1B complexes. WDR82 depletion in several human cell types leads to a drastic reduction in H3K4me3 levels whereas H3K4me1/me2 are unaffected [111]. Moreover, WDR82 loss affects the stability of the entire SET1A complex and SET1A levels also drops upon RNAi. Finally, WDR82 targeting to chromatin is strictly dependent on the monoubiquitination status of H2B, providing a direct link between H2BK120 ubiquitination and H3K4 methylation. Interestingly, Swd2 interact in vitro and in vivo with Dot1 and partial loss of Swd2 results in a significant reduction of H3K79me3 levels in yeast [111]. One intriguing possibility is that the cross-talk between H2BK120ub and H3K79me in mammals also depends on Swd2/WDR82. However this model requires further studies.
While H2B ubiquitination is linked to active transcription, H2A ubiquitination is considered as a repressive mark. H2AK119 ubiquitination is an abundant modification present in most eukaryotes with the exception of S. cerevisiae and S. pombe. RING1B, a subunit of the PRC1 polycomb complex is the E3 ubiquitin ligase catalyzing H2AK119 ubiquitination [113]. While H2BK120ub controls H3K4 and H3K79 methylation, H3K27me3 set by the PRC2 complex is a prerequisite for H2AK119ub, indicative of a role in HOX gene silencing in mammals [114, 115]. In line with these results, depletion of UTX, the specific H3K27me3 demethylase, results in increased occupancy of PRC1 at the promoter of target HOX genes and concomitant enhancement of H2AK119 ubiquitination [116]. The function of H2AK119ub in transcriptional repression has been reinforced by the finding that H2A deubiquitination by USP21 positively modulates H3K4 methylation at the promoter of several genes induced during liver regeneration [117]. Additionally, in Drosophila H2AK119 ubiquitination is stimulated by H3K36 demethylation catalyzed by dRING associated factors (dRAF), an alternative Polycomb repressive complex involved in transcriptional silencing [117]. dRAF shares with the PRC1 complex both dRING and PSC subunits, but it specifically contains dKDM2, a specific H3K36me2 demethylase. dKDM2 plays a pivotal role in this dynamic trans-histone mechanism by directly coupling H3K36me2 demethylation with stimulation of H2AK119 ubiquitination by dRING. Since H3K36me2 is a mark for transcriptional elongation it has been proposed that dRAF mediated silencing acts via blocking RNA Pol II progression through chromatin [118].
CONCLUDING REMARKS
In an era when rapidly advancing tools are available to perform high throughput genomic screenings, the knowledge about histone modifications interdependence and cross-regulation is crucial, in particular for a comprehensive analysis of histone genomic data in the context of chromatin functions. Moreover, considering the enormous number of histone modifications that had been discovered, their broad combinatorial potential, their cross-talk, as well as the precise and fascinating network built up by the enzymes regulating their chromatin deposition and/or removal, it is logical to assume that the simplistic view of one modification equals one readout equals one specific function needs to be updated, if not reconsidered. So far, we focused on the cross-talk between two modifications, however there is no reason to exclude cross-talk between multiple modifications. We also know still very little about the trans-nucleosome cross-talks contributing to the establishment and maintenance of chromatin domains. Soon it will not be possible to depict these cross-talks in simple models or tables as in this review and the complete picture of the interdependence of histone modifications will be even more complex than originally predicted by the ‘histone code’ hypothesis. This does not account for a lack in specificity, but rather increasing layers of complexity could have been evolved by higher eukaryotes to further control the functional outputs of combination of modifications. Finally, we also need to keep in mind that not only histones are post-translationally modified, but that also the enzymes setting the marks are subjected to the same modifications. The cross-talk and the intense ‘chat’ between post-translation modifications and histone modifying enzymes as well as histone modifications and their readers is just starting to be appreciated and will be for sure one of the main focus of researchers in the field for the future.
Key Points.
Histone post-translational modifications regulate chromatin dynamics.
Lysine and arginine methylation within histones have distinct functions in chromatin dependent processes.
Combinations of different modifications are recognized by specific effector proteins and increase the epigenetic information.
Histone modification can cross-talk in cis and in trans.
FUNDING
Max Planck Society; the Deutsche Forshungsgemeinschaft (DFG, SFB 746); the European Union (EU, the Epigenome); European Research Council (ERC) starting grant.
Acknowledgements
We thank E. Metzger for helpful discussions.
Biographies
Annalisa Izzo, PhD is a postdoctoral scientist at the Max Planck Institute for Immunobiology in Freiburg, where her research focus is the functional role of human H1 in chromatin.
Robert Schneider, PhD is a group leader at the Max Planck Institute for Immunobiology in Freiburg. His scientific interests include histone modifications, histone variants and epigenetic regulation of chromatin dependent processes.
References
- 1.Luger K, Rechsteiner TJ, Flaus AJ, et al. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272(3):301–11. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 2.Kunert N, Brehm A. Novel Mi-2 related ATP-dependent chromatin remodelers. Epigenetics. 2009;4(4):209–11. doi: 10.4161/epi.8933. [DOI] [PubMed] [Google Scholar]
- 3.Talbert PB, Henikoff S. Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11(4):264–75. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–99. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 6.Beck HC, Nielsen EC, Matthiesen R, et al. Quantitative proteomic analysis of post-translational modifications of human histones. Mol Cell Proteomics. 2006;5(7):1314–25. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Wisniewski JR, Zougman A, Kruger S, et al. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics. 2007;6(1):72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Su X, Ren C, Freitas MA. Mass spectrometry-based strategies for characterization of histones and their post-translational modifications. Expert Rev Proteomics. 2007;4(2):211–25. doi: 10.1586/14789450.4.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young NL, DiMaggio PA, Plazas-Mayorca MD, et al. High throughput characterization of combinatorial histone codes. Mol Cell Proteomics. 2009;8(10):2266–84. doi: 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee AY, Paweletz CP, Pollock RM, et al. Quantitative analysis of histone deacetylase-1 selective histone modifications by differential mass spectrometry. J Proteome Res. 2008;7(12):5177–86. doi: 10.1021/pr800510p. [DOI] [PubMed] [Google Scholar]
- 11.Choi JK, Howe LJ. Histone acetylation: truth of consequences? Biochem Cell Biol. 2009;87(1):139–50. doi: 10.1139/O08-112. [DOI] [PubMed] [Google Scholar]
- 12.Robinson PJ, An W, Routh A, et al. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381(4):816–25. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner BM. Decoding the nucleosome. Cell. 1993;75(1):5–8. [PubMed] [Google Scholar]
- 14.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 15.Adams-Cioaba MA, Min J. Structure and function of histone methylation binding proteins. Biochem Cell Biol. 2009;87(1):93–105. doi: 10.1139/O08-129. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Develop. 2009;12(5):659–65. [PMC free article] [PubMed] [Google Scholar]
- 17.Winter S, Fischle W, Seiser C. Modulation of 14-3-3 interaction with phosphorylated histone H3 by combinatorial modification patterns. Cell Cycle. 2008;7(10):1336–42. doi: 10.4161/cc.7.10.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins RE, Northrop JP, Horton JR, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15(3):245–50. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins R, Cheng X. A case study in cross-talk: the histone lysine methyltransferases G9a and GLP. Nucleic Acids Res. 2010;38(11):3503–11. doi: 10.1093/nar/gkq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tropberger P, Schneider R. Going global: novel histone modifications in the globular domain of H3. Epigenetics. 2010;5(2):112–7. doi: 10.4161/epi.5.2.11075. [DOI] [PubMed] [Google Scholar]
- 21.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol. 2004;11(11):1037–43. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 22.Dillon SC, Zhang X, Trievel RC, et al. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6(8):227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min J, Feng Q, Li Z, et al. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112(5):711–23. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 24.Feng Q, Wang H, Ng HH, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–8. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 25.Hublitz P, Albert M, Peters AH. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol. 2009;53(2–3):335–54. doi: 10.1387/ijdb.082717ph. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen SJ, Schneider R, Bauer UM, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412(6846):561–5. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 27.Peters AH, Mermoud JE, O’Carroll D, et al. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet. 2002;30(1):77–80. doi: 10.1038/ng789. [DOI] [PubMed] [Google Scholar]
- 28.Plath K, Fang J, Mlynarczyk-Evans SK, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300(5616):131–5. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 29.Cao R, Wang H, He J, et al. Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol. 2008;28(5):1862–72. doi: 10.1128/MCB.01589-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Cao R, Wang L, et al. Mechanism of Polycomb group gene silencing. Cold Spring Harb Symp Quant Biol. 2004;69:309–17. doi: 10.1101/sqb.2004.69.309. [DOI] [PubMed] [Google Scholar]
- 31.Daujat S, Weiss T, Mohn F, et al. H3K64 trimethylation marks heterochromatin and is dynamically remodeled during developmental reprogramming. Nat Struct Mol Biol. 2009;16(7):777–81. doi: 10.1038/nsmb.1629. [DOI] [PubMed] [Google Scholar]
- 32.Huen MS, Sy SM, van Deursen JM, et al. Direct interaction between SET8 and proliferating cell nuclear antigen couples H4-K20 methylation with DNA replication. J Biol Chem. 2008;283(17):11073–7. doi: 10.1074/jbc.C700242200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pesavento JJ, Yang H, Kelleher NL, et al. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol. 2008;28(1):468–86. doi: 10.1128/MCB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Mizzen CA. The multiple facets of histone H4-lysine 20 methylation. Biochem Cell Biol. 2009;87(1):151–61. doi: 10.1139/O08-131. [DOI] [PubMed] [Google Scholar]
- 35.Vakoc CR, Sachdeva MM, Wang H, et al. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol. 2006;26(24):9185–95. doi: 10.1128/MCB.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Jia S. Degrees make all the difference: the multifunctionality of histone H4 lysine 20 methylation. Epigenetics. 2009;4(5):273–6. doi: 10.4161/epi.4.5.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schotta G, Lachner M, Sarma K, et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18(11):1251–62. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santos-Rosa H, Schneider R, Bernstein BE, et al. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell. 2003;12(5):1325–32. doi: 10.1016/s1097-2765(03)00438-6. [DOI] [PubMed] [Google Scholar]
- 40.Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–71. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morillon A, Karabetsou N, Nair A, et al. Dynamic lysine methylation on histone H3 defines the regulatory phase of gene transcription. Mol Cell. 2005;18(6):723–34. doi: 10.1016/j.molcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Krogan NJ, Dover J, Wood A, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell. 2003;11(3):721–9. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120(2):169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Bolland DJ, Wood AL, Johnston CM, et al. Antisense intergenic transcription in V(D)J recombination. Nat Immunol. 2004;5(6):630–7. doi: 10.1038/ni1068. [DOI] [PubMed] [Google Scholar]
- 45.Gribnau J, Diderich K, Pruzina S, et al. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell. 2000;5(2):377–86. doi: 10.1016/s1097-2765(00)80432-3. [DOI] [PubMed] [Google Scholar]
- 46.Bannister AJ, Kouzarides T. Histone methylation: recognizing the methyl mark. Methods Enzymol. 2004;376:269–88. doi: 10.1016/S0076-6879(03)76018-2. [DOI] [PubMed] [Google Scholar]
- 47.Lee JS, Shilatifard A. A site to remember: H3K36 methylation a mark for histone deacetylation. Mutat Res. 2007;618(1–2):130–4. doi: 10.1016/j.mrfmmm.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 48.Schubeler D, MacAlpine DM, Scalzo D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18(11):1263–71. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 50.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 51.Wyrick JJ, Parra MA. The role of histone H2A and H2B post-translational modifications in transcription: a genomic perspective. Biochim Biophys Acta. 2009;1789(1):37–44. doi: 10.1016/j.bbagrm.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Daujat S, Zeissler U, Waldmann T, et al. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J Biol Chem. 2005;280(45):38090–5. doi: 10.1074/jbc.C500229200. [DOI] [PubMed] [Google Scholar]
- 53.Kuzmichev A, Jenuwein T, Tempst P, et al. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14(2):183–93. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 54.Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 55.Lee MG, Wynder C, Cooch N, et al. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437(7057):432–5. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 56.Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–9. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 57.Lee MG, Wynder C, Bochar DA, et al. Functional interplay between histone demethylase and deacetylase enzymes. Mol Cell Biol. 2006;26(17):6395–402. doi: 10.1128/MCB.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karytinos A, Forneris F, Profumo A, et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284(26):17775–82. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 60.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–27. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 61.Agger K, Cloos PA, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–4. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 62.Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006;11:344–55. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- 63.Litt M, Qiu Y, Huang S. Histone arginine methylations: their roles in chromatin dynamics and transcriptional regulation. Biosci Rep. 2009;29(2):131–41. doi: 10.1042/BSR20080176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner S, Weber S, Kleinschmidt MA, et al. SET-mediated promoter hypoacetylation is a prerequisite for coactivation of the estrogen-responsive pS2 gene by PRMT1. J Biol Chem. 2006;281(37):27242–50. doi: 10.1074/jbc.M605172200. [DOI] [PubMed] [Google Scholar]
- 65.Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24(2):233–43. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang S, Litt M, Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005;19(16):1885–93. doi: 10.1101/gad.1333905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le Guezennec X, Vermeulen M, Brinkman AB, et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. 2006;26(3):843–51. doi: 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Q, Rank G, Tan YT, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16(3):304–11. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Majumder S, Alinari L, Roy S, et al. Methylation of histone H3 and H4 by PRMT5 regulates ribosomal RNA gene transcription. J Cell Biochem. 2010;109(3):553–63. doi: 10.1002/jcb.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dacwag CS, Ohkawa Y, Pal S, et al. The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol Cell Biol. 2007;27(1):384–94. doi: 10.1128/MCB.01528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ancelin K, Lange UC, Hajkova P, et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol. 2006;8(6):623–30. doi: 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- 72.Koh SS, Chen D, Lee YH, et al. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276(2):1089–98. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- 73.Torres-Padilla ME, Parfitt DE, Kouzarides T, et al. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature. 2007;445(7124):214–8. doi: 10.1038/nature05458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guccione E, Bassi C, Casadio F, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449(7164):933–7. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 75.Hyllus D, Stein C, Schnabel K, et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21(24):3369–80. doi: 10.1101/gad.447007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cuthbert GL, Daujat S, Snowden AW, et al. Histone deimination antagonizes arginine methylation. Cell. 2004;118(5):545–53. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 77.Chang B, Chen Y, Zhao Y, et al. JMJD6 is a histone arginine demethylase. Science. 2007;318(5849):444–7. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 78.Dormann HL, Tseng BS, Allis CD, et al. Dynamic regulation of effector protein binding to histone modifications: the biology of HP1 switching. Cell Cycle. 2006;5(24):2842–51. doi: 10.4161/cc.5.24.3540. [DOI] [PubMed] [Google Scholar]
- 79.Prigent C, Dimitrov S. Phosphorylation of serine 10 in histone H3, what for? J Cell Sci. 2003;116(Pt 18):3677–85. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- 80.Lachner M, O'Carroll D, Rea S, et al. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 81.Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 82.Fischle W, Tseng BS, Dormann HL, et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438(7071):1116–22. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 83.Hirota T, Lipp JJ, Toh BH, et al. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438(7071):1176–80. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 84.Crosio C, Heitz E, Allis CD, et al. Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci. 2003;116(Pt 24):4905–14. doi: 10.1242/jcs.00804. [DOI] [PubMed] [Google Scholar]
- 85.Rea S, Eisenhaber F, O’Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 86.Metzger E, Imhof A, Patel D, et al. Phosphorylation of histone H3T6 by PKCbeta(I) controls demethylation at histone H3K4. Nature. 2010;464(7289):792–6. doi: 10.1038/nature08839. [DOI] [PubMed] [Google Scholar]
- 87.Metzger E, Yin N, Wissmann M, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008;10(1):53–60. doi: 10.1038/ncb1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shimada M, Niida H, Zineldeen DH, et al. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132(2):221–32. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 89.Strahl BD, Briggs SD, Brame CJ, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11(12):996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 90.Wang H, Huang ZQ, Xia L, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293(5531):853–7. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 91.Chen D, Huang SM, Stallcup MR. Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J Biol Chem. 2000;275(52):40810–6. doi: 10.1074/jbc.M005459200. [DOI] [PubMed] [Google Scholar]
- 92.Daujat S, Bauer UM, Shah V, et al. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol. 2002;12(24):2090–7. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 93.Pal S, Vishwanath SN, Erdjument-Bromage H, et al. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24(21):9630–45. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gartenberg M. Heterochromatin and the cohesion of sister chromatids. Chromosome Res. 2009;17(2):229–38. doi: 10.1007/s10577-008-9012-z. [DOI] [PubMed] [Google Scholar]
- 95.Vaquero A, Scher M, Lee D, et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16(1):93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 96.Vaquero A, Scher M, Erdjument-Bromage H, et al. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450(7168):440–4. doi: 10.1038/nature06268. [DOI] [PubMed] [Google Scholar]
- 97.Wirth M, Jedrusik-Bode MA. Interplay between histone deacetylase SIR-2, linker histone H1 and histone methyltransferases in heterochromatin formation. Epigenetics. 2009;4(6):353–6. doi: 10.4161/epi.4.6.9710. [DOI] [PubMed] [Google Scholar]
- 98.Ng HH, Feng Q, Wang H, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16(12):1518–27. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matthews JM, Bhati M, Lehtomaki E, et al. It takes two to tango: the structure and function of LIM, RING, PHD and MYND domains. Curr Pharm Des. 2009;15(31):3681–96. doi: 10.2174/138161209789271861. [DOI] [PubMed] [Google Scholar]
- 100.Yue WW, Hozjan V, Ge W, et al. Crystal structure of the PHF8 Jumonji domain, an Nepsilon-methyl lysine demethylase. FEBS Lett. 2010;584(4):825–30. doi: 10.1016/j.febslet.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 101.Feng W, Yonezawa M, Ye J, et al. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat Struct Mol Biol. 2010;17:445–50. doi: 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- 102.Iwase S, Lan F, Bayliss P, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128(6):1077–88. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 103.Kleine-Kohlbrecher D, Christensen J, Vandamme J, et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. 2010;38(2):165–78. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim J, Daniel J, Espejo A, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7(4):397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wen H, Li J, Song T, et al. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J Biol Chem. 2010;285(13):9322–6. doi: 10.1074/jbc.C109.097667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kirmizis A, Santos-Rosa H, Penkett CJ, et al. Distinct transcriptional outputs associated with mono- and dimethylated histone H3 arginine 2. Nat Struct Mol Biol. 2009;16(4):449–51. doi: 10.1038/nsmb.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhu B, Zheng Y, Pham AD, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20(4):601–11. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 108.Pavri R, Zhu B, Li G, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125(4):703–17. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 109.Xiao T, Kao CF, Krogan NJ, et al. Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol Cell Biol. 2005;25(2):637–51. doi: 10.1128/MCB.25.2.637-651.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 111.Nakanishi S, Lee JS, Gardner KE, et al. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J Cell Biol. 2009;186(3):371–7. doi: 10.1083/jcb.200906005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee JS, Shukla A, Schneider J, et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131(6):1084–96. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 113.Stock JK, Giadrossi S, Casanova M, et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9(12):1428–35. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- 114.Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20(6):845–54. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 115.Osley MA. Regulation of histone H2A and H2B ubiquitylation. Brief Funct Genomic Proteomic. 2006;5(3):179–89. doi: 10.1093/bfgp/ell022. [DOI] [PubMed] [Google Scholar]
- 116.Lee MG, Villa R, Trojer P, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318(5849):447–50. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 117.Nakagawa T, Kajitani T, Togo S, et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008;22(1):37–49. doi: 10.1101/gad.1609708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lagarou A, Mohd-Sarip A, Moshkin YM, et al. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev. 2008;22(20):2799–810. doi: 10.1101/gad.484208. [DOI] [PMC free article] [PubMed] [Google Scholar]