

Abstract
BACKGROUND AND PURPOSE
We previously reported that the activation of the nuclear factor of activated T-lymphocyte-3 (NFAT3) by carboplatin leads to renal apoptosis as a result of oxidative stress, which is reversed by N-acetylcysteine. Herein, we extend our previous work to provide evidence of the molecular mechanisms of haem oxygenase (HO)-1 in protecting against injury.
EXPERIMENTAL APPROACH
Protective mechanisms of HO-1 in carboplatin-mediated renal apoptosis were examined in C57BL/6 mice and rat renal tubular cells (RTC) with HO-1 induction or inactivation/knockdown.
KEY RESULTS
The HO-1, induced by cobalt protoporphyrin, protected against carboplatin-induced renal injury in vivo. This protection was decreased by an inhibitor of HO-1 action, tin protoporphyrin. In cultures of RTC, carboplatin-induced apoptosis was similarly affected by HO-1 overexpression or knockdown. Carboplatin-mediated NFAT3 activation and apoptosis involve activation of the signalling kinases, extracellular signal regulated kinase, Jun N-terminal kinase and protein kinase C, and such activation was reversed in cells overexpressing HO-1. Both products of the HO-1 reaction, CO and bilirubin, inhibited (by 30–40%) NFAT3 activation and production of the pro-apoptotic proteins Bcl-XS/Bax. Additionally, the activation of NFκB was markedly decreased by HO-1 induction.
CONCLUSION AND IMPLICATIONS
HO-1 and its reaction products show anti-apoptotic effects in carboplatin-mediated renal injury. A novel functional NFAT3 binding site identified in the rat HO-1 promoter region was involved in producing a 1.5-fold to 2.5-fold increase in HO-1 induction by carboplatin. Nevertheless, only HO-1 overexpression and activation prior to the carboplatin challenge provided protection against carboplatin-induced injury.
Keywords: carboplatin, haem oxygenase-1, nuclear factor of activated T-lymphocytes, ERK/JNK, PKC, reactive oxygen species, apoptosis, renal tubular cells
Introduction
Haem oxygenase (HO) is a rate-limiting enzyme in haem degradation which produces equimolar amounts of carbon monoxide iron, and biliverdin. Biliverdin is further converted to the antioxidant, bilirubin by biliverdin reductase (Maines, 1988; Ponka, 1999). Two HO isozymes were identified with distinct genes (Maines, 1997) and one of them HO-1, a stress-response protein, can be induced by various oxidation-inducing agents, including haem, heavy metals, UV radiation, cytokines and endotoxins (Platt and Nath, 1998; Otterbein and Choi, 2000). Recently, numerous in vitro and in vivo studies showed that the induction of HO-1 is an important cellular protective mechanism against oxidative injury (Maines, 1997). Several lines of evidence indicate that not only HO-1 per se but also its products, such as CO and bilirubin, enhance the anti-apoptotic and anti-oxidant functions in various cells and animal models (Kwak et al., 1991; Taille et al., 2004; Abraham and Kappas, 2008). The biological actions of bilirubin have been shown to scavenge reactive oxygen species (ROS) in vitro (Kushida et al., 2002) and inhibit NADPH oxidase (Kwak et al., 1991), thereby reducing oxidant-mediated cellular damage and attenuating oxidant stress in vivo (Stocker et al., 1987). Furthermore, CO has been demonstrated to contribute significantly to the anti-inflammatory properties of HO-1 by suppressing inflammatory cytokines through activation of both soluble guanylate cyclase and p38 mitogen activated protein kinase (MAPK) (Otterbein et al., 2000; Zhan et al., 2003).
Carboplatin (cis-diammine-1,1-cyclobutanedicarboxylate platinum II), a second-generation platinum-containing anticancer drug, is currently being clinically used against lung, ovarian, and head and neck cancers (Pivot et al., 2001; Fujiwara et al., 2003). Carboplatin is more water-soluble and has fewer adverse effects than its analogue, cisplatin. Carboplatin, however, has a cancer DNA-damaging activity that is equal to cisplatin at similar toxic doses (Alberts, 1995). The antitumour action of carboplatin is mediated by alkylation of DNA, which can lead to the death of cancerous cells. Carboplatin has less toxic adverse effects than cisplatin and increased doses of carboplatin are commonly used to achieve optimal antitumour effects. The predominant dose-limiting toxicities of carboplatin are bone marrow suppression and ototoxicity caused by free radical oxidative injury to such organs (Husain et al., 2001). Previously, using both gain- and loss-of-function strategies, we showed that activation of a transcription factor, nuclear factor of activated T-lymphocyte-3 (NFAT3), is responsible for renal tubular cell (RTC) apoptosis. Interestingly, pretreatment with the ROS scavenger, N-acetylcysteine (NAC), reversed NFAT3 activation and RTC apoptosis (Lin et al., 2010).
Moderate levels of ROS were reported to modulate the functions of transcriptional factors through many signalling pathways, although excess ROS can cause oxidative damage and induce cytotoxic effects in cells. H2O2 was shown to increase intracellular Ca2+ and phosphorylation of protein kinase C (PKC), p44/42, p38 MAPK, and Jun N-terminal kinase (JNK)/SAPK. An increase in ROS is also a common mechanism for activating NFκB. This was supported by the observation that H2O2 exposure can rapidly activate NFκB (Chandel et al., 2000). We previously demonstrated that ROS, increased by carboplatin, activated NFAT3 through the extracellular signal regulated kinase (ERK), JNK, and PKC signalling pathways. Herein, we extend our previous work to assess the therapeutic implications of the anti-apoptotic and anti-inflammatory protein, HO-1.
The nuclear factor of activated T-cells, cytoplasmic (NFATc) transcription factors contain a conserved calcineurin (CNA)-binding domain and a Rel-homologous DNA-binding domain. The NFAT are activated by the protein phosphatase, CNA, in response to increased intracellular Ca2+. The NFAT dephosphorylated by calcineurin are translocated to the nucleus, bind DNA, and regulate gene expression (Bushdid et al., 2003). Five NFAT genes, NFATc1–4 and NFAT5, were identified, each with distinct temporally and spatially regulated expression patterns (Rao et al., 1997; Bushdid et al., 2003). The NFAT1–4 activate gene transcription by integrating inputs from the calcium/CNA and PKC/MAPK signalling pathways. The NFAT3 is mainly expressed outside the immune system with high levels in the kidneys, lungs, and placenta. The NFAT3 is also required for cardiac development and mitochondrial functioning (Bushdid et al., 2003). The NFAT activation was mediated by the generation of ROS by asbestos, nickel subsulphide and vanadium. The scavenging of these compound-mediated ROS with NAC resulted in the inhibition of NFAT activation (Huang et al., 2001a,b; Li et al., 2002).
The physiological role of NFAT3 is important for embryonic and cardiovascular development, whereas knockdown of NFATn3 and NFATn4 resulted in cardiac hypertrophy. We also found (Lin et al., 2010) that the activation of NFAT3 is attributable to the carboplatin-mediated alternative Bcl-X splicing which generates a high level of the pro-apoptotic molecule, Bcl-XS. NFAT3, a nuclear transcription factor, may be beneficial to downstream targets by turning them on for self-protection when encountering oxidative stress. However, this remains to be investigated.
The NFAT3 was shown to be one of the gene expression profiles which identified patients with a higher risk of recurrence (Rosell et al., 2007). The involvement of NFAT3 in human lung cancer development was supported by the finding that high NFAT expression was present in Chinese non-small cell lung cancers (Zhang et al., 2007). However, the role of NFAT in cancer progression is still a matter of debate. For example, the activation of NFAT3 by arsenite in human bronchial epithelial cells was shown to exert an anti-apoptotic effect through induction of cyclooxygenase-2 expression. Conversely, mice lacking NFAT2 exhibited a decreased extent of central tumour necrosis, which was associated with a reduced tumour necrosis factor-α and interleukin-2 production by CD8+ cells in a murine model of bronchoalveolar adenocarcinoma.
We previously showed that carboplatin-mediated ROS generation induced NFAT3 activation, leading to RTC apoptosis. Here, we have used HO-1 as a therapeutic agent, based on its antioxidative and anti-apoptotic properties, to attenuate carboplatin-mediated renal toxicity. We also examined the molecular mechanism of endogenous HO-1 induction by carboplatin and its effect on carboplatin-mediated renal injury.
Methods
Histological and biochemical assessments of mice treated with carboplatin or with an additional HO-1 inducer/inhibitor
All animal care and experimental procedures were conducted in conformity with the protocols of the Animal Center, Taipei Medical University. Male C57BL/6 mice (8 weeks old, 25–30 g) were used in all studies. Mice were housed under standard conditions on a 12 h light : 12 h dark cycle. Experiments were carried out under general anaesthesia following an intraperitoneal (i.p.) injection of a mixture of ketamine (Pfizer, Taipei, Taiwan; 40 mg·kg−1) and xylazine (Sigma, St. Louis, MO, USA; 8 mg·kg−1). Animals were divided into five groups each receiving various treatments (n = 6): (i) control; (ii) 100 mg·kg−1 carboplatin (from Sigma); (iii) 5 mg·kg−1 cobalt protoporphyrin (CoPP; Sigma) + 100 mg·kg−1 carboplatin; (iv) 10 mg·kg−1 tin(IV) protoporphyrin (SnPP; Frontier Scientific, Salt Lake city, UT, USA) + 100 mg·kg−1 carboplatin; and (v) 10 mg·kg−1 SnPP + 5 mg·kg−1 CoPP + 100 mg·kg−1 carboplatin. Mice received CoPP at a dose of 5 mg·kg−1 (dissolved in 0.2 M NaOH with the pH adjusted to 7.4 and diluted in 0.85% NaCl) by an i.p. injection 24 h prior to the carboplatin challenge. This dose (10 mg·kg−1) of SnPP effectively reversed a CoPP-mediated increase in HO-1 activity, even though SnPP is a nonselective HO inhibitor (Hartsfield et al., 2004). The SnPP was given for 1 day, 2 h ahead of the CoPP treatment. This was followed by a single dose of 100 mg·kg−1 of carboplatin (i.p.) for 3 days. Mice were challenged with carboplatin to cause renal damage, whereas control mice were injected with an equal amount of double-distilled water (ddH2O) (n = 6/group). Blood urea nitrogen and serum creatinine levels were measured using Fuji Dri-Chem Slides (Fujifilm, Kanagawa-ken, Japan) at the indicated times. For histological analysis, kidneys were removed from the mice, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5–10 µm for histological staining with haematoxylin and eosin and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Apoptosis in renal tissues was identified by a TUNEL assay with an in situ Cell Death Detection kit (Roche, Mannheim, Germany) used according to the manufacturer's instructions. Five fields per section and two sections per kidney were examined in each experimental group. For immunohistochemical staining of HO-1, tissue sections were pretreated with 3% H2O2 for 10 min at room temperature to exhaust the endogenous peroxidase activities. After incubation in PBS containing 1% bovine serum albumin and 1% goat serum at 37°C for 30 min, sections were first treated with an anti-HO-1 antibody (Stressgen, Victoria, BC, Canada or Assay Designs, Ann Arbor, MI, USA) for 30 min at 37°C, washed three times in PBS, and then incubated with Texas red-conjugated goat-anti-mouse horseradish immunoglobulin (IgG) at 37°C for 30 min. After a solution of 0.1% 3,3-diaminobenzidine tetrahydrochloride/0.01% H2O2 had developed colour, sections were counterstained in 4′,6-diamidino-2-phenylindole (DAPI) by using previously described procedures (Chang et al., 2009).
Cell culture and establishment of HO-1 transfectants
The NRK-52E rat renal proximal tubular cells (RTC) were purchased from the Bioresource Collection and Research Center (Hsinchu, Taiwan)and used in this study. The RTC were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with an antibiotic/antifungal solution and 10% fetal bovine serum (FBS) (pH 7.2). They were grown until the monolayer became confluent. The medium for the cultured cells was then changed to a serum-free medium, and cells were incubated overnight before the experiment. A constitutive expression vector, pcDNA-HO-1, carries full-length human HO-1 complementary (c)DNA under the control of the cytomegalovirus promoter/enhancer sequence. A short 771 hairpin (sh)RNA against rat HO-1 was generated in the pSM2 vector (Open Biosystems, Huntsville, AL, USA) and amplified in an Escherichia coli system. Confirmation was verified by restriction site analysis and sequencing. Using the jetPEI™ (Polyplus-transfection, San Marcos, CA, USA), pcDNA-HO-1, pcDNA or pSM2-shHO-1 (4 µg/3.5 cm Petri dish) was transfected into RTC. After transfection, cells were plated in DMEM with 10% FBS and 400 µg·mL−1 of G418 for pcDNA variants, or 2 µg·mL−1 of puromycin for pSM2-shHO-1 as selective pressures. G418- or puromycin-resistant cells were selected and expanded. The level of HO-1 was analysed by Western blotting.
HO-1 enzymatic activity
The generation of bilirubin was used to estimate the activity of the HO-1 enzyme (Abraham et al., 1995). Livers were pooled and homogenized on ice in a Tris-HCl lysis buffer (pH 7.4, containing 0.5% Triton X-100 and protease inhibitors). Homogenates (100 µL) were further mixed with 0.8 mM NADPH, 0.8 mM glucose-6-phosphate, 1.0 unit of glucose-6-phosphate dehydrogenase, 1 mM MgCl2, haemin (0.25 mM; from Sigma), and mouse liver cytosol containing biliverdin reductase which was at 4°C. The reaction was conducted at 37°C for 15 min in the dark, and terminated by the addition of chloroform. Insoluble material was removed by centrifugation and the bilirubin concentration of the supernatant was analysed by comparing the absorbances at 464 and 530 nm. Controls included samples prepared without the NADPH-generating system.
Construction of the rat HO-1 promoter and luciferase activity assays of the NFAT and NFkB enhancers and rat HO-1 promoter
The pGL3/rat HO-1 reporter plasmid contains a 642-bp fragment located −667 to −26 bp relative to the transcription start site of the rat HO-1 gene. The method of obtaining it and the reporter activity assay were described previously (Juan et al., 2005). A DNA fragment of the rat HO-1 gene promoter sequence from −667 to −26 was obtained by polymerase chain reaction (PCR) using mouse genomic DNA as the template. The respective sense and antisense primers were 5′-AAGCTTCATCCTCCAGCTCA-3′ and 5′-GCTCCGCTCGAGATGGCTCTGCTC-3′. The PCR product was flanked by dA and subcloned into the TA cloning vector, which was digested with KpnI and BglII restriction enzymes and subcloned into the KpnI and BglII sites of the pGL3-basic vector.
For the reporter activity assay, cell variants were seeded in 24-well plates at a density of 1 × 105 cells per well. In brief, cell variants were transiently transfected with 1.02 µg of plasmid DNA containing 0.02 µg of the Renilla luciferase construct, pRL-TK (Promega), to control the transfection efficiency, and 1 µg of the pGL4.10-NFAT (Promega, Madison, WI, USA), pNFkB-luciferase (TGGGGACTTTCCGC)5 (Stratagene, La Jolla, CA,USA) or pGL3-rat HO-1 promoter. Cell variants were transfected with these vectors using LipofectAMINE 2000™ (Invitrogen, Carlsbad, CA, USA). After transfection for 4 h, the medium was replaced with complete medium, and incubated for another 20 h. Transfected cells were then treated with carboplatin for 4 h, after which, cell lysates were collected. Luciferase activities were recorded in a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA) using a dual luciferase assay kit (Promega) according to the manufacturer's instructions. Luciferase activities of the reported plasmids were normalized to luciferase activities of pRL-TK.
Analysis of gene expression by reverse-transcription PCR and Western blot analysis
RNA for the by reverse-transcription PCR analysis was obtained as previously described (Pang et al., 2008) with minor modifications. Sequences of primer pairs for amplification of each gene were 5′-TCTTCAGGACCTCTGCCCTA-3′ and 5′-AGCCTAGGAGCTTGACCACA-3′ for the NFAT3 gene (180 bp); 5′-CACGCATATACCCGCTACCT-3′ and 5′-AAGGCGGTCTTAGCCTCTTC for the HO-1 gene (227 bp); and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for the GAPDH gene (451 bp). Total RNA, at 5 µg, of extracts from RTC was used. The level of the housekeeping gene, GAPDH, was analysed and used to identify the presence of the same amount of total complementary (c)DNA in each RNA sample.
Antibodies for NFAT3, p47-PHOX, Bcl-XL/XS, Bax, NFκB-p65, NFκB-p50, Lamin A/C, total and phosphorylated MAPK, PKC, cadherin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), HO-1 (Stressgen or Assay Designs), caspase-9 (Cell Signaling, Beverly, MA, USA), cleaved caspase-3 (Cayman Chemical, Ann Arbor, MI, USA), and GAPDH (Ab Frontier, Seoul, Korea) were included in the assay. The RTC in 10 cm2 dishes was harvested after treatment with 200 µM carboplatin at the indicated times and partitioned into cytosolic and nuclear fractions using NE-PER™ nuclear extraction reagents (Pierce, Rockford, IL, USA) with the addition of protease inhibitors according to the manufacturer's instructions. To prepare the membrane-cytosolic fractions, after the indicated treatment, cells were collected and incubated in 0.1 mL of hypotonic buffer [10 mM Tris (pH 7.5), 0.5 mM EDTA, and 2 mM phenylmethylsulphonyl fluoride] at 4°C for 30 min. After centrifugation, the supernatant (cytosolic fraction) was collected, and the pellet was resuspended in 0.1 mL of radioimmune precipitation assay (RIPA) buffer and incubated at 4°C for 30 min. The resulting fractions were sheared 100 times through an insulin syringe with a 29 Gauss needle. After centrifugation, the supernatant (membrane fraction) was collected for analysis. Cell lysates (50 µg) were analysed by electrophoresis on a 10% sodium dodecyl sulphate (SDS)-polyacrylamide gel and then transblotted onto a Hybond-P membrane (GE Healthcare, Hong Kong, China). Subsequent procedures are described elsewhere (Lin et al., 2008).
Electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) assay
The EMSA was performed as described previously (Shih et al., 2004) with minor modifications. To prepare the nuclear protein extracts, RTC in 10 cm2 dishes were subjected to various indicated pretreatments for 1 h, followed by an increasing concentration of carboplatin from 0 to 400 µM for another hour. Then, NE-PER™ nuclear extraction reagents (Pierce) and protease inhibitors were added. The subsequent procedures for the nuclear protein extraction followed the manufacturer's instructions. The MOTIF Search found a putative NFAT in the rat HO-1 promoter region. The NFAT fragment spanning from −614 to −595 bp of the HO-1 promoter was TGAACCTTGGAAAAGTCAGT (the putative NFAT is boxed). This was synthesized as a probe for the EMSA and end-labelled with biotin according to the manufacturer's protocol (Pierce). Briefly, unlabeled oligonucleotides (1 µM) were incubated in TdT reaction buffer containing biotin-11-dUTP (0.5 µM) and TdT (0.2 U·µL−1) at 37°C for 30 min, followed by the addition of 2.5 µL EDTA (0.2 M, pH 8.0) to stop each reaction and 50 µL chloroform/isoamyl alcohol to extract the TdT. Extracted nuclear proteins (10 µg) were incubated with biotin-labelled (1 pmol) probes at 15°C for 30 min in a binding buffer containing 1 µg of poly-deoxyinosinedeoxycytidine (Panomics, Redwood City, CA, USA). A 100-fold molar excess of unlabeled oligonucleotides, relative to the biotin-labelled probes, was added to the binding assay. The mixture was separated on a 6% non-denaturing polyacrylamide gel at 4°C in 1 × TBE (90 mM Tris borate and 2 mM EDTA; pH 8.3) and then transblotted onto a Hybond N+ membrane (Amersham Pharmacia Biotech, Freiburg, Germany). Blots were incubated with blocking buffer, followed by the addition of streptavidin-horseradish peroxidase conjugates. Blots were imaged by means of an enhanced chemiluminescence system.
A ChIP assay was performed according to the instructions of Upstate Biotechnology (Lake Placid, NY, USA) with minor modifications. Briefly, 6 × 105 cells cultured in 100 mm dishes with previously indicated treatments were harvested. The resulting supernatant was subjected to overnight co-immunoprecipitationusing an anti-NFAT3 antibody, or the same amount of a nonspecific antibody (α-Tu) as a negative control. This was followed by incubation with a salmon sperm DNA/protein G agarose slurry (Upstate) to immobilize the DNA-protein-antibody complex. DNA-protein complexes were then eluted with 200 µL of elution buffer (Tris-EDTA buffer containing 1% SDS) for 30 min, and the cross-links were reversed by incubating overnight at 65°C. DNA was purified with a PCR purification kit (Qiagen, Hilden, Germany). The DNA filtrates were amplified by PCR with primers flanking the promoter of the HO-1 gene containing the putative NFAT: HO-1 forward 5′-AAGCTTCATCCTCCAGCTCA-3′ and reverse primers 5′-ACCTTCCCGGAACTCTAAGG-3′. The template was replaced with H2O as a negative internal control. The PCR products were analysed by electrophoresis on a 2% agarose gel, and PCR products of the expected size of 176 bp were visualized, quantified using the Image analysis system, and eluted from the agarose gel for sequencing to verify the site of amplification.
NFAT3 siRNA preparation
An NFAT3 siRNA (CCAAGGUGGUGUUCAUUGATT and CUACAGAUGUUCAUCGGCATT) duplex was chemically synthesized by Ambion (Austin, TX, USA). RTC were seeded in six-well plates and transfected with either 100 pmol of NFAT3 siRNA (nos. s9483 and s9484, Ambion), or a scrambled control siRNA (no. 4611, Ambion) in a 100 µL volume with siPORT NeoFX. The efficiency of siRNA silencing was analysed by Western blotting after transfection for 48 h, followed by carboplatin treatment for the indicated times.
The NADPH oxidase activity and protein oxidation were assessed by measuring the amount of protein carbonyls.
The RTC were treated with 200 µM of carboplatin for various periods of time, and NADPH oxidase was measured as previously described (Clark et al., 2000; Sue et al., 2009). To measure protein carbonyls, cells pretreated with NAC followed by a 24 h carboplatin challenge were extracted in RIPA buffer which contained 2% 2-mercaptoethanol to inhibit oxidation. Carbonyl groups formed as a result of oxidative stress. This was achieved with the exposure of 20 µg of protein to 2,4-dinitrophenylhydrazine for 15 min using an OxyBlot kit following the manufacturer's instructions (Millipore, Bedford, MA, USA).
Determination of ROS generation and apoptosis in RTC
The RTC from each treatment group were grown on a coverglass loaded with 10 µM of the non-fluorescent dye, 20,70-dichlorodihydrofluorescein diacetate (H2DCFDA, Molecular Probes, Eugene, OR, USA) at 37°C for 30 min in the dark. This was followed by a carboplatin challenge (30 min, 200 µM). Subsequent procedures were as described previously (Cheng et al., 2008). Apoptosis of RTC after a 24 h carboplatin challenge was identified by a TUNEL assay with an in situ Cell Death Detection kit (Roche, Mannheim, Germany) used according to the manufacturer's instructions. Fluorescence was viewed with a CCD camera (DP72, Olympus, Melville, NY, USA) attached to a microscope system (BX51, Olympus) at 100 × magnification. Four coverslips in each experimental group were examined.
Statistical analysis
Values are expressed as the mean ± SEM of at least three experiments. The significance of the difference from the control groups was analysed by a Student's t-test or one-way anova. A value of P < 0.05 was considered statistically significant.
Materials
Glucose-6-phosphate, glucose-6-phosphate dehydrogenase, bilirubin, [Ru(CO)3Cl2]2/tricarbonyldichloro-ruthenium(II) dimer (CORMII), NAC, phorbol 12-myristate 13-acetate (TPA) were purchased from Sigma Chemical. U0126 and SP600125 were obtained from Tocris Cookson Inc. (Bristol, UK). RO318220 was from Calbiochem.
Results
Therapeutic effects and mechanisms of HO-1 in protecting mice against a carboplatin challenge
In light of the antioxidative and anti-apoptotic activities of HO-1, the HO-1 inducer, CoPP, was used to examine the therapeutic effects of HO-1 on carboplatin-mediated renal damage. SnPP, an inhibitor of the HO-1 reaction, was used to confirm the importance of HO-1 in this effect. One day prior to the carboplatin challenge, 8-week-old C57BL/6J mice were given SnPP (10 mg·kg−1 i.p.) 2 h before treatment with CoPP (5 mg·kg−1). After 4 days, these animals were killed, and blood samples and kidneys were collected for further analyses. Figure 1A shows the total nuclei in kidney sections revealed as bright spots stained with DAPI. The scattered and bright nuclei stained by TUNEL staining could be detected over the entire cortex of the carboplatin-treated animals, but were rarely detected in specimens of the control or CoPP-treated animals. Most of the TUNEL-labelled nuclei were seen in the proximal tubules. This result demonstrated that mice receiving CoPP showed a marked decrease (80–90%) of carboplatin-induced cell apoptosis in the kidneys, compared to those receiving carboplatin alone. This was reversed in mice with additional SnPP treatment. Additionally, mice challenged with carboplatin showed substantial histological changes, such as severe tubular apoptosis. In contrast, most of the tubular structure and integrity were maintained, and histological damage was milder in mice additionally treated with CoPP compared to those given carboplatin alone or with additional SnPP treatment. Furthermore, in the renal and liver functional assessment shown in Figure 1B, mice given carboplatin showed increased serum levels of urea, creatinine, and alanine transaminase (GPT), which suggests renal and liver dysfunction. However, levels of serum urea, creatinine, and GPT in mice additionally treated with CoPP were significantly lower than those observed in the carboplatin-alone group. These levels were reversed with additional SnPP administration, which suggests the importance of HO-1 activity in the prevention of renal or liver malfunction associated with carboplatin toxicity. Furthermore, CoPP treatment caused a significant induction of HO-1 expression and activity and rescued carboplatin-mediated NFAT3 activation (Figure 1C). The additional SnPP that inhibited HO-1 activity reversed the protective effect of HO-1 by CoPP according to a Western blot analysis. Although the carboplatin treatment alone also induced HO-1 expression and activity, this was insufficient to reverse the detrimental effect of carboplatin on the kidneys in terms of the extent of renal injury, NFAT3 activation, and levels of blood urea nitrogen and creatinine. This suggests that the timing of HO-1 expression and its activity are critical for exerting its protection against carboplatin challenge.
Figure 1.
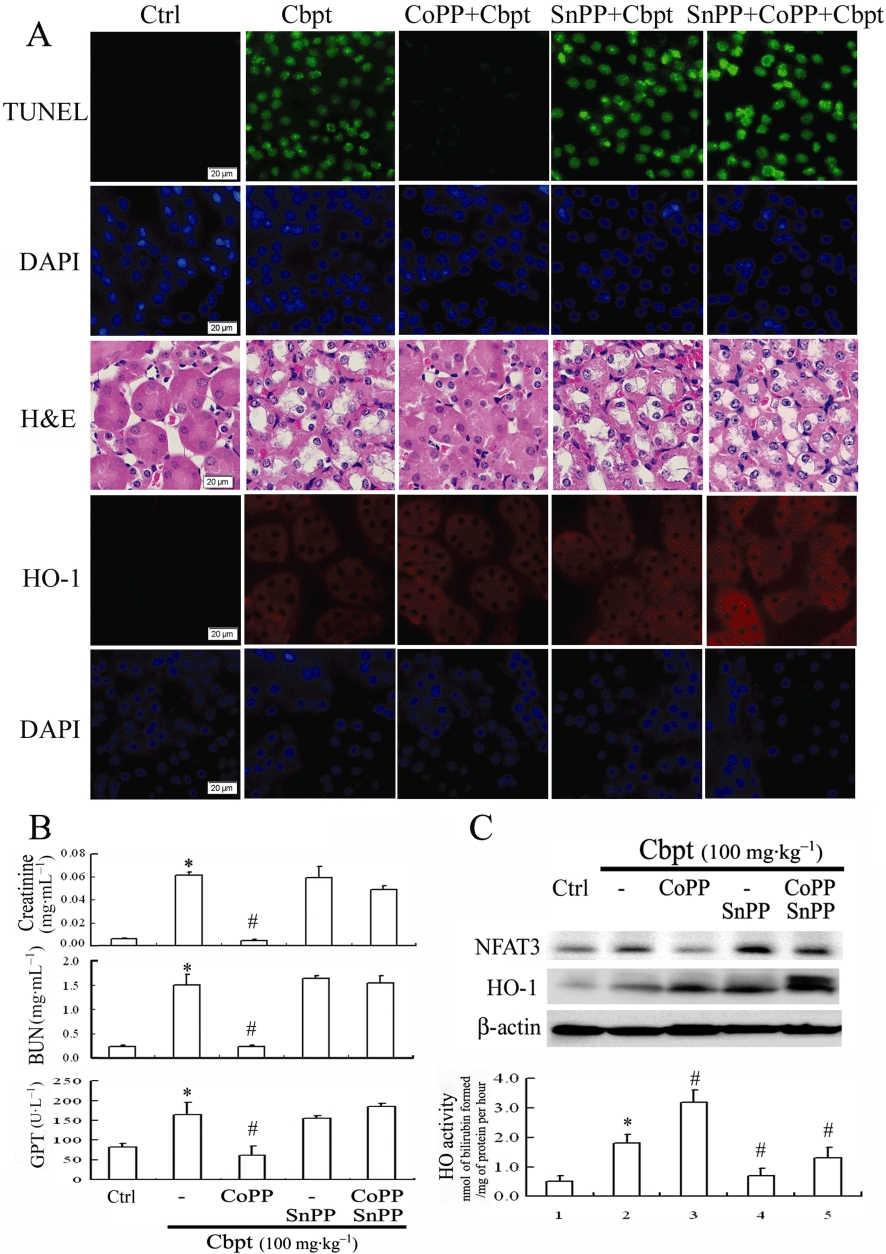
Therapeutic effect of haem oxygenase (HO)-1 in protecting against carboplatin (Cbpt)-mediated renal injury. (A) Animals were pretreated with cobalt protoporphyrin (CoPP) alone or in combination with tin protoporphyrin (SnPP). This modulated the expression or activity of HO-1 before giving carboplatin (100 mg·kg−1) once daily, over a 3 day period. Animals were divided into five groups (n = 6): control (Ctrl); carboplatin alone; and carboplatin plus additional pretreatment with CoPP, SnPP, or both. Kidneys were embedded and sectioned for apoptotic and histological evaluations by terminal deoxynucleotidyl transferase dUTP nick end labeling and haematoxylin and eosin staining and immunochemical staining of HO-1. (B) Serum from treated animals was collected to assess kidney and liver functions, including levels of creatinine, blood urea nitrogen (BUN), and alanine transaminase (GPT). (C) Lysates of kidney homogenization were analysed for protein levels of nuclear factor of activated T-lymphocyte-3 (NFAT3) (140 kDa; dephosphorylated NFAT3) and HO-1 in response to various treatments, and β-actin was used as the internal control to indicate equal amounts of protein loading. A representative result of three separate experiments is shown. Additionally, the activity of the induced-HO-1 expression in each group was evaluated. This is shown in the lower panel of Figure 1C. Data shown in Figure 1B,C are presented as the mean ± SD of three independent experiments. *P < 0.05, significantly different from control group; #P < 0.05, significantly different from carboplatin-treatment alone.
Carboplatin-mediated ROS generation was reversed by HO-1 overexpression in RTC
The protective molecular mechanism of the therapeutic effect of HO-1 in vivo was further examined in RTC alone, with HO-1 overexpression or with knockdown (RTC variants). The RTC overexpressing HO-1 showed a decreased carboplatin-mediated p47PHOX activation and its activity by approximately 80%, whereas RTC with HO-1 knockdown showed an increased effect (Figure 2A,B), suggesting a role for the antioxidative effect of HO-1 in preventing carboplatin-mediated oxidative stress. Carboplatin-mediated ROS production (Figure 2C), as measured by NADPH oxidase activity and ROS-mediated carbonyl formation (Figure 2D) were consistently reduced in RTC overexpressing HO-1, but were increased in RTC with HO-1 knockdown.
Figure 2.
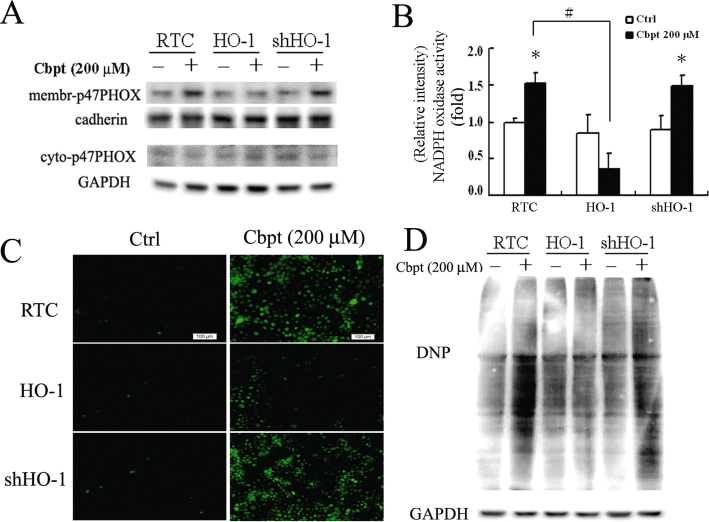
Elimination of carboplatin (Cbpt)-mediated p47PHOX membrane translocation, NADPH oxidase activity, reactive oxygen species (ROS) production, and protein carbonyl group formation in renal tubular cells (RTC) overexpressing haem oxygenase (HO)-1. (A) RTC variants were challenged with 200 µM carboplatin for 1 h, and cell lysates were partitioned into membrane-cytosolic fractions for a Western blot analysis of activated p47PHOX. (B) NADPH oxidase in RTC variants challenged with 200 µM carboplatin for 1 h. Four samples were analysed in each group, and values are presented as the mean ± SEM of three independent experiments. *P < 0.05, significantly different from its respective cell variants alone; #P < 0.05, significantly different from RTC with additional carboplatin challenge. (C) ROS production in cell variants was subjected to 1 h of 200 µM carboplatin administration as previously described. (D) Cell variants were treated with 200 µM carboplatin for 24 h, and cell lysates were harvested and analysed for oxidative stress-mediated carbonyl formation by Western blotting. Representative CM-H2DCFDA fluorescent photomicrographs of carboplatin-mediated ROS production and Western blot analysis of carbonyl group formation induced by carboplatin in RTC variants with the above-mentioned treatments are shown. Ctrl, control.
Inhibition of carboplatin-mediated NFAT3 activation in RTC overexpressing HO-1 by inactivation of the ERK/JNK/PKC signalling pathways
The NFAT activity is responsible for carboplatin-mediated RTC apoptosis through activation of the ERK/JNK/PKC pathway as a result of oxidative stress. The effects of HO-1 overexpression on these signalling pathways in protecting against carboplatin-mediated RTC apoptosis were examined. Figure 3A shows that RTC overexpressing HO-1 reduced carboplatin-mediated activation of PKC/ERK/JNK, whereas knockdown of HO-1 in RTC increased these signalling pathways. Likewise, carboplatin-mediated NFAT3 activation was suppressed in RTC overexpressing HO-1, but was increased in RTC with HO-1 knockdown according to a Western blot analysis of the cytosol-nuclear fractions of NFAT3 (Figure 3B) and by a luciferase assay driven by an NFAT enhancer (Figure 3C).
Figure 3.
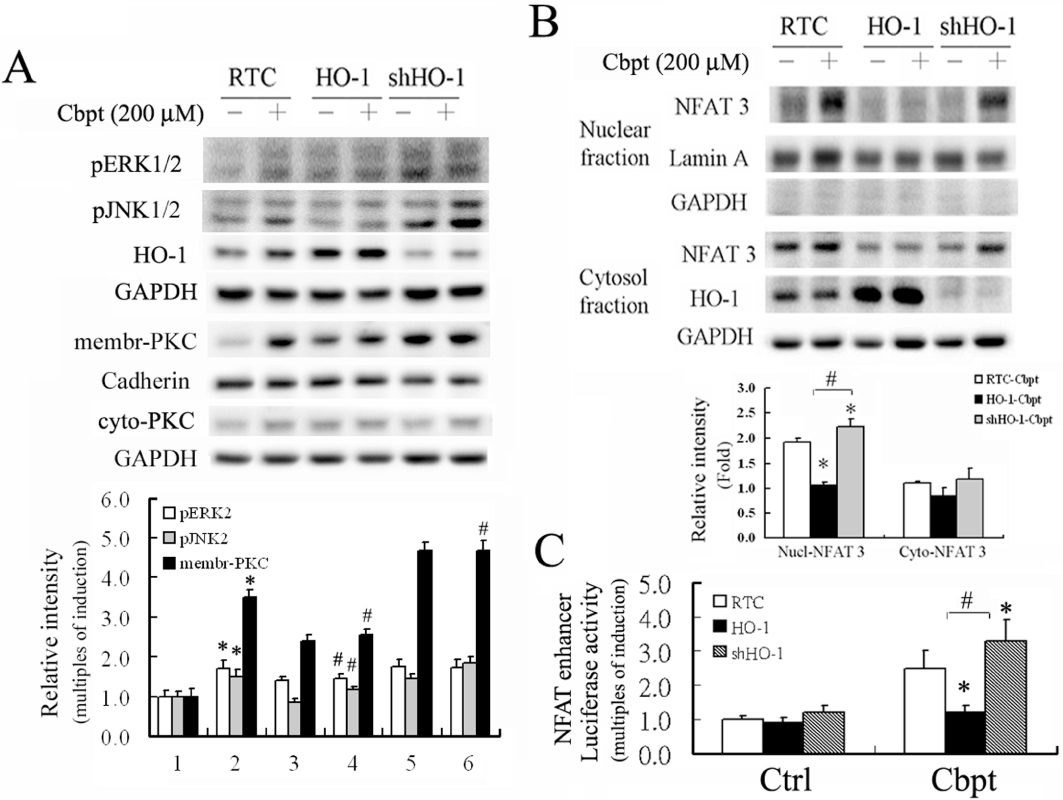
Decrease of nuclear factor of activated T-lymphocyte-3 (NFAT3) activation, its related signalling pathways, and NFAT-driven luciferase activity induced by carboplatin (Cbpt) in renal tubular cells (RTC) overexpressing haem oxygenase (HO)-1 and their increase in RTC with HO-1 knockdown. (A) Cell variants were treated with 200 µM carboplatin to examine activation of the extracellular signal regulated kinase (ERK)/Jun N-terminal kinase (JNK)/protein kinase C (PKC) pathways involved in NFAT3 activation by Western blot analysis. (B) carboplatin-treated RTC variants were partitioned into nuclear-cytosolic fractions for the Western blot analysis of NFAT3 activation. (C) Cell variants were transiently transfected with the NFAT3-reported vector and pRL-TK for 24 h, followed by a 200 µM carboplatin treatment for 4 h. Results show the mean ± SEM of four independent experiments (*P < 0.05, significantly different from pGL3 alone and #P < 0.05, significantly different from pGL3 with additional carboplatin treatment). Ctrl, control.
Anti-apoptotic effect of HO-1 in rescuing carboplatin-mediated RTC apoptosis
Figure 4A shows that RTC overexpressing HO-1 produced an approximately 20% increase in the anti-apoptotic molecule, Bcl-XL, and 25–30% and 35–50% decreases in the pro-apoptotic molecules of Bax and Bcl-XS at both the mRNA and protein levels respectively. A 65–85% concomitant decrease in the amounts of cleaved caspases-3 and -9 occurred with RTC overexpressing HO-1. Conversely, these effects were potentiated in RTC with HO-1 knockdown. Additionally, PKC activation was shown to be involved in Bcl-X alternative splicing (Revil et al., 2007). A carboplatin-mediated alteration in Bcl-X alternative splicing was effectively reversed by the PKC inhibitor, R0318220, but to a slightly lesser extent than by the respective ERK and JNK inhibitors, U0126 and SP600125 (Figure 4B). Therefore, the PKC activator, TPA, was used to reverse the protective effects of HO-1 in carboplatin-mediated RTC apoptosis. R0318220 and NAC, a PKC inhibitor and ROS scavenger, respectively, were used to provide protection against injury in RTC with HO-1 knockdown. Figure 4C shows that TPA reversed the protective effect of HO-1 with an increasing ratio of Bcl-XL to Bcl-XS, but in contrast, the PKC inhibitor and antioxidant NAC restored the pro-apoptotic effect of carboplatin in RTC with HO-1 knockdown. The HO-1-mediated rescue of carboplatin-induced increases in the activated forms of caspases-3 and -9 lead to decreased renal apoptosis, as seen in the TUNEL assay (Figure 4D) and the cell viability assay (Figure 4E). As expected, RTC overexpressing HO-1 showed decreased cell apoptosis with a concomitant increase in cell viability by approximately 25% compared to RTC alone. A 30% decrease in the extent of cell survival was observed in RTC with HO-1 knockdown as compared to RTC alone.
Figure 4.
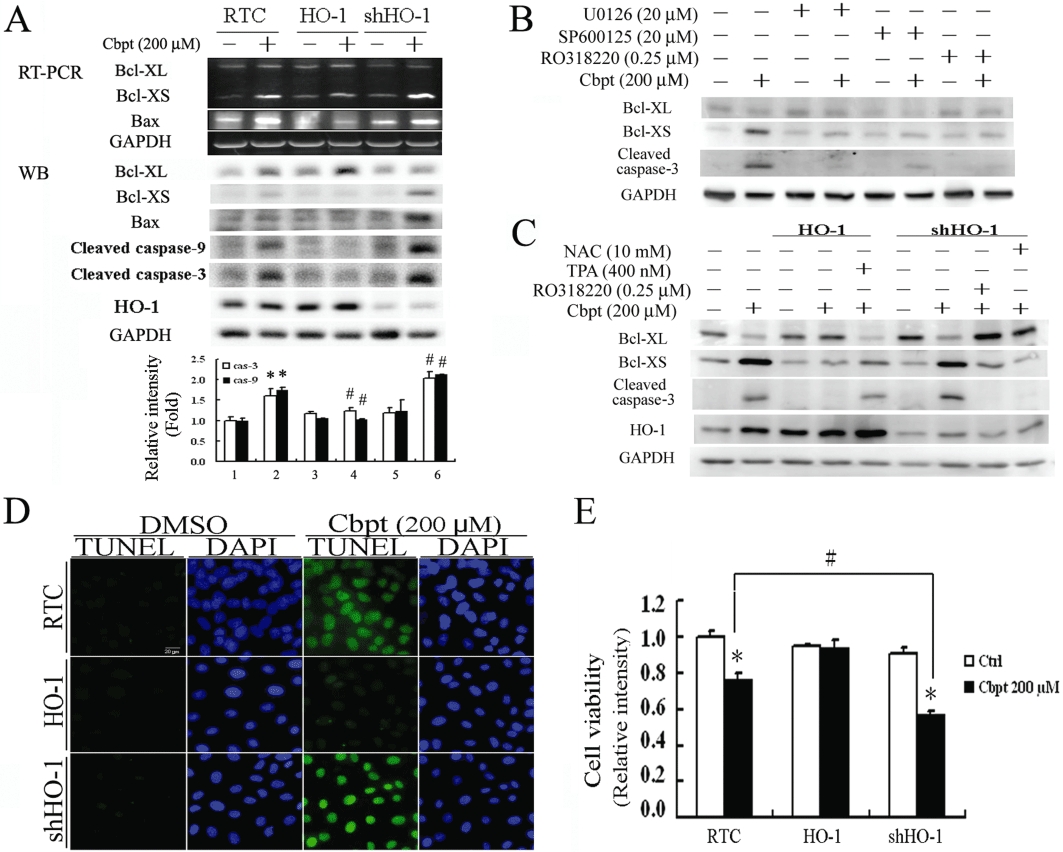
Rescue of carboplatin (Cbpt)-mediated apoptosis by haem oxygenase (HO)-1 overexpression through inhibition of apoptosis-related molecules. (A) Renal tubular cell (RTC) variants were treated with 200 µM carboplatin for 4 h for the reverse-transcription polymerase chain reaction (RT-PCR) analysis of Bcl-XL/XS and Bax, and for 18 h for the Western blot analysis of cleaved caspses-3 and -9. The lower panel shows the intensity of bands from the Western blots by densitometry. Data are presented as the mean ± SEM of three independent experiments. (B) RTC were pretreated with U0126, SP600125 and R0318220, which are inhibitors of the extracellular signal regulated kinase (ERK)/Jun N-terminal kinase (JNK)/protein kinase C (PKC) pathways involved in nuclear factor of activated T-lymphocyte-3 activation. The treatment continued for 1 h prior to a 200 µM carboplatin treatment for 18 h. Cell lysates were harvested for the Western blot analysis of Bcl-XL/Bcl-XS, and the cleaved form of caspase-3. (C) The effect of the PKC pathway in carboplatin-mediated Bcl-X alternative splicing was examined in cell variants pretreated with a PKC activator or inhibitor. RTC with either HO-1 overexpression or knockdown were pretreated with TPA (a PKC activator) or R0318220 (a PKC inhibitor), for 1 h prior to a 200 µM carboplatin treatment for 18 h. Cell lysates were analysed by Western blots for levels of Bcl-XL and Bcl-XS. A representative result of three separate experiments is shown. Cell variants treated with 200 µM carboplatin for 24 or 36 h were analysed for cell apoptosis (D) and cell viability (E) respectively. A representative result of three separate experiments is shown. Data are presented as the mean ± SEM (*P < 0.05, significantly different from its respective RTC variants; #P < 0.05, significantly different from RTC with additional carboplatin treatment). Ctrl, control; DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulphoxide; NAC, N-acetylcysteine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Molecular mechanism of HO-1 in protecting against carboplatin-mediated RTC apoptosis
The actual mechanism of the anti-apoptotic effect of HO-1 in RTC challenged with carboplatin was examined by including the end-products of the HO-1 reaction, CO (as the CO releasing compound CORMII; Scragg et al., 2008) and bilirubin. Cells were pretreated with CORMII or bilirubin for 1 h prior to the carboplatin challenge and, as shown in Figure 5, either treatment significantly suppressed carboplatin-mediated NFAT3 activation by 30–40%. It also suppressed caspase-3 activation. A concomitant 2.6-fold to 4.1-fold increase in Bcl-XL occurred with decreases in Bcl-XS and Bax by 43–50% and 48–67% respectively. This mimicked the protective effect of RTC overexpressing HO-1. This suggests that the anti-apoptotic effects of HO-1 include effects derived from both products of the HO-1 reaction, CO and bilirubin.
Figure 5.
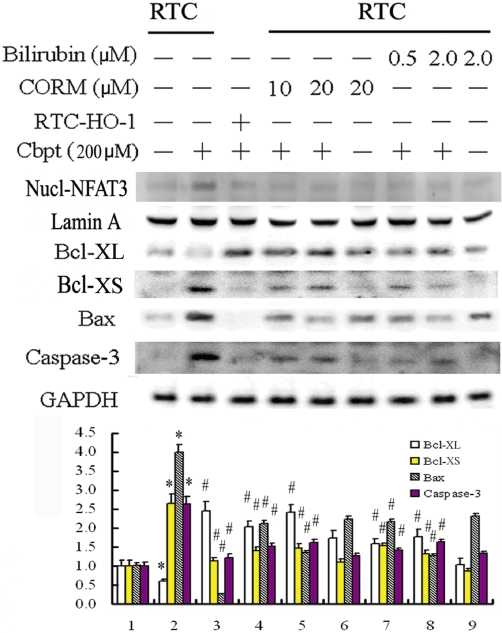
The protective mechanism of haem oxygenase (HO)-1 in carboplatin (Cbpt)-mediated apoptosis by the administration of carbon monoxide (from CORMII) or bilirubin, which are both products of HO-1 action. Cells were pretreated with [Ru(CO)3Cl2]2/Tricarbonyldichlororuthenium(II) dimer (CORMII) or bilirubin for 1 h to mimic the anti-apoptotic effect of HO-1. This was followed by a carboplatin challenge for 18 h for the Western blot analysis of nuclear factor of activated T-lymphocyte-3 (NFAT3) from nuclear fractions, Bcl-XL/-XS, Bax, and cleaved caspase-3. Equal loading or transfer was confirmed by incubation with anti-Lamin A, for nuclear protein, and an anti-GAPDH antibody, for total protein. A representative result of three separate experiments is shown. The lower panel shows the intensity of bands from Western blots by densitometry. Data are presented as the mean ± SEM of three independent experiments. *P < 0.05, significantly different from RTC alone; #P < 0.05, significantly different from RTC with additional carboplatin treatment.
Anti-inflammatory effect of HO-1 in carboplatin-mediated renal apoptosis
The pro-inflammatory effects of platinum-based chemotherapeutic agents such as carboplatin, induce RTC injury (Mukhopadhyay et al., 2010a,b;). Thus, apart from the antioxidative effect of HO-1, the anti-inflammatory effect of HO-1 in carboplatin-mediated RTC apoptosis was also examined. To assess activation of pro-inflammatory molecules, such as NFκB-p65 and NFκB-p50, RTC with NFAT3 knockdown and cell variants treated with or without 200 µM carboplatin were partitioned into cytosolic and nuclear fractions. Cells with NFAT3 knockdown exhibited approximately 50% and 30% reductions in NFκB-p65 and NFκB-p50 activation, respectively (Figure 6A). Additionally, Figure 6B shows that HO-1 overexpression decreased carboplatin-mediated nuclear translocation of the pro-inflammatory nuclear transfection factors, p65/p50. In contrast, these transfection factors are increased by approximately 62–75% in cells with HO-1 knockdown. Furthermore, the effect of carboplatin on the NFκB enhancer-driven luciferase activity and the relation of carboplatin to cells with NFAT3 knockdown and RTC variants were examined. Figure 6C shows that the carboplatin-induced increase in NFκB-driven luciferase activity in RTC alone was significantly inhibited in RTC with NFAT3 knockdown or with HO-1 overexpression by 50–56%. The same activity was increased in RTC with HO-1 knockdown by approximately 21%.
Figure 6.
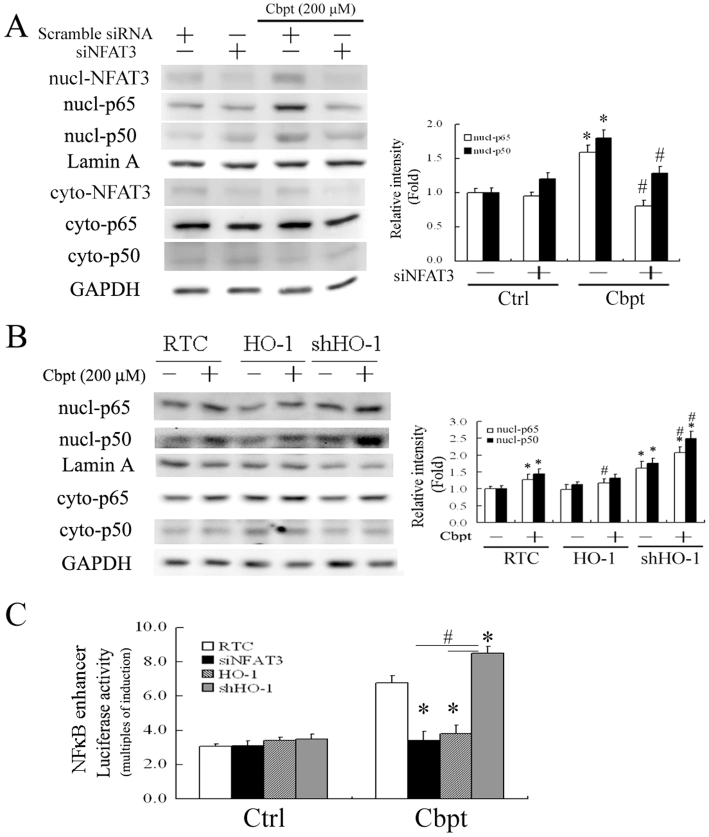
Protective effect of haem oxygenase (HO)-1 in carboplatin (Cbpt)-mediated renal tubular cell (RTC) apoptosis by blocking NFκB activation. (A,B) RTC with nuclear factor of activated T-lymphocyte-3 (NFAT3) knockdown and RTC variants were treated with 200 µM carboplatin for 1 h and partitioned into nuclear-cytosolic fractions to activate the proinflammatory factors of NFκB-p65 and -p50 (*P < 0.05, significantly different from RTC alone; #P < 0.05, significantly different from RTC with additional carboplatin treatment). (C) Cells with various treatments were transiently transfected with the NFκB enhancer vector and pRL-TK for 24 h, followed by a 200 µM carboplatin treatment for 4 h. Results are presented as the mean ± SEM of four independent experiments (*P < 0.05, significantly different from RTC with additional carboplatin challenge; #P < 0.05, significantly different from RTC with NFAT3 knockdown and overexpressing HO-1 with additional carboplatin treatment).
Effects and molecular mechanisms of HO-1 induction by carboplatin
As noted, RTC with carboplatin challenge slightly increased HO-1 induction (Figures 1A,C and 3A) while RTC with HO-1 knockdown exhibited more extensive carboplatin injury than RTC alone (Figures 3, 4 and 6). HO-1 induction by carboplatin was hypothesized to be a cellular protective mechanism. Nevertheless, Figure 7A shows that carboplatin concentration-dependently induced HO-1 at both the mRNA and protein levels and this induction was positively correlated with increasing activation of caspase-3. This suggests the importance of timing for HO-1 induction to exert its protection against carboplatin-mediated renal injury. Whether HO-1 contributed to any self-protection in cells with carboplatin challenge was further examined using RTC with HO-1 knockdown. The results demonstrated that RTC alone decreased the extent of caspase-3 activation by 30–50% as compared with HO-1 knockdown RTC with additional carboplatin challenge. Therefore, the rat HO-1 promoter region spanning −667 to −26 bp was cloned into the pGL3 basic luciferase vector. Figure 7B shows that carboplatin, at 50–200 µM concentrations, concentration-dependently increased native HO-1 promoter-driven luciferase activities by 1.5-fold to 2.5-fold in RTC, except at the highest concentration of 400 µM carboplatin. This might have been due to its severe toxicity to cells. The MOTIF analysis found a highly conserved NFAT response element spanning −614 to −595 bp of the rat HO-1 promoter region. An EMSA was performed in RTC with carboplatin challenge to substantiate the binding of NFAT3 to the predicted binding site derived from the rat HO-1 promoter region. Figure 7C shows the results of the EMSA. Increased DNA-binding activity of NFAT3 in cells treated with carboplatin was found. The increased binding activity of NFAT3 to the NFAT-binding site in cells treated with carboplatin was eliminated by competition with a 100-fold molar excess, relative to the biotin-labeled probe, of unlabeled oligonucleotides. The association of NFAT3 with the NFAT region of the HO-1 promoter was further confirmed using a ChIP assay. A carboplatin-induced association with the NFAT3-DNA complex was created by pulling down the NFAT fragment of the HO-1 gene promoter using an anti-NFAT3 antibody with an anti-α-tubulin antibody serving as a negative control. The immunoprecipitated NFAT fragments were amplified by PCR to examine the binding of NFAT3 to the fragment. Figure 7D shows that carboplatin concentration-dependently increased the association of NFAT3 recruitment to NFAT after 1 h of treatment to approximately 1.3-fold to 2.2-fold at concentrations of 50–200 µM The control group with an anti-α-tubulin antibody showed no apparent target band, which is in agreement with the findings in Figure 7C. Together, these results suggest that carboplatin increased NFAT-mediated up-regulation of HO-1 by increasing the binding activity of NFAT3 to NFAT, which suggests the transcriptional regulation of HO-1 induction by carboplatin. To further clarify the importance of NFAT3 in carboplatin-mediated HO-1 induction, knockdown of NFAT3 in cells transfected with siNFAT3 was performed to examine its effect on HO-1 levels in response to a carboplatin challenge. Knockdown of NFAT3 in RTC effectively reversed carboplatin-mediated induction of HO-1 at both the mRNA and protein levels and also reduced caspase-3 activation (Figure 7E). This suggests that carboplatin can induce HO-1 through an NFAT3-dependent mechanism and that NFAT3 is essential for caspase-3 activation.
Figure 7.
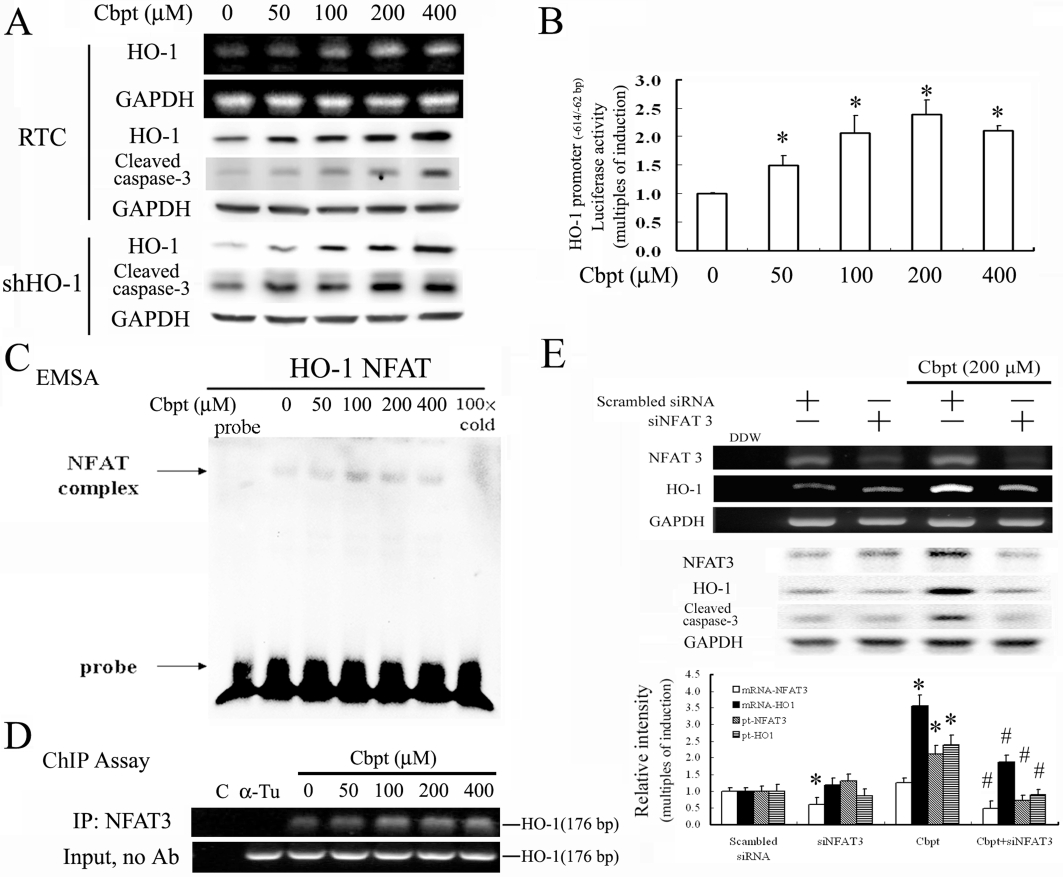
Carboplatin (Cbpt) transcriptionally up-regulates haem oxygenase (HO)-1 through a nuclear factor of activated T-lymphocyte-3 (NFAT3)-dependent mechanism. (A) Renal tubular cells (RTC) were treated with increasing concentrations of carboplatin as indicated for 6 h for the reverse-transcription polymerase chain reaction (RT-PCR) analysis of HO-1 induction and 18 h for the Western blot analysis of HO-1 and cleaved caspase-3. (B) RTC were transiently transfected for 24 h with the pGL3/rat HO-1 promoter containing NFAT binding sites. This was followed by the indicated concentrations of carboplatin treatment for 4 h. Data are presented as the mean ± SEM of three independent experiments (*P < 0.05, significantly different from RTC alone). (C) The putative NFAT-binding activity derived from the HO-1 promoter region of nuclear proteins was assayed for 1 h by electrophoretic mobility shift assay (EMSA) in cells with the indicated concentrations of carboplatin treatment. The 100× cold denotes a 100-fold molar excess of unlabeled oligonucleotides relative to the biotin-labeled probe. This was added to the binding assay for competition with the unlabeled oligonucleotide. The mobility of specific NFAT complexes is indicated. (D) Cells with 1 h of indicated carboplatin treatment were subjected to a chromatin immunoprecipitation (ChIP) assay. The DNA associated with the NFAT3 was immunoprecipitated with an anti-NFAT3 antibody, and PCR amplification was used to determine the extent of NFAT3 association with the functional NFAT3 in an HO-1 promoter fragment of 176 bp. Double-distilled water and anti-Tu were, respectively, used as negative controls for the ChIP assay. Representative results of three separate experiments are shown, and data are presented as the mean ± SEM. *P < 0.05, significantly different from the control; #P < 0.05, significantly different from carboplatin alone). (E) Cells were transfected with siNFAT3 for 24 h, followed by 1 or 6 h of 200 µM carboplatin treatment for the RT-PCR and Western blot analysis of NFAT3 and HO-1, respectively. Data are presented as the mean ± SEM of three independent experiments. *P < 0.05, significantly different from RTC alone; #P < 0.05, significantly different from knockdown of NFAT3 in RTC with additional carboplatin treatment.
Discussion
We previously observed that activation of NFAT3 mediated by oxidative stress was responsible for carboplatin-mediated renal apoptosis. In this paper, we demonstrated that the antioxidative and anti-inflammatory effects of HO-1 reversed carboplatin-mediated renal apoptosis through suppression of NFAT3 activation. We also demonstrated that cells under carboplatin-mediated oxidative stress exhibited a self-defence mechanism to induce HO-1 as a result of NFAT3-mediated transcriptional regulation. Oxidative stress leads to HO-1 induction in various cell types. We showed that NFAT3 activation by carboplatin-mediated oxidative stress is responsible for HO-1 up-regulation. HO-1 is up-regulated to protect cells by increasing the DNA-binding activity in the HO-1 promoter region. Although carboplatin-mediated HO-1 induction was insufficient to reverse the resulting injury when compared to RTC overexpressing HO-1, cell viability was significantly improved in untreated RTC, compared to RTC with HO-1 knockdown.
Based on the antioxidative, anti-apoptotic, and anti-inflammatory effects of HO-1, we explored the therapeutic potential of HO-1 in renal toxicity caused by using carboplatin as a chemotherapeutic drug. Both gain and loss of function of HO-1 were used to show prevention of renal injury by carboplatin, even though CoPP-mediated inhibition with an anti-inflammatory effect has been shown to be dependent on STAT3 phosphorylation, but independent of HO-1 activity (Mashreghi et al., 2008). The results presented in this study show that the antioxidative activity of HO-1 effectively reversed carboplatin-mediated p47PHOX activation and subsequent ROS production. This effect then decreased carboplatin-mediated renal apoptosis through suppression of the PKC/JNK/ERK signalling pathways leading to NFAT3 activation. Inhibition of MAPK calcineurin/NFAT signalling by HO-1 has also been implicated in protecting against hypertrophy in cardiac myocytes (Tongers et al., 2004).
The results presented in this study are supported by a recent publication showing that carboplatin-mediated renal toxicity was prevented by pravastatin as a result of HO-1 induction through a PPARα-dependent mechanism. However, the authors did not clarify exactly how HO-1 operated in pravastatin-mediated protection against carboplatin-induced renal toxicity (Chen et al., 2010). The results presented in this study provide evidence that the protective effect of HO-1 against carboplatin-induced apoptosis is through the anti-apoptotic and anti-inflammatory effects of bilirubin and CO (Figure 5). CO mediates anti-inflammatory, anti-apoptotic, and vasodilatory effects, some of which are mediated via cGMP-dependent mechanisms (Otterbein and Choi, 2000). In contrast, oxidative stress and bilirubin, which is rapidly generated by biliverdin reductase, were identified as cytoprotective, antioxidant moieties. In the heart, HO-1 confers protection against ischaemia/reperfusion injury. Biliverdin suppresses interleukin-2 production via inhibiting NFAT and NFκB activation which induces tolerance to cardiac allografts (Yamashita et al., 2004).
The RTC overexpressing HO-1 not only inhibited NFAT3 activation, but also decreased the apoptotic signalling by attenuating carboplatin-mediated induction of Bcl-X alternative splicing to Bcl-XS. The results presented in our study showed that HO-1 overexpression effectively reversed carboplatin-mediated PKC activation, and, to a lesser extent, ERK and JNK activation. Intriguingly, TPA, a PKC activator, reversed the protective effect of HO-1 in carboplatin-mediated RTC apoptosis. The PKC inhibitor, R0318220, rescued the pro-apoptotic effect of carboplatin in RTC with HO-1 knockdown in terms of the levels of Bcl-XL and Bcl-XS. It also activated caspase-3 (Figure 4C). This suggests that PKC activation is one of the important pathways to elicit carboplatin-mediated Bcl-X alternative splicing. Such a finding is supported by a study showing PKC-dependent control of Bcl-X alternative splicing (Revil et al., 2007).
The pro-inflammatory effect of platinum-based chemotherapeutic agents, such as carboplatin also induced RTC injury. A cannabinoid CB2 receptor agonist attenuated the cisplatin-induced inflammatory response, oxidative/nitrosative stress, and cell death in the kidneys and improved renal function (Mukhopadhyay et al., 2010a), whereas CB1 receptor agonists enhanced inflammation and tissue injury (Mukhopadhyay et al., 2010b). Apart from the antioxidative effect of HO-1, the anti-inflammatory effect of HO-1 in carboplatin-mediated RTC apoptosis was examined. We demonstrated that HO-1 overexpression or NFAT3 knockdown significantly decreased carboplatin-mediated nuclear translocation of the pro-inflammatory nuclear transfection factors, p65/p50, which, in contrast, was enhanced in cells with HO-1 knockdown (Figure 6).
Although there is increasing interest in the pro-oxidant activity of HO in diabetes, this activity is possibly due to increased redox-active iron in disease states. The enhancement of HO-1 expression with diabetes mellitus, hypercholesterolaemia, and smoking may be related to the severity of atherosclerosis (Song et al., 2009). Additionally, diabetes-induced oxidative damage to the heart was eliminated by SnPP, an inhibitor of HO-1 action, suggesting that the injury is intimately associated with up-regulation of HO-1 expression and activity (Farhangkhoee et al., 2003). However, the possibility that the induction of HO-1 is a consequence rather than the cause of diabetes-mediated oxidative damage to the heart cannot be eliminated. Tissues in a pathological condition might not properly respond to the increasing free iron accompanying the increase in HO-1 activity. Furthermore, significant positive correlations were reported (Calabrese et al., 2007) between the degree of renal failure and HO-1 levels in diabetic uremic patients; nevertheless, these authors speculated that HO-1 is induced to counteract the intracellular pro-oxidant status in diabetic nephropathy. Additionally, HO-1 overexpression following ischaemia is believed to be an endogenous means of self-protection (Zhu et al., 2007). However, the levels of HO-1 induced in pathological states, such as in atherosclerotic lesions, were insufficient to overcome oxidative challenge but boosting HO-1 expression in vascular cells by gene transfection successfully attenuated the formation of atherosclerosis (Wang et al., 1998). This agrees with our findings that the induction of HO-1 by carboplatin resulted in a decreased extent of cell death compared to cells with HO-1 knockdown, even though carboplatin-mediated RTC apoptosis could not be effectively reversed in cells with HO-1 induction by carboplatin. Therefore, our data suggest that HO-1 induction by carboplatin might add to the natural defence mechanisms.
This is the first study to report that carboplatin-mediated renal apoptosis can be effectively prevented by HO-1 overexpression, before carboplatin administration. The HO-1 exerts its therapeutic protective effects by virtue of its antioxidative and anti-inflammatory properties and its inhibition of ERK/JNK/PKC and NFAT3 activation in RTC subjected to carboplatin challenge, both in vitro and in vivo. However, the timing of HO-1 induction might be critical for it to be effective.
In summary, the data presented provide evidence to support the protective role of HO-1 in carboplatin-mediated NFAT3 activation and subsequent renal apoptosis. Identifying the protective effect of HO-1 and its underlying mechanisms will provide an important molecular basis for the design of new therapeutic strategies to treat complications or side-effects of carboplatin chemotherapy. Determining whether HO-1 can serve as an alternative gene therapy for patients with carboplatin chemotherapy requires further investigation.
Acknowledgments
This study was sponsored by a grant from Taipei Medical University-associated Wan Fang Hospital (99TMU-WFH-07).
Glossary
Abbreviations
- CoPP
cobalt protoporphyrin
- CORMII
[Ru(CO)3Cl2]2/Tricarbonyldichlororuthenium(II) dimer
- DAPI
4′,6-diamidino-2-phenylindole
- ERK
extracellular signal regulated kinase
- GTP
alanine transaminase
- HO-1
haem oxygenase-1
- JNK
Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- NAC
N-acetylcysteine
- NFAT3
nuclear factor of activated T-lymphocyte-3
- PKC
protein kinase C
- ROS
reactive oxygen species
- RTC
renal tubular cells
- SnPP
tin(IV) protoporphyrin
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Conflicts of interest
None to declare.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- Abraham NG, da Silva JL, Lavrovsky Y, Stoltz RA, Kappas A, Dunn MW, et al. Adenovirus-mediated heme oxygenase-1 gene transfer into rabbit ocular tissues. Invest Ophthalmol Vis Sci. 1995;36:2202–2210. [PubMed] [Google Scholar]
- Alberts DS. Carboplatin versus cisplatin in ovarian cancer. Semin Oncol. 1995;22:88–90. [PubMed] [Google Scholar]
- Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–1313. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Mancuso C, Sapienza M, Puleo E, Calafato S, Cornelius C, et al. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones. 2007;12:299–306. doi: 10.1379/CSC-270.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol. 2000;165:1013–1021. doi: 10.4049/jimmunol.165.2.1013. [DOI] [PubMed] [Google Scholar]
- Chang CC, Tsai SY, Lin H, Li HF, Lee YH, Chou Y, et al. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cell Mol Life Sci. 2009;66:3193–3205. doi: 10.1007/s00018-009-0102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HH, Chen TW, Lin H. Pravastatin attenuates carboplatin-induced nephrotoxicity in rodents via peroxisome proliferator-activated receptor alpha-regulated heme oxygenase-1. Mol Pharmacol. 2010;78:36–45. doi: 10.1124/mol.109.061101. [DOI] [PubMed] [Google Scholar]
- Cheng CF, Juan SH, Chen JJ, Chao YC, Chen HH, Lian WS, et al. Pravastatin attenuates carboplatin-induced cardiotoxicity via inhibition of oxidative stress associated apoptosis. Apoptosis. 2008;13:883–894. doi: 10.1007/s10495-008-0214-9. [DOI] [PubMed] [Google Scholar]
- Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J. 2000;348(Pt 3):615–619. [PMC free article] [PubMed] [Google Scholar]
- Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, et al. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol. 2003;35:1439–1448. doi: 10.1016/j.yjmcc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Sakuragi N, Suzuki S, Yoshida N, Maehata K, Nishiya M, et al. First-line intraperitoneal carboplatin-based chemotherapy for 165 patients with epithelial ovarian carcinoma: results of long-term follow-up. Gynecol Oncol. 2003;90:637–643. doi: 10.1016/s0090-8258(03)00377-9. [DOI] [PubMed] [Google Scholar]
- Hartsfield CL, McMurtry IF, Ivy DD, Morris KG, Vidmar S, Rodman DM, et al. Cardioprotective and vasomotor effects of HO activity during acute and chronic hypoxia. Am J Physiol Heart Circ Physiol. 2004;287:H2009–H2015. doi: 10.1152/ajpheart.00394.2002. [DOI] [PubMed] [Google Scholar]
- Huang C, Ding M, Li J, Leonard SS, Rojanasakul Y, Castranova V, et al. Vanadium-induced nuclear factor of activated T cells activation through hydrogen peroxide. J Biol Chem. 2001a;276:22397–22403. doi: 10.1074/jbc.M010828200. [DOI] [PubMed] [Google Scholar]
- Huang C, Li J, Costa M, Zhang Z, Leonard SS, Castranova V, et al. Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel subsulfide. Cancer Res. 2001b;61:8051–8057. [PubMed] [Google Scholar]
- Husain K, Scott RB, Whitworth C, Somani SM, Rybak LP. Dose-response of carboplatin-induced hearing loss in rats: antioxidant defense system. Hear Res. 2001;151:71–78. doi: 10.1016/s0300-2977(00)00081-4. [DOI] [PubMed] [Google Scholar]
- Juan SH, Cheng TH, Lin HC, Chu YL, Lee WS. Mechanism of concentration-dependent induction of heme oxygenase-1 by resveratrol in human aortic smooth muscle cells. Biochem Pharmacol. 2005;69:41–48. doi: 10.1016/j.bcp.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Kushida T, Quan S, Yang L, Ikehara S, Kappas A, Abraham NG. A significant role for the heme oxygenase-1 gene in endothelial cell cycle progression. Biochem Biophys Res Commun. 2002;291:68–75. doi: 10.1006/bbrc.2002.6403. [DOI] [PubMed] [Google Scholar]
- Kwak JY, Takeshige K, Cheung BS, Minakami S. Bilirubin inhibits the activation of superoxide-producing NADPH oxidase in a neutrophil cell-free system. Biochim Biophys Acta. 1991;1076:369–373. doi: 10.1016/0167-4838(91)90478-i. [DOI] [PubMed] [Google Scholar]
- Li J, Huang B, Shi X, Castranova V, Vallyathan V, Huang C. Involvement of hydrogen peroxide in asbestos-induced NFAT activation. Mol Cell Biochem. 2002;234–235:161–168. [PubMed] [Google Scholar]
- Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. J Cell Physiol. 2008;214:434–441. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- Lin H, Sue YM, Chou Y, Cheng CF, Chang CC, Li HF, et al. Activation of a nuclear factor of activated T-lymphocyte-3 (NFAT3) by oxidative stress in carboplatin-mediated renal apoptosis. Br J Pharmacol. 2010;161:1661–1676. doi: 10.1111/j.1476-5381.2010.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- Mashreghi MF, Klemz R, Knosalla IS, Gerstmayer B, Janssen U, Buelow R, et al. Inhibition of dendritic cell maturation and function is independent of heme oxygenase 1 but requires the activation of STAT3. J Immunol. 2008;180:7919–7930. doi: 10.4049/jimmunol.180.12.7919. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Pan H, Rajesh M, Batkai S, Patel V, Harvey-White J, et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol. 2010a;160:657–668. doi: 10.1111/j.1476-5381.2010.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Pan H, Patel V, Mukhopadhyay B, Batkai S, et al. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med. 2010b;48:457–467. doi: 10.1016/j.freeradbiomed.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- Pivot X, Cals L, Cupissol D, Guardiola E, Tchiknavorian X, Guerrier P, et al. Phase II trial of a paclitaxel-carboplatin combination in recurrent squamous cell carcinoma of the head and neck. Oncology. 2001;60:66–71. doi: 10.1159/000055299. [DOI] [PubMed] [Google Scholar]
- Platt JL, Nath KA. Heme oxygenase: protective gene or Trojan horse. Nat Med. 1998;4:1364–1365. doi: 10.1038/3947. [DOI] [PubMed] [Google Scholar]
- Ponka P. Cell biology of heme. Am J Med Sci. 1999;318:241–256. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Revil T, Toutant J, Shkreta L, Garneau D, Cloutier P, Chabot B. Protein kinase C-dependent control of Bcl-x alternative splicing. Mol Cell Biol. 2007;27:8431–8441. doi: 10.1128/MCB.00565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell R, Skrzypski M, Jassem E, Taron M, Bartolucci R, Sanchez JJ, et al. BRCA1: a novel prognostic factor in resected non-small-cell lung cancer. PLoS One. 2007;2:e1129. doi: 10.1371/journal.pone.0001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg JL, Dallas ML, Wilkinson JA, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem. 2008;283:24412–24419. doi: 10.1074/jbc.M803037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih CM, Lin H, Liang YC, Lee WS, Bi WF, Juan SH. Concentration-dependent differential effects of quercetin on rat aortic smooth muscle cells. Eur J Pharmacol. 2004;496:41–48. doi: 10.1016/j.ejphar.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Song J, Sumiyoshi S, Nakashima Y, Doi Y, Iida M, Kiyohara Y, et al. Overexpression of heme oxygenase-1 in coronary atherosclerosis of Japanese autopsies with diabetes mellitus: hisayama study. Atherosclerosis. 2009;202:573–581. doi: 10.1016/j.atherosclerosis.2008.05.057. [DOI] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Sue YM, Cheng CF, Chang CC, Chou Y, Chen CH, Juan SH. Antioxidation and anti-inflammation by haem oxygenase-1 contribute to protection by tetramethylpyrazine against gentamicin-induced apoptosis in murine renal tubular cells. Nephrol Dial Transplant. 2009;24:769–777. doi: 10.1093/ndt/gfn545. [DOI] [PubMed] [Google Scholar]
- Taille C, El-Benna J, Lanone S, Dang MC, Ogier-Denis E, Aubier M, et al. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J Biol Chem. 2004;279:28681–28688. doi: 10.1074/jbc.M310661200. [DOI] [PubMed] [Google Scholar]
- Tongers J, Fiedler B, Konig D, Kempf T, Klein G, Heineke J, et al. Heme oxygenase-1 inhibition of MAP kinases, calcineurin/NFAT signaling, and hypertrophy in cardiac myocytes. Cardiovasc Res. 2004;63:545–552. doi: 10.1016/j.cardiores.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Wang LJ, Lee TS, Lee FY, Pai RC, Chau LY. Expression of heme oxygenase-1 in atherosclerotic lesions. Am J Pathol. 1998;152:711–720. [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, McDaid J, Ollinger R, Tsui TY, Berberat PO, Usheva A, et al. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 2004;18:765–767. doi: 10.1096/fj.03-0839fje. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, et al. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li N, Chen Z, Shao K, Zhou F, Zhang C, et al. High expression of nuclear factor of activated T cells in Chinese primary non-small cell lung cancer tissues. Int J Biol Markers. 2007;22:221–225. doi: 10.1177/172460080702200310. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zhang Y, Ojwang BA, Brantley MA, Jr, Gidday JM. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-1alpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.