

The small intestine is an incredibly adaptable, “plastic” organ, the tissues of which are involved intimately in endocrine regulation, immune surveillance, multiple aspects of metabolism, excretion of metabolites, symbiosis with the enteric microbiome, and, of course, absorption of ingested nutrients. As the primary site of nutrient absorption, the gut must be adaptable to the quality, quantity, and timing of nutrient availability, in order to optimize nutrient absorption and maintain health. Indeed, intestinal adaptation, as evident by changes in gene expression as the neonate grows older and transitions from breast milk to oral nutrients, (i.e., ontogeny) are well described. Similarly, chronic changes in diet/nutrient supply alter gene expression resulting in different levels of nutrient transporters by the enterocytes. A dramatic example of this remodeling of the gut mucosa was demonstrated by Secor and Diamond in studying the onset of hyperplasia/hypertrophy in Burmese pythons in response to ingestion of a meal.1 Indeed, these forms of classic, genomic-mediated adaptation result in a change not only in function but also in structure of the mucosa, and typically occur over days. Similarly, when major parts of the intestinal absorptive surface area are lost (via resection for trauma, neoplasm, or disease) or bypassed (jejunoileal bypass), a remarkable hypertrophy/hyperplasia occurs, again through a complex set of changes in gene expression and cellular proliferation leading to villus hypertrophy and cellular hyperplasia.
A less well understood and less well appreciated “acute adaptation,” deemed by some to be more of a physiologic effect, also occurs in the enterocyte itself, whereby its cellular absorptive capacity is altered acutely in response to luminal presence of specific nutrients – a form of cell biology that does not, at least initially, involve a change in genomic expression and reflects a post-transcriptional process. This acute adaptation involves a rapid alteration (occurring within minutes) in nutrient transporters in the apical membrane secondary to acute translocation of pre-formed transporter from the cytoplasm to the apical membrane of the enterocyte; this acute translocation of pre-formed transporters matches intestinal absorptive capacity to nutrient availability. This review will address (focusing on hexose transporters) this remarkable capacity of the enterocyte to undergo a rapid, post-transcriptional, increase in absorptive capacity, a process that appears different and complementary to the classic genomic-based forms of intestinal adaptation.
Classic Model of Hexose and Glucose Absorption
In the past, glucose absorption was believed to occur primarily via two different hexose transporters with defined locations within the enterocyte (Figure 1). Sodium-glucose cotransporter 1 (SGLT1) is a secondary active symporter located in the apical membrane of the enterocyte. SGLT1 has a high affinity for glucose but a relatively low capacity for its transport and utilizes the intracellular Na+ gradient created by the basilar Na+/K+ ATPase to transport glucose with sodium ions across a concentration gradient from the intestinal lumen into the cytosol of the enterocyte. Once in the cytosol, glucose is then transported out of the cell and into the portal venous system via glucose transporter 2 (GLUT2), a high capacity, low affinity, facilitated transporter believed at one time to be limited to the basolateral membrane of the enterocyte. GLUT2 is also responsible for the transport of fructose across the basolateral membrane. Glucose transporter 5 (GLUT5), also a facilitated transporter but located in the apical membrane, moves fructose from the intestinal lumen into the cytosol (Figure 1). The limitation of this classic model of hexose uptake has always been that this model cannot account adequately for the substantial amount of glucose absorbed immediately after a meal, even with saturation of SGLT1. With an apparent Km of ~5 mM (i.e. the glucose concentration leading to half the maximum velocity of uptake), SGLT1 is saturated at 30–50 mM glucose; however, overall glucose absorption from the gut does not saturate even at concentrations of glucose exceeding 100 mM.2 To early investigators, this “unsaturable” component appeared to be simple diffusion and seemed to call into question the teleologic need for a glucose transporter if large amounts of glucose could be absorbed simply via diffusion. A clue that led to the discovery of the transporter responsible for the “unsaturable” component and necessitated a change in the working model of hexose absorption was the finding that GLUT2 was located in the apical membrane as well as the basolateral membrane of enterocytes in diabetic rats.
Figure 1. Classic Model of Hexose Absorption by the Enterocyte.
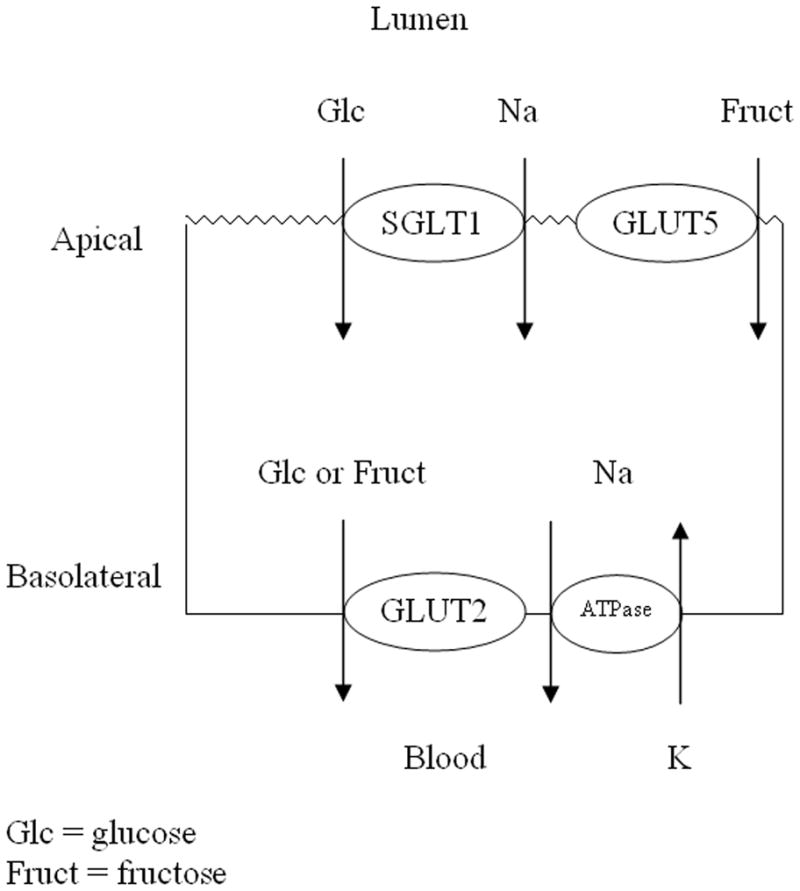
SGLT1 and GLUT5 located in the apical membrane and GLUT2 located only basolaterally. Basal Na+/K+ ATPase generates the apical gradient of sodium ions.
Proposed New Model of Glucose Absorption
Apically-located GLUT2, what was once thought to be a pathologic aberration of diabetes mellitus, is now thought to be a normal, albeit transient, physiologic phenomenon that occurs in response to high luminal concentrations of glucose. Through a series of elegant experiments over the past decade by Kellett and colleagues, preformed cytosolic stores of GLUT2 have been shown to “traffic” reversibly from cytosolic vesicles to the apical membrane to increase the glucose absorptive capacity of the enterocyte2, 3 –so called apical translocation.
SGLT1 is thought to be vital to the initiation of the translocation process. SGLT1 transports simultaneously glucose and Na+ into the enterocyte in a ratio of 1:2 (Figure 2). This co-transport of Na+ into the cell causes depolarization which activates the voltage-gated, L-type calcium channel Cav1.3 promulgating an influx of Ca2+. This increase in intracellular Ca2+ results in remodeling of the terminal web and cytoskeletal structures necessary for the trafficking of GLUT2 to the apical membrane. Increases in intracellular Ca2+ also “prime” but do not fully activate the βII isoform of protein kinase C (PKCβII) by increasing its affinity for phosphatidylserine. In this manner, Ca2+ has the effect of increasing the amount of PKCβII at the apical membrane.4 The influx of calcium is greatest at glucose concentrations that just begin to saturate SGLT1 (~30 mM).2–4 Other stimuli that increase PKC activity also appear to affect GLUT2 translocation. PMA (phorbol 12-myristate 13-acetate) increases PKC activity non-specifically and, in doing so, augments glucose absorption via stimulation of apical translocation of GLUT2 despite otherwise low concentrations of luminal glucose.
Figure 2. Proposed New Model of Glucose Transport.

High luminal concentrations of glucose saturate SGLT1 causing influx of Na+, stimulating Cav1.3 causing influx of Ca2+. Through remodeling of the terminal web PKCβII is activated. Lumenal glucose also stimulates apical sweet taste receptors which activate PLCβ2 which further activates PKCβII via PIP2 and DAG. This bimodal activation of PKCβII induces apical translocation of pre-formed GLUT2 to markedly augment cellular uptake of luminal glucose. Whether PKCβII also induces basolateral translocation of GLUT2 is unknown.
In addition, at glucose concentrations near the saturating levels of SGLT1, a newly appreciated set of “taste receptors” located apically on enterocytes and enteroendocrine cells is activated.5 The sweet taste receptor, which “senses” glucose concentrations exceeding 30 mM, is a heterodimer composed of the T1R2+T1R3 proteins coupled to gustducin, a heterotrimeric G-protein composed of α, β, and γ subunits. The sweet taste receptor initiates the remaining intracellular signals necessary for GLUT2 to be trafficked and inserted into the apical membrane. When activated, the receptor complex is internalized, and the α subunit of gustducin is released into the cytosol. Subunits β and γ remain at the apical membrane, where they activate the B2 isoform of phospholipase C (PLCβ2). PLCβ2 cleaves diacyglycerol (DAG) from phosphatidyl inositol-bisphosphate (PIP2), which further activates PKCβII by facilitating the removal of the N-terminal pseudosubstrate from the active site. The now activated PKCβII, which appears to be the main effector of GLUT2 trafficking, causes GLUT2 to be targeted and inserted into the apical membrane from a cytosolic pool of preformed GLUT2.4
The process of apical translocation of preformed cytosolic vesicles containing GLUT2 is a complex process involving the sorting of GLUT2 vesicles from the Golgi apparatus, mobilizing these vesicles along the cellular cytostructure/microtubules, a process that involves “motors,” “priming,” and “docking” of GLUT2 to the terminal web area of the cellular membrane. A similar process of translocation of cytosolic vesicles is used with many other cellular proteins and is a well-orchestrated, complex process that is directed not only apically but also to the basolateral membrane, depending on the substrate to be translocated.
Sweet taste receptors are involved in the regulation of both SGLT1 and GLUT2. Recent data demonstrate that stimulation of sweet taste receptors leads to a rapid (<3 h) post-transcriptional increase in whole-cell levels of SGLT1, through a process that involves vagal afferent neurons and 5-HT receptors.6
Because the translocation into (and out of) the apical membranes is so rapid and dependent on local conditions, the presence of apical GLUT2 was overlooked for many years. Under “normal” dietary conditions, a minor and as of yet poorly defined amount of GLUT2 is expressed constitutively in the apical enterocyte membrane.7 Increased amounts of apical GLUT2 are detectable at glucose concentrations >30–40 mM, but much greater levels become evident at glucose concentrations in the gut lumen ≥75 mM.2, 4 Given a typical Western diet and the presence of disaccharidases at the brush border membrane, local concentrations of glucose at this level and greater appear to occur frequently after a meal of polysaccharides. Interestingly, several commercial artificial sweeteners (i.e. saccharine, sucralose, acesulfame) used commonly in “diet” soft drinks have been shown (ironically) to activate the sweet taste receptor complex independent of glucose concentration.4, 6 This activation of sweet taste receptors by artificial “sweeteners” even when the concentration of glucose is low (i.e. ~ 10mM) triggers trafficking of GLUT2 to the apical membrane resulting in an increased capacity to absorb glucose (energy). This overall process of GLUT2 trafficking is quick, occurring within minutes.2, 3
Such rapid post-transcriptional regulation of proteins allows the enterocyte to modulate transporter levels very rapidly based on the nutrient milieu present in the intestinal lumen and thus “adapt” swiftly in real-time. Such an “adaptive” response by the enterocyte does not appear to be limited only to GLUT2 and SGLT1. Changes in the amount of transport have been described for transporters of non-glucose nutrients as well. Peptide transporter 1 (PepT1), a secondary active symporter that relies on a proton gradient to transport di- (predominantly) and tri-peptides, also appears to traffic to the apical membrane in a manner similar to that of GLUT2. To date, under all conditions studied, PepT1 levels in the apical membrane appear to decrease when GLUT2 is increased and to increase when GLUT2 is decreased.4 PepT1 appears to be regulated in the same manner, although reciprocally, as GLUT2 with activated PKCβII being the final mediator. Ultimately, any stimulus that activates PKCβII will increase the apical levels of GLUT2 in the enterocyte while simultaneously decreasing the levels of PepT1 (Figure. 2A-D).
Caveats to the GLUT2 hypothesis
Of the theories proposed above, the one related to apical translocation of GLUT2 has caused significant debate. The late discovery of the translocation process has been attributed to several factors. Much of the early work on glucose absorption was performed in vitro. Several groups have demonstrated a very rapid loss of apical GLUT2 when the intestine is excised under normal conditions.3, 7 For GLUT2 to be trafficked and to remain in the apical membrane, it appears to require some heretofore unidentified component(s) (i.e. paracrine hormone, permissive intrinsic nervous system signal, etc.) not present in the intestine when studied under many in vitro conditions.7 The practice of fasting animals (or humans prior to endoscopy) the evening prior to experimentation has also been cited as a factor that caused apical GLUT2 to be overlooked initially, because even brief periods of starvation decrease markedly the levels of apical GLUT2; the enterocyte uses the apical levels of constitutive SGLT1, a high affinity, low capacity transporter, to capture what little glucose may be present in the intestinal lumen between meals. Initially, immunocytochemical techniques directed at the C-terminus detected GLUT2 only at the basolateral membrane; however, when a peptide sequence in the first extracellular loop was the targeted epitope, and the intestine was harvested rapidly, kept cold, and placed in an appropriate medium that increased intracellular calcium and prevented inhibition of PKCβII, constitutive levels of GLUT2 and increased levels of translocated GLUT2 were found in the apical membrane and were determined to be regulated as described above.8
Although the GLUT2 hypothesis makes biologic “sense” and appears appealing, several groups have questioned its validity, specifically in humans. Patients suffering with glucose-galactose malabsorption, a disorder that results from mutations in the SGLT1 gene, cannot absorb glucose or galactose. Fructose, however, can be absorbed normally, indicating the presence of a functional transcellular route via luminal GLUT5 and basolateral GLUT2 for the absorption of fructose. The presence of GLUT2 on the luminal membrane would forestall the chronic diarrhea experienced by these children, suggesting that the expression of GLUT2 on the luminal membrane may not extend to humans.9, 10 Others studies have also shown relatively normal intestinal glucose absorption in mice with a form of GLUT2 knockout suggesting a different transcellular mechanism of glucose uptake.11
In addition, several groups, including both of ours have been unable to demonstrate GLUT2 translocation in our studies, which could be due to rapid translocation of GLUT2 out of the membrane during our harvest, experimental setup, or our antibody selection.
Cellular Mechanisms Regulating Apical Translocation of GLUT2
Much or most of the studies demonstrating the presence of apical translocation of GLUT2 have been carried out in vivo in the rat, possibly because of difficulty showing a similar phenomenon in ex vivo small intestine. Interestingly, little if any prior work in cell culture models has explored this mechanism regulating GLUT2 translocation. Because of this concept, we have developed several cell culture models of enterocyte-like cells–Caco-2 cells, RIE-1 cells, and IEC-6 cells to study the GLUT2 hypothesis. Caco-2 is a human cell line developed from colon cancer which, when grown under specific conditions, differentiates into enterocyte-like cells with apical microvilli and high levels of apical alkaline phosphatase. Both RIE-1 and IEC-6 cells are derived from rat enterocytes and also differentiate with enterocyte characteristics. All three cell lines take up glucose by a carrier-mediated process inhibited by the specific SGLT1 inhibitor phlorezin. In contrast, in Caco-2 and RIE-1 cells, but not in IEC-6 cells, glucose uptake is also partially inhibited by phloretin, a specific GLUT2 inhibitor, suggesting constitutive expression of some apical GLUT2 in Caco-2 and RIE-1 cells, but not in IEC-6 cells, giving us a nice model to examine regulation of GLUT2 translocation. Interestingly, at short durations of exposure to glucose in the culture medium, uptake becomes saturated at 10–25 mM glucose. In contrast, when exposed for ≥5 minutes, glucose uptake no longer saturates, indicative of the presence of a different apical transporter with a much greater Km. This glucose uptake is decreased by GLUT2 inhibition, PKC inhibition, and disruption of the cell cytostructure; conversely, uptake is increased by PMA which activates PKC non-specifically. Consistent with the in vivo experiments, inhibiting SGLT1 also prevents the GLUT2-augmented uptake. When studied in IEC-6 cells that lack functional GLUT2, these GLUT2-directed stimulatory or inhibitory approaches had no effect on glucose uptake (unpublished data). In contrast, when we transfected IEC-6 cells with a plasmid containing GLUT2 (inducing the cells to express GLUT2), we were able to reproduce the GLUT2-specific effects, suggesting the presence of cellular machinery able to translocate GLUT2 apically. These experiments outline a mechanism whereby the enterocyte functionally “senses” the luminal content of glucose and adapts acutely via an intracellular signaling pathway to augment glucose uptake.
Summary
The function of the small intestine has evolved to maximize uptake of glucose (and other nutrients) to maintain homeostasis. While most concepts of intestinal “adaptation” involve genomically-mediated hypertrophy/hyperplasia, the enterocyte itself has an adaptive mechanism that responds rapidly (within minutes) to changes in luminal nutrients that does not require gene expression (hours to days). Investigating the cell biology of this process of “acute adaptation” has led to insights into a new receptor system (sweet taste receptors), and possibly the beginnings of further understanding of several aspects of diabetes mellitus and obesity. Much remains to be learned about this adaptive mechanism and how it is related to other cellular systems and other intracellular processes of cell biology. In particular, little is known about how apical GLUT2, SGLT1, and other transporters are modulated by various local and systemic, metabolic, hormonal, and paracrine effects.
References
- 1.Secor SM, Diamond J. Adaptive responses to feeding in Burmese pythons: pay before pumping. J Exp Biol. 1995;198:1313–25. doi: 10.1242/jeb.198.6.1313. [DOI] [PubMed] [Google Scholar]
- 2.Kellett GL, Helliwell PA. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J. 2000;350(Pt 1):155–62. [PMC free article] [PubMed] [Google Scholar]
- 3.Helliwell PA, Richardson M, Affleck J, Kellett GL. Stimulation of fructose transport across the intestinal brush-border membrane by PMA is mediated by GLUT2 and dynamically regulated by protein kinase C. Biochem J. 2000;350(Pt 1):149–54. [PMC free article] [PubMed] [Google Scholar]
- 4.Mace OJ, Lister N, Morgan E, et al. An energy supply network of nutrient absorption coordinated by calcium and T1R taste receptors in rat small intestine. J Physiol. 2009;587:195–210. doi: 10.1113/jphysiol.2008.159616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mace OJ, Affleck J, Patel N, Kellett GL. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J Physiol. 2007;582:379–92. doi: 10.1113/jphysiol.2007.130906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stearns AT, Balakrishnan A, Rhoads DB, Tavakkolizadeh A. Rapid upregulation of sodium-glucose transporter SGLT1 in response to intestinal sweet taste stimulation. Ann Surg. 2010;251:865–71. doi: 10.1097/SLA.0b013e3181d96e1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scow JS, Iqbal CW, Jones TW, 3rd, et al. Absence of evidence of translocation of GLUT2 to the apical membrane of enterocytes in everted intestinal sleeves. J Surg Res. doi: 10.1016/j.jss.2010.04.026. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Affleck JA, Helliwell PA, Kellett GL. Immunocytochemical detection of GLUT2 at the rat intestinal brush-border membrane. J Histochem Cytochem. 2003;51:1567–74. doi: 10.1177/002215540305101116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turk E, Zabel B, Mundlos S, Dyer J, Wright EM. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature. 1991;350:354–6. doi: 10.1038/350354a0. [DOI] [PubMed] [Google Scholar]
- 10.Wright EM, Turk E, Martin MG. Molecular basis for glucose-galactose malabsorption. Cell Biochem Biophys. 2002;36:115–21. doi: 10.1385/CBB:36:2-3:115. [DOI] [PubMed] [Google Scholar]
- 11.Kellett GL, Brot-Laroche E. Apical GLUT2: a major pathway of intestinal sugar absorption. Diabetes. 2005;54:3056–62. doi: 10.2337/diabetes.54.10.3056. [DOI] [PubMed] [Google Scholar]