

Abstract
BACKGROUND AND PURPOSE
Exposure to mercury is known to increase cardiovascular risk but the underlying mechanisms are not well explored. We analysed whether chronic exposure to low mercury doses affects endothelial modulation of the coronary circulation.
EXPERIMENTAL APPROACH
Left coronary arteries and hearts from Wistar rats treated with either HgCl2 (first dose 4.6 µg·kg−1, subsequent doses 0.07 µg·kg−1 day−1, 30 days) or vehicle were used. Endothelial cells from pig coronary arteries incubated with HgCl2 were also used.
KEY RESULTS
Mercury treatment increased 5-HT-induced vasoconstriction but reduced acetylcholine-induced vasodilatation. It also reduced nitric oxide (NO) production and the effects of NO synthase inhibition with L-NAME (100 µmol·L−1) on 5-HT and acetylcholine responses. Superoxide anion production and mRNA levels of NOX-1 and NOX-4 were all increased. The superoxide anion scavenger tiron (1 mmol·L−1) reduced 5-HT responses and increased acetylcholine responses only in vessels from mercury-treated rats. In isolated hearts from mercury-treated rats, coronary perfusion and diastolic pressure were unchanged, but developed isovolumetric systolic pressure was reduced. In these hearts, L-NAME increased coronary perfusion pressure and diastolic pressure while it further reduced developed systolic pressure.
CONCLUSIONS AND IMPLICATIONS
Chronic exposure to low doses of mercury promotes endothelial dysfunction of coronary arteries, as shown by decreased NO bioavailability induced by increased oxidative stress. These effects on coronary function increase resistance to flow, which under overload conditions might cause ventricular contraction and relaxation impairment. These findings provide further evidence that mercury, even at low doses, could be an environmental risk factor for cardiovascular disease.
Keywords: mercury, oxidative stress, coronary arteries, endothelial dysfunction
Introduction
Mercury is a toxic metal considered to be a hazard, and exposure to mercury might produce toxicological consequences (ATSDR, 1997; Guzzi & La Porta, 2008). Recent reports have shown that human exposure to environmental contamination, including mercury, seems to be more common than otherwise expected. Professional exposure to mercury vapour (Langworth et al., 1997; Sandborgh-Englund et al., 1998), mercury release from or during removal of amalgam fillings (Hahn et al., 1990; Björkman et al., 1997; Halbach et al., 1998) and ingestion of contaminated fish (Salonen et al., 1995), among several other causes, increase its concentration in blood. In the last 10 years, several reports have focused more attention on the toxic effects of mercury in the cardiovascular system and its association with hypertension, carotid atherosclerosis, myocardial infarction and coronary heart disease (Salonen et al., 2000; Virtanen et al., 2005; Houston, 2007).
Endothelial integrity is crucial for the maintenance of blood flow and anti-thrombotic capacity. These effects involve the release of humoral factors that control relaxation and contraction, thrombogenesis and fibrinogenesis and platelet activation and inhibition. Several studies have revealed that mercury generates oxygen radicals (Houston, 2007). Although the exact mechanisms of free radical generation are not yet completely understood, it has been hypothesized that depletion of reduced glutathione (GSH) by mercury may trigger the production of reactive oxygen species (ROS). Vascular endothelium is highly sensitive to oxidative stress and this stress is the main cause of the endothelial dysfunction observed in cardiovascular diseases such as hypertension and atherosclerosis (Paravicini and Touyz, 2008). Recently, we have demonstrated that chronic administration of low doses of mercury induced endothelial dysfunction in rat aorta and mesenteric resistance arteries as a result of decreased nitric oxide (NO) bioavailability, which in turn is caused by higher NADPH oxidase-derived O2.− production (Wiggers et al., 2008). The importance of endothelial NO in the control of both vascular tone in large epicardial coronary arteries and the coronary microcirculation is well known. Previous reports show that the regulatory function of endothelium in coronary vessels is altered by cardiovascular risk factors or disorders such as hypercholesterolaemia, chronic smoking, hypertension and chronic heart failure (Drexler and Hornig, 1999; Osto et al., 2007).
The purpose of the present study was to investigate whether chronic exposure to low doses of mercury, approximating those found in the blood of exposed humans (Gupta et al., 1996; Langworth et al., 1997), affects endothelial modulation of the coronary circulation. We therefore studied the effect of such mercury exposure on: (i) vasoconstrictor and endothelium-dependent vasodilator responses of coronary arteries; (ii) vascular NO and ROS production and its participation in these vascular responses; (iii) coronary circulation and cardiac function of isolated perfused hearts; and (iv) the structure of coronary arteries. Our observations provide, for the first time, evidence that mercury causes endothelial dysfunction in coronary arteries mainly due to reduction of NO bioavailability and increased oxidative stress. This reduction could have important consequences for cardiac function.
Methods
Animals
All animal care and experimental procedures complied with current Spanish and European laws (RD 233/88 Ministerio de Agricultura, Pesca y Alimentación and 609/86). Three-month-old male normotensive Wistar rats (280–300 g, n = 38) were obtained from colonies maintained at the Animal Quarters of the Universidad Autónoma de Madrid. During treatment, rats were housed at a constant room temperature, humidity and light cycle (12:12 h light-dark). Rats had free access to tap water and were fed with standard chow ad libitum. Rats were divided into two groups: control (vehicle – saline solution, i.m.) and rats treated with mercuric chloride (HgCl2) for 30 days (first dose 4.6 µg·kg−1, subsequent dose 0.07 µg·kg−1·day−1, i.m. to cover daily loss). As previously reported, this treatment produces mercury concentrations of 8 ± 0.6 ng·mL−1 in blood (Wiggers et al., 2008).
Vascular reactivity measurements of coronary arteries
After 30 days of treatment with either HgCl2 or vehicle, rats were killed by decapitation. Left coronary arteries were isolated, and segments, 2 mm in length, were mounted in a small vessel dual chamber myograph for measurement of isometric tension according to the method described by Mulvany and Halpern (1977). After a 30 min equilibration period in oxygenated Krebs–Henseleit solution (KHS, in mmol·L−1: 115 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4.7H2O, 2.5 CaCl2, 1.2 KH2PO4, 11.1 glucose and 0.01 Na2EDTA) at 37°C and pH 7.4, segments were stretched to their optimal lumen diameter for active tension development. Left coronary arteries were exposed to a high K+ solution (120 mmol·L−1 K+-KHS, which was identical to KHS except that NaCl was replaced by KCl on an equimolar basis) in order to check their functional integrity. Concentration–response curves to acetylcholine (ACh) were then performed in arteries previously contracted with 5-HT at a concentration that produced approximately 50% of the contraction induced by K+-KHS in each case. After a washout period of 60 min, concentration–response curves to 5-HT were constructed. A single concentration-dependent curve to ACh and 5-HT was performed in each segment. A parallel study was done of the effects of the nitric oxide synthase (NOS) inhibitor N-nitro-L-arginine methyl ester (L-NAME, 100 µmol·L−1), the ROS scavengers superoxide dismutase (SOD, 150 U·mL−1) and 4,5-dihydroxy-1,3-benzene-disulphonic acid (tiron, 1 mmol·L−1), the COX inhibitor indomethacin (10 µmol·L−1) and the KCa channel blocker tetraethylammonium (TEA, 1 mmol·L−1) by the addition of these to the medium 30 min prior to that of ACh or 5-HT.
In another set of experiments, concentration–response curves to diethylamine NONOate (DEA-NO) were performed in segments contracted with 5-HT at a concentration that produced approximately 50% of the contraction induced by K+-KHS in each case.
Endothelial cell culture
Left and right coronary arteries from 7- to 8-month-old pigs were obtained from the Experimental Surgery Service of Research Center of La Paz Hospital (Madrid, Spain). They were cleaned of myocardial and adipose tissues in Nutrient Mixture F-12-HAM containing antibiotics and fungicide to prevent contamination. Coronary arteries were opened along their long axes, and fragments 2–3 cm in length were placed on six multi-well plates luminal surface down with 0.2% collagenase (Worthington Biochemical Corporation, Lakewood, NJ, USA) in Nutrient Mixture F-12-HAM for 5 min at 37°C. The cells were then scraped off and cultured in Iscove-modified Dulbecco's culture medium (Gibco, Invitrogen, Carlsbad, CA, USA) plus 20% fetal bovine serum (FBS, Biological Industries, Kibbutz Beit Haemek, Israel) and antibiotics (penicillin 100 U·mL−1, streptomycin 100 µg·mL−1). Cultures were incubated at 37°C in a humidified 5% CO2 incubator and the medium was changed every 2–3 days. Confluent cell monolayers were harvested by a brief incubation with 0.1% trypsin (Biological Industries) and 0.1% EDTA and cells were resuspended in Iscove-modified Dulbecco's medium (Dufresne and Warocquier-Clérout, 2001). Endothelial cell phenotype was confirmed by the expression of the specific marker von Willebrand factor (antibody dilution 1:100, Dako, Glostrup, Denmark). An appropriate secondary antibody labelled with fluorescein isothiocyanate (FITC) was used (antibody dilution 1:200). The cells were examined with a fluorescence microscope (Nikon Eclipse T300) and images were obtained with a digital spot camera (Diagnostic Instruments, Inc.).
Isolated heart preparation
As chronic treatment with HgCl2 affected the reactivity of isolated coronary vessels, we performed experiments to determine whether mercury might be affecting coronary circulation of isolated perfused hearts. Rats were anaesthetized with sodium pentobarbital (60 mg·kg−1, i.p.) and heparinized (40 U). After thoracotomy, the heart was removed and the ascending aorta was perfused with gassed (95% O2 and 5% CO2) KHS according to the Langendorff technique, under constant flow (10 mL·min−1), at 37°C. The right atrium was removed in order to take out the sinoatrial pacemaker. The heart was then stimulated with a fixed rate of 200 bpm through two platinum electrodes. The left atrium was opened to introduce a soft distensible balloon mounted at the tip of a rigid plastic tube into the left ventricular cavity through the atrioventricular valve. To avoid liquid accumulation in the ventricular cavity, the ventricle was perforated with a puncture needle. The balloon was connected to a pressure transducer (TSD 104A- Biopac Systems, Inc; Goleta, CA, USA) and to a syringe so that the diastolic pressure of the left ventricle could be adjusted to predetermined values by injecting water into the balloon. Developed left ventricle isovolumetric systolic pressure (systolic pressure minus diastolic pressure – LVISP) was measured with a pressure amplifier (MP 100 Biopac) and recorded with a data acquisition system (Biopac MP100WSW, including a software Acqknowledge III). Coronary perfusion pressure (CPP) was also measured at the aortic cannula. As a constant flow was maintained, changes in the CPP represented changes in vascular resistance. Measurements were initiated after a stabilization period of 20 min.
The basic protocol was performed at a constant initial diastolic pressure of 5 mmHg by adjusting balloon volume. A constant balloon volume maintained throughout the experiments permitted measurements of diastolic and systolic pressure changes. LVISP and CPP were recorded for 60 min under control conditions and after treatment with 100 µmol·L−1 L-NAME.
Nitric oxide release
Nitric oxide release was measured as previously described (Martín et al., 2005). The septal coronary artery was dissected and equilibrated for 60 min in HEPES buffer (in mmol·L−1: 119 NaCl; 20 HEPES; 1.2 CaCl2; 4.6 KCl; 1 MgSO4; 0.4 KH2PO4; 5 NaHCO3; 5.5 glucose; 0.15 NaH2PO4; pH 7.4) at 37°C; arteries were then incubated with the fluorescent probe 4,5-diaminofluorescein (2 µmol·L−1) for 1 h and the medium was collected to measure basal NO release. 5-HT (1 µmol·L−1) and ACh (10 µmol·L−1) was applied and the medium collected to measure stimulated NO production. The fluorescence of the medium was measured at room temperature using a spectrofluorometer (LS50 Perkin Elmer instruments, FL WINLAB Software) with excitation wavelength set at 492 nm and emission wavelength at 515 nm. Blank measurement samples were similarly collected but without arteries to subtract background emission. The amount of NO released was expressed as arbitrary units·mg−1 tissue.
Nitrite assay
Endothelial cells were plated into six-well plates, cultured until confluence and then incubated 24 h before HgCl2 treatment in Iscove-modified Dulbecco's culture medium plus 5% FBS and antibiotics. Cells were then exposed to 0.05, 0.5 and 5 µg·mL−1 HgCl2 for 24 h. Apocynin (300 µmol·L−1) and tiron (1 mmol·L−1) were applied 30 min before exposure to 5 µg·mL−1 HgCl2. Finally, treated cells were incubated for 30 min with bradykinin (1 µmol·L−1) before collecting supernatants, and cells were washed and scraped in RIPA buffer. NO production was determined by measuring nitrite contents of 225 µL of each well with 75 µL of Griess reagent. Absorbance was measured and nitrite concentration was determined by interpolation of a calibration curve of standard sodium nitrite concentrations against absorbance. Nitrite levels were corrected by protein concentration measured by Lowry assay and using BSA as a standard; data are shown as µg·mg−1 protein.
ROS production
Lucigenin chemiluminescence
Superoxide anion (O2.−) levels were measured in septal coronary arteries using lucigenin chemiluminescence, as previously described (Martín et al., 2005). Briefly, arteries were equilibrated for 30 min in HEPES buffer at 37°C, transferred to test tubes containing 1 mL HEPES buffer (pH = 7.4) with lucigenin (250 mmol·L−1) and then kept at 37°C. The luminometer was set to report arbitrary units of emitted light; repeated measurements were collected during 5 min at 10 s intervals and averaged. Tiron (1 mmol·L−1), a cell permeant non-enzymatic scavenger of O2.−, was added to quench the O2.−-dependent chemiluminescence. Blank samples were similarly collected from medium without arteries in order to subtract background emission.
Detection of ROS by fluorescence microscopy
The oxidative fluorescent dye dihydroethidium (DHE) was used to evaluate O2.− production in situ in endothelial cells. Hydroethidine freely permeates cells and is oxidized in the presence of O2.− to ethidium bromide, which is trapped by intercalation with DNA. Ethidium bromide is excited at 546 nm and has an emission spectrum of 610 nm. Primarily isolated endothelial cells from passages 2 to 4 were used. Cells were incubated 24 h before HgCl2 treatment in Iscove-modified Dulbecco's culture medium plus 5% FBS and antibiotics; they were then exposed to 0.05, 0.5 and 5 µg·mL−1 HgCl2 for 24 h. Tempol (10 µmol·L−1), apocynin (300 µmol·L−1), tiron (1 mmol·L−1) and L-NAME (10 µmol·L−1) were applied 30 min before exposure to 5 µg·mL−1 HgCl2. Images were acquired with a fluorescence microscope (Nikon Eclipse T300), captured using a digital spot camera (Diagnostic) and processed using Metamorph 7.1.0.0.
Western blot analysis
eNOS protein expression was evaluated in primarily isolated endothelial cells from passages 2 to 4 after exposure to different concentrations of HgCl2. Cells were seeded and grown for 48 h and then incubated prior to treatment with HgCl2 in Iscove-modified Dulbecco's culture medium with 5% FBS and antibiotics. Cells were then exposed to 0.05 and 5 µg·mL−1 of HgCl2 for 24 h and non-treated cells were used as control. After treatment, cells were washed in ice-cold PBS buffer and whole-cell lysates were prepared in RIPA buffer containing a protease inhibitor cocktail (Complete C, Roche, Barcelona, Spain). 40 µg of total protein from each sample was separated in a 7.5% SDS-PAGE and transferred to polyvinyl difluoride membranes (Amersham International plc, Little Chalfont, UK) that were incubated with a mouse monoclonal antibody against eNOS (1:1000, BD Biosciences, San Jose, CA, USA). After washing, membranes were incubated with anti-mouse (1:5000, StressGen, Ann Arbor, MI, USA) immunoglobulin antibody conjugated to horseradish peroxidase followed by several washes. Immunocomplexes were detected using an enhanced horseradish peroxidase/luminal chemiluminiscence system (ECL Plus, Amersham International plc) and after exposure to X-ray AX film (Konica Minolta). Signals on the immunoblot were quantified with a National Institutes of Health Image V1.56 computer program. The same membrane was used to determine β-actin expression using a mouse monoclonal antibody conjugated to horseradish peroxidase (1:200 000, Sigma Chemical Co, St. Louis, MO, USA).
RT-PCR real-time assay
NOX-1, NOX-4, SOD-1 and SOD-2 mRNA levels were determined in coronary segments from control and mercury-treated rats. Total RNA was obtained using Tri Reagent (Sigma Chemical Co). A total of 1 µg of DNase I-treated RNA was reverse transcribed into cDNA using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) with random hexamers in a 10 µL reaction. PCR was performed in duplicate for each sample using 1 µL of cDNA as a template, 1× of TaqMan Universal PCR Master Mix (Applied Biosystems) and 10× of Taqman Gene Expression Assays (Applied Biosystems) in a 20 µL reaction. Assays-on-Demand (Applied Biosystems) of TaqMan fluorescent real-time PCR primers and probes were used for NOX-1 (Rn 00586652_m1), NOX-4 (Rn 00585380_m1), SOD-1 (Rn00566938_m1), SOD-2 (Rn00566942_g1) and 18S rRNA (4319413E), which was used as endogenous control to normalize results. Quantitative RT-PCR was carried out in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) using the following conditions: 2 min at 50°C, 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Relative NOX-1, NOX-4, SOD-1 and SOD-2 levels were determined using the 2−ΔΔCt method. Results are expressed as the relative expression of mRNA in mercury-treated compared with untreated rats.
Morphometry of coronary vessels
A total of eight animals from each group were analysed. Ventricular tissue was fixed in 4% sodium-buffered formaldehyde. The samples were then dehydrated and embedded in paraffin. Serial sections (4 µm) were stained with Masson's trichrome. A total of 48 intramyocardial small vessels (three vessels per rat) were observed and individually analysed using a high-resolution monochromatic photocamera CCD (Sony XC-75CE) attached to a photomicroscope (Leica DMRB). The morphometric analyses were performed using an Image System Analysis (Leica Q500MC) with 8 bits of images in grey gradation (256 levels of grey: 0 representing black as blank and 255 colours). The binary edition was used to remove artefacts that did not correspond to stained coronary vessel area. Analyses were performed with 40× lenses in which the vessels were clearly distinguished. The same illumination conditions were used for all measurements. Calibration of the system was carried out using a stage micrometer (Leitz) that allowed computation of the object area in units of µm2. Media area was obtained by subtracting the lumen area from the area encompassed by the external elastic lamina. A single researcher unaware of the experimental groups performed the analysis.
Data analysis and statistics
Vasoconstrictor responses induced by 5-HT were expressed as percentages of tension generated by 120 mmol·L−1 KCl (control, 2.1 ± 0.15 mN·mm−1, n = 19; HgCl2, 1.99 ± 0.16 mN·mm−1, n = 19; P > 0.05). Vasodilator responses are expressed as percentage of previous contraction. To compare the effect of drugs on response to 5-HT in coronary segments from both treatments, some results were expressed as differences of area under the concentration–response curves (dAUC) in control and experimental situations. AUCs were calculated from individual concentration–response curve plots; the differences were expressed as percentage of AUC of the control situation.
All values are expressed as mean ± SEM of the number of animals used in each experiment, or independent experiment in the case of cell cultures. Results were analysed using either Student's t-test, completely randomized two-way anova for comparison between groups, repeated-measure two-way anova for comparing treatments within the same group or the Mann–Whitney non-parametric test. When anova showed a significant treatment effect, Bonferroni's post hoc test was used to compare individual means. Differences were considered statistically significant at P-values < 0.05.
Materials
L-NAME, ACh, apocynin, 5-HT, EDTA, FITC, HEPES, DAF, DEANO, lucigenin, SOD, Nutrient Mixture F-12-HAM, tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl), indomethacin, TEA and tiron were obtained from Sigma Chemical Co. DHE, streptomycin and penicillin were obtained from Invitrogen (Carslbad, CA, USA). All drugs were dissolved in distilled water except tiron and lucigenin, which were dissolved in Krebs-HEPES, and indomethacin, which was dissolved in 1.5 mmol·L−1 NaHCO3. Nomenclature of channels, receptors and ligands follows Alexander et al. (2009).
Results
As previously described (Wiggers et al., 2008), rats exposed to 30 day HgCl2 treatment had similar body weight [control: before 293 ± 6.2 g and after 375 ± 8.9 g (n = 16); Hg-treated: before 287 ± 4.8 g and after 366 ± 6.1 g (n = 22) P > 0.05]. Systolic blood pressure also remained unchanged by treatment (control: 129 ± 10.4 mmHg, n = 6; Hg-treated: 128 ± 9.3 mmHg, n = 6, P > 0.05).
Effect of mercury treatment on vasoconstrictor and vasodilator responses of coronary arteries
Mercury treatment increased vasoconstrictor responses of coronary arteries to 5-HT (10 nmol·L−1–100 µmol·L−1) and decreased endothelium-dependent responses induced by ACh (1 nmol·L−1–10 µmol·L−1) (Figure 1). However, the vasodilator response induced by the NO donor DEA-NO (1 nmol·L−1–10 µmol·L−1) was unaffected by mercury treatment (Figure 1). This result suggests that mercury treatment induces endothelial dysfunction in coronary arteries.
Figure 1.
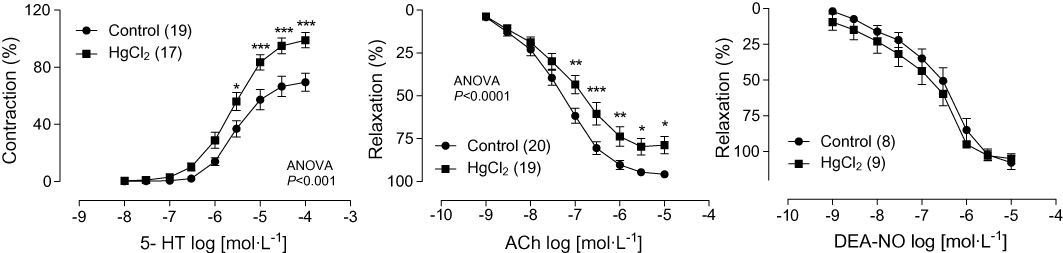
Concentration–response curves to 5-HT, acetylcholine (ACh) and diethylamine (DEA)-NONOate (NO) in left coronary arteries from control and HgCl2-treated rats. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control. Number of animals used is indicated in parentheses.
Effect of mercury treatment on NO and ROS production and its participation in vascular responses
In order to evaluate if mercury treatment alters NO modulation of coronary responses, the effect of the NOS inhibitor L-NAME on vasoconstrictor and vasodilator responses was investigated. Figure 2A shows that the negative modulation by NO of 5-HT responses was reduced in segments of coronary arteries from rats treated with mercury. Thus, L-NAME (100 µmol·L−1) shifted the concentration–response curve to 5-HT to the left, in segments from either group, but this effect was smaller in preparations from mercury-treated than in those from control rats, as shown by dAUC values (Figure 2A, insert). The participation of NO in relaxation induced by ACh was also reduced in coronary arteries from mercury-treated rats, as shown by the reduced inhibitory effect of L-NAME (Figure 2B). These findings suggest that NO production or bioavailability could be reduced after mercury treatment. In agreement, NO release was reduced in arteries from mercury-treated rats both in basal and after 5-HT and ACh stimulation (Figure 2C). On the other hand, endothelial cells exposed to HgCl2 shows reduced nitrite production, which was restored by both apocynin (300 µmol·L−1) and tiron (1 mmol·L−1) (Figure 2D). However, eNOS expression did not change (or even increased) in endothelial cells exposed to 0.05 or 5 µg·mL−1 HgCl2 respectively (Figure 2E).
Figure 2.

Effect of chronic treatment with mercury chloride (HgCl2) on the modulation of vascular response by NO in coronary arteries. (A) Effect of L-NAME (100 µmol·L−1) on the vasoconstrictor responses to 5-HT in left coronary arteries from untreated and HgCl2-treated rats. (B) Effect of L-NAME on the vasodilator responses to ACh in untreated and HgCl2-treated rats. The insert graphs show differences in area under the concentration–response curve (dAUC) in the presence and absence of L-NAME. (C) Vascular production of NO in septal coronary arteries from control and HgCl2-treated rats. (D) Effect of apocynin (300 µmol·L−1) and tiron (1 mmol·L−1) on nitrite production in endothelial cells exposed to 5 µg·mL−1 HgCl2; the effect of 0.05 and 0.5 µg·mL−1 HgCl2 is also shown. (E) Representative blot and densitometric analysis showing eNOS protein expression normalized with β-actin expression in endothelial cells exposed to HgCl2*P < 0.05, **P < 0.01, ***P < 0.001 versus the corresponding control situation; #P < 0.05 versus HgCl2. Number of animals used is indicated in parentheses. Ctrl: control.
The participation of ROS in vascular responses was evaluated using the superoxide anion scavengers, tiron and SOD. Tiron (1 mmol·L−1) did not modify 5-HT responses in coronary arteries from the control group but reduced the increased reactivity to 5-HT produced by mercury treatment (Figure 3A). Similarly, tiron did not affect endothelium-dependent ACh-induced relaxation in arteries from control rats but improved impaired vasodilator response observed in segments from mercury-treated rats (Figure 3B). The effect of SOD (150 U·mL−1) on 5-HT contraction was similar to that found for tiron; however, SOD did not affect ACh-induced relaxation in any group (results not shown).
Figure 3.

Effects of chronic treatment with HgCl2 on the participation of reactive oxygen species (ROS) in vascular responses in coronary arteries. Effect of tiron (1 mmol·L−1) on the vasoconstrictor responses to 5-HT (A) and the vasodilator responses to ACh (B) in left coronary arteries from untreated and HgCl2-treated rats. *P < 0.05, *P < 0.01 versus the corresponding control situation. Number of animals used is indicated in parentheses.
To evaluate if mercury treatment alters the vascular production of ROS we measured their levels in endothelial cells from coronary arteries exposed to different concentrations of HgCl2 and in coronary arteries from control and mercury-treated rats. Cultured endothelial cells exposed for 24 h to increasing HgCl2 concentrations (0.05, 0.5 and 5 µg·mL−1) display a progressive increase of superoxide production (Figure 4A). This increased production was diminished by the SOD mimetic tempol (1 mmol·L−1), the NADPH oxidase inhibitor apocynin (300 µmol·L−1), tiron (1 mmol·L−1) and L-NAME (100 µmol·L−1) (Figure 4B). In coronary arteries from mercury-treated rats, basal O2.− production was greater than in arteries from controls (Figure 4C). Mercury treatment also increased mRNA levels of the NADPH oxidase subunits NOX-1 and NOX-4 (Figure 4D) and of SOD-2 (Figure 4E) in these arteries; however, mRNA levels of SOD-1 were not significantly affected by mercury treatment.
Figure 4.

Effects of HgCl2 on vascular superoxide anion production. (A) Images and quantification of superoxide anion production measured by fluorescence microscopy in untreated (Ctrl) and HgCl2-treated endothelial cells from porcine coronary arteries. (B) Images and quantification of production of superoxide anion measured by fluorescence microscopy in endothelial cells exposed to HgCl2 (5 µg·mL−1) in the absence or the presence of tempol (1 mmol·L−1), apocynin (0.3 mmol·L−1), tiron (1 mmol·L−1) or L-NAME (100 µmol·L−1). (C) Vascular production of superoxide anion, measured by lucigenin chemiluminescence, in septal coronary arteries from control and HgCl2-treated rats. Quantitative RT-PCR assessment of (D) NOX-1 and NOX-4 and (E) SOD-1 and SOD-2 mRNA levels in coronary arteries from control and HgCl2-treated rats; results are expressed as the relative expression of mRNA compared with control. *P < 0.05, **P < 0.01 versus Ctrl, #P < 0.05 versus HgCl2. Number of animals used or independent experiment in the case of cell cultures is indicated in parentheses.
Participation of prostanoids and KCa channels on vascular responses: effect of mercury treatment
To evaluate if mercury treatment alters the participation of prostanoids on coronary responses, the COX inhibitor indomethacin (10 µmol·L−1) was used. This drug did not change 5-HT or ACh responses in arteries from control rats. However, in arteries from mercury-treated rats, indomethacin reduced 5-HT contraction (Figure 5A) and potentiated ACh responses (Emax, HgCl2: 64.0 ± 9.4 %, n = 11; HgCl2+ indomethacin: 93.5 ± 2.3 %, n = 9; P < 0.05).
Figure 5.

Effect of chronic treatment with HgCl2 on the participation of prostanoids and KCa channels in vascular responses in coronary arteries. Effect of indomethacin (10 µmol·L−1) (A) and tetraethylammonium (TEA, 1 mol·L−1) (B) on the vasoconstrictor responses to 5-HT in left coronary arteries from untreated and HgCl2-treated rats. The inset graph shows differences in area under the concentration–response curve (dAUC) in the presence and absence of TEA. *P < 0.05 versus the corresponding control situation. Number of animals used is indicated in parentheses.
In order to analyse the role of KCa channels on vascular responses, the effect of TEA was analysed. TEA (1 mmol·L−1) potentiated 5-HT responses in segments from both groups but this effect was smaller in preparations from mercury-treated than in control rats (Figure 5B). However, TEA did not change ACh response in coronary arteries from either group (results not shown).
Effect of mercury treatment on coronary circulation of isolated perfused hearts
To investigate whether increased coronary reactivity could affect heart function, isolated hearts, perfused according to the Langendorff technique, were used. Isolated hearts were used to avoid nervous or humoral mechanisms that could blunt or mask the effects of chronic Hg treatment. Figure 6A shows that at 5 mmHg of diastolic pressure, developed left ventricle isovolumetric systolic pressure (LVISP) was reduced in the Hg-treated group. When L-NAME was used, LVISP was further reduced in the Hg-treated group. CPP was also measured (Figure 6B); in hearts from Hg-treated rats, CPP was similar to controls. However, CPP increased after L-NAME treatment, and this increase was greater in the Hg-treated group. Developed left ventricle isovolumetric diastolic pressure (LVIDP) was similar in both control and HgCl2-treated rats (Figure 6C). When using L-NAME, LVIDP increased only in HgCl2-treated rats (Figure 6C). This increment is parallel to the reduction of coronary blood flow and the developed systolic pressure, which normally reduces coronary blood flow by reducing metabolic needs.
Figure 6.

(A) Developed left ventricle isovolumetric systolic pressure (LVISP), (B) coronary perfusion pressure (CPP) and (C) left ventricle isovolumetric diastolic pressure (LVIDP) from control (Ctrl) and HgCl2-treated rats before and after perfusion with L-NAME (100 µmol·L−1) in isolated hearts. *P < 0.05 versus Control, #P < 0.05 versus HgCl2, $P < 0.05 versus control + L-NAME. Number of animals used is indicated in parentheses.
Effect of mercury treatment on coronary morphology
To analyse putative actions of mercury treatment on intramyocardial vessel morphology, morphometric measurements of total, luminal and media areas were performed. Results and representative photomicrographs are presented in Figure 7. The morphometric analysis demonstrated that although no differences in media area were observed between both groups, vessel and luminal areas were reduced in mercury-treated rats as compared with controls. This pattern was maintained even after correcting the data (data not shown) with the following criteria. To determine luminal or vessel area, the cross-sectional area enclosed by internal or external elastic lamina, respectively, was corrected to a circle by applying the form factor l2/4π to the measurement of the lamina, where l is the length of the lamina. This method avoids miscalculations of areas caused by the eventual collapse of immersion-fixed arteries (de las Heras et al., 1999).
Figure 7.

Morphometric analysis of coronary vessels of control and HgCl2-treated rats. (A) Representative microphotographs (original magnification × 40) of cross sections of intramyocardial vessels from control (Ctrl; on left) and HgCl2-treated rats (on right) stained with Masson's trichrome. (B) Total vessel area. (C) Vascular luminal area. (D) Vascular media area. *P < 0.05. Number of animals used is indicated in parentheses.
Discussion
Results presented herein show for the first time that treatment with low doses of mercury for 30 days increased reactivity to 5-HT in isolated coronary arterial rings. Accordingly, CPP in isolated hearts was also increased. The present study also demonstrates that chronic mercury treatment induced endothelial dysfunction in coronary arteries, probably due to a decreased NO bioavailability caused by increased endothelial superoxide production.
Cardiovascular risk factors or disorders such as hypercholesterolaemia, chronic smoking, hypertension or chronic heart failure alter the regulatory function of endothelium both in coronary and peripheral conduit and resistance vessels. In addition to these risk factors, attention has recently been given to toxic effects of mercury in the cardiovascular system (Evans and Weingarten, 1990; Salonen et al., 1995; 2000; Virtanen et al., 2005; Houston, 2007). Chronic exposure to mercury is reported to increase vascular resistance and to induce hypertension (Torres et al., 2000; García Gómez et al., 2007). On the other hand, a concentration-dependent cytotoxicity in endothelial cells, as well as a reduction in NOS activity, has been described after treatment with methyl mercury (Kishimoto et al., 1995). Moreover, ROS accumulation induced by high concentrations of HgCl2 also results in cytotoxicity of endothelial cell monolayers (Wolf and Baynes, 2007). Despite the number of studies showing that mercury increases vascular resistance and induces oxidative stress, as well as risk for cardiovascular diseases, the effect of in vivo chronic exposure to mercury on endothelial modulation of coronary vascular responses is unknown. We have recently described a model of chronic (30 day) exposure to low doses of HgCl2 that did not induce changes in blood pressure, despite important endothelial dysfunction observed in both aorta and mesenteric resistance arteries (Wiggers et al., 2008). The blood concentrations reached with this exposure, 8 ng·mL−1, are similar to high blood levels frequently reported to be an environmental risk factor (Gupta et al., 1996). In such conditions, coronary reactivity to 5-HT increased, whereas relaxation to acetylcholine was reduced; similar findings were reported for aorta and mesenteric resistance arteries (Wiggers et al., 2008). It is well known that endothelial NO participates in the control of vascular tone in large epicardial coronary arteries and in the coronary microcirculation. In fact, changes observed in the reactivity of coronary arteries after mercury treatment might be explained by a decreased modulation of such responses by endothelial NO, as suggested by the reduced effect of L-NAME on 5-HT and acetylcholine responses. It is established that EDHF induces hyperpolarization and vasodilatation through activation of K+ channels (Feletou and Vanhoutte, 1996). We found that the KCa channel blocker TEA did not change ACh response; however, it potentiated 5-HT responses in segments from both groups, albeit to a greater extent in preparations from mercury-treated rats. These results suggest that HgCl2 treatment reduces the involvement of one factor, which opens KCa channels. Our group has recently described that mercury treatment increases the release of vasoconstrictor prostanoids and its participation in phenylephrine response in rat aorta (Peçanha et al., 2010). Accordingly, indomethacin reduced 5-HT contraction and potentiated ACh responses, albeit only in segments from mercury-treated animals. Both mechanisms would also contribute to increased response to 5-HT found in coronary arteries from mercury-treated rats.
Reduction of endothelial NO-dependent modulation might be due to decreased production or increased NO metabolism. We observed that vascular NO release was diminished in coronary arteries from mercury-treated rats, as well as in endothelial cells from coronary arteries exposed to HgCl2. A reduction in NO production and NOS activity has also been described in cultured human umbilical vascular endothelial cells exposed to methyl mercury (Kishimoto et al., 1995). We have previously reported unchanged or even increased eNOS protein expression in aorta and mesenteric resistance arteries respectively (Wiggers et al., 2008). In endothelial cells exposed to low concentration of mercury (0.05 µg·mL−1), no changes in eNOS expression was found; moreover, high concentrations (5 µg·mL−1) increased eNOS expression, thus excluding reduction in the expression of this enzyme as being responsible for the reduced NO production and endothelial dysfunction observed in coronary arteries after mercury treatment. However, we cannot discount a possible reduction in the activity of this isoform, or that uncoupled eNOS due to tetrahydrobiopterin (BH4) deficiency might result in a switch from NO to O2.− production (Förstermann and Münzel, 2006). In this sense, the inhibition of NOS slightly reduced the increased superoxide anion production observed in endothelial cells exposed to HgCl2. Alterations in the NO relaxation mechanism might also explain the inhibitory effect of mercury on endothelial function. As mercury treatment did not modify relaxation induced by the NO donor, DEA-NO, a direct effect of Hg on vascular smooth muscle cells is unlikely. These findings so far suggested that increased NO metabolism was the putative mechanism by which Hg treatment affected coronary reactivity.
Toxicity from mercury is associated with in vivo oxidative stress. Mercury exposure of animals or humans thus induces the generation of ROS with subsequent oxidative damage in several organs and systems, as well as altering the antioxidant defence system in cells (Miller and Woods, 1993; Huang et al., 1996; Mahboob et al., 2001; Reus et al., 2003; Chen et al., 2005). We have previously found that vascular O2.− production, plasma malondialdehyde levels and total antioxidant status increased after mercury treatment in rats (Wiggers et al., 2008). We likewise found an increased O2.− production in coronary arteries from mercury-treated rats and in endothelial cells from porcine coronary arteries exposed to increasing concentrations of mercury for 24 h. This increased production was abolished by the superoxide anion scavengers, tiron and tempol, as well as by apocynin, suggesting that NADPH oxidase would be the source of increased superoxide production, although several authors have reported different actions of apocynin independently of its ability to inhibit NADPH oxidase (Heumüller et al., 2008; Riganti et al., 2008). In a previous study, we found that mercury treatment did not change NOX-1 mRNA levels in mesenteric resistance arteries from mercury-treated rats, although an increasing trend was found (Wiggers et al., 2008). In coronary arteries, we have found that mercury treatment increased both NOX-1 and NOX-4 subunits, supporting the involvement of NADPH oxidase in the effect of mercury on oxidative stress. A decrease in antioxidant defences would also contribute to the increased superoxide production observed after mercury treatment. However, SOD-1 mRNA levels were similar in vessels from both groups, and SOD-2 mRNA levels even increased after mercury treatment. An increase in the total antioxidant status of plasma, as well as in aortic SOD-3 protein expression, after mercury exposure has been previously described (Wiggers et al., 2008). It is therefore possible that antioxidant mechanisms might become activated in mercury-exposed rats, in order to protect cells against increased oxidative stress.
Free radicals are known to increase sensitivity to phenylephrine in other vessels (Hao et al., 2006), and this might be due to the generation of peroxynitrite reducing NO bioavailability (Frisbee et al., 2002). We found that tiron and apocynin increased nitrite levels reduced by HgCl2 in endothelial cells, thus supporting the possibility that free radicals might promote reduction of NO bioavailability. In addition, both tiron and SOD reduced increased reactivity to 5-HT observed in coronary arteries from mercury-treated rats, with tiron also improving the impaired relaxation to ACh. These findings reinforce previous reports that show the relationship between the effects of mercury and free radical production.
Evidence in recent years has suggested that increased oxidative stress plays a pathophysiological role in cardiovascular diseases. In fact, ROS have been shown to play a critical role in remodelling in the heart and vasculature (Xu and Touyz, 2006; Wei et al., 2007; Paravicini and Touyz, 2008). As an enhanced superoxide production after mercury treatment was confirmed, the possibility of changes in coronary morphology has been considered. We carried out in vitro morphometric analyses, which disclosed a reduction in total coronary area and luminal area. The wall area did not change after mercury treatment, suggesting eutrophic remodelling that allows small arteries to maintain high resistance to blood flow without activation (Mulvany, 2002). However, we cannot confirm whether the morphological changes described here are a consequence of functional changes.
Several reports have focused attention on the possible association between toxic effects of mercury and myocardial infarction and coronary heart disease (Salonen et al., 2000; Virtanen et al., 2005; Houston, 2007). Because HgCl2 treatment affects coronary reactivity, we have performed experiments with isolated perfused hearts to determine whether the increased coronary reactivity could affect heart function. Indeed, hearts from mercury-treated rats in steady-state conditions had a lower systolic pressure development. Although mercury treatment did not change diastolic pressure and CPP, CPP increased more in the mercury-treated group when L-NAME was used. These results are in contrast to those found in isolated vessels. However, we have to consider that isolated coronary vessels could behave differently in situ. The isolated arteries we examined in the myograph are those arising from the aortic trunk and may behave as conductance vessels, different from those that determine coronary resistance. LVISP was further reduced in perfused heart in the HgCl2+ L-NAME group suggesting that the residual NO activity, although diminished, was of greater importance for maintaining myocardial perfusion in mercury-exposed hearts than in normal hearts. The increased coronary vasoconstriction also induced an increase of diastolic pressure, which might be a consequence of reduced oxygen uptake, leading to calcium overload (Endoh, 2008).
In conclusion, we demonstrate for the first time that treatment with low doses of mercury altered coronary vascular reactivity, because of endothelial dysfunction. This might be attributable, at least in part, to reduction in NO bioavailability due to increased ROS production, probably as a result of increased NADPH oxidase activation. The present findings also show that chronic treatment with mercury has an important effect on coronary artery function, inducing an increased resistance to flow. Under normal load conditions, coronary resistance was just enough to allow adequate flow, even though NO production was reduced. However, coronary flow might be insufficient when the heart is under overload conditions, thereby causing myocardial contraction and relaxation impairment. Therefore, these observations also offer further evidence that chronic exposure to low concentrations of mercury is hazardous and can be considered an important risk factor for cardiovascular disease and development of cardiovascular events.
Acknowledgments
This study was supported by MCINN (SAF 2009-07201, PHB2005-0008-PC) and ISCIII (Red RECAVA, RD06/0014/0011 and RD06/0014/0007), Spain, and by grants from CAPES and CNPq/FAPES/FUNCITEC (39767531/07), Brazil. We thank Mariana Boechat de Abreu for her help with the endothelial cell cultures.
Glossary
Abbreviations
- CPP
coronary perfusion pressure
- DEA-NO
diethylamine NONOate
- DHE
dihydroethidium
- FITC
fluorescein isothiocyanate
- KHS
Krebs–Henseleit solution
- L-NAME
N-nitro-L-arginine methyl ester
- LVISP
left ventricle isovolumetric systolic pressure
- NOS
nitric oxide synthase
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TEA
tetraethylammonium
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATSDR. Agency for Toxic Substances and Disease Registry. National Alert. A warning about continuing patterns of metallic mercury exposure. 1997. Available at: http://www.atsdr.cdc.gov/alerts/970626.html.
- Björkman L, Sandborgh-Englund G, Ekstrand J. Mercury in saliva and feces after removal of amalgam fillings. Toxicolol Appl Pharmacol. 1997;144:156–162. doi: 10.1006/taap.1997.8128. [DOI] [PubMed] [Google Scholar]
- Chen C, Qu L, Li B, Xing L, Jia G, Wang T, et al. Increased oxidative DNA damage, as assessed by urinary 8-hydroxy-2′-deoxyguanosine concentrations, and serum redox status in persons exposed to mercury. Clin Chem. 2005;51:759–767. doi: 10.1373/clinchem.2004.042093. [DOI] [PubMed] [Google Scholar]
- Drexler H, Hornig B. Endothelial dysfunction in human disease. J Mol Cell Cardiol. 1999;31:51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- Dufresne M, Warocquier-Clérout R. Explants of porcine coronary artery in culture: a paradigm for studying the influence of heparin on vascular wall cell proliferation. Cytotechnology. 2001;37:13–22. doi: 10.1023/A:1016195029359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M. Cardiac Ca2+ signaling and Ca2+ sensitizers. Circ J. 2008;72:1915–1925. doi: 10.1253/circj.cj-08-0838. [DOI] [PubMed] [Google Scholar]
- Evans DH, Weingarten K. The effect of cadmium and other metals on vascular smooth muscle of the dogfish shark, Squalus acanthias. Toxicology. 1990;61:275–281. doi: 10.1016/0300-483x(90)90177-i. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor. Clin Exp Pharmacol Physiol. 1996;23:1082–1090. doi: 10.1111/j.1440-1681.1996.tb01174.x. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Maier KG, Stepp DW. Oxidant stress-induced increase in myogenic activation of skeletal muscle resistance arteries in obese Zucker rats. Am J Physiol Heart Circ Physiol. 2002;283:H2160–H2168. doi: 10.1152/ajpheart.00379.2002. [DOI] [PubMed] [Google Scholar]
- García Gómez M, Boffetta P, Caballero Klink JD, Español S, Gómez Quintana J. Cardiovascular mortality in mercury miners. Med Clin (Barc) 2007;128:766–771. doi: 10.1157/13106327. [DOI] [PubMed] [Google Scholar]
- Gupta M, Bansal JK, Khanna CM. Blood mercury in workers exposed to the preparation of mercury cadmium telluride layers on cadmium telluride base. Ind Health. 1996;34:421–425. doi: 10.2486/indhealth.34.421. [DOI] [PubMed] [Google Scholar]
- Guzzi G, La Porta CAM. Molecular mechanisms triggered by mercury. Toxicology. 2008;244:1–12. doi: 10.1016/j.tox.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Hahn LJ, Kloiber R, Leininger RW, Vimy MJ, Lorscheider FL. Whole-body imaging of the distribution of mercury released from dental fillings into monkey tissues. FASEB J. 1990;4:3256–3260. doi: 10.1096/fasebj.4.14.2227216. [DOI] [PubMed] [Google Scholar]
- Halbach S, Kremers L, Willruth H, Mehl A, Welzl G, Wack FX, et al. Systemic transfer of mercury from amalgam fillings before and after cessation of emission. Environ Res. 1998;77:115–123. doi: 10.1006/enrs.1998.3829. [DOI] [PubMed] [Google Scholar]
- Hao L, Nishimura T, Wo H, Fernandez-Patron C. Vascular responses to alpha1-adrenergic receptors in small mesenteric arteries depend on mitochondrial reactive oxygen species. Arterioscler Thromb Vasc Biol. 2006;26:819–825. doi: 10.1161/01.ATV.0000204344.90301.7c. [DOI] [PubMed] [Google Scholar]
- de las Heras N, Aragoncillo P, Maeso R, Vazquez-Pérez S, Navarro-Cid J, DeGasparo N, et al. AT(1) receptor antagonism reduces endothelial dysfunction and intimal thickening in atherosclerotic rabbits. Hypertension. 1999;34:969–975. doi: 10.1161/01.hyp.34.4.969. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Houston MC. The role of mercury and cadmium heavy metals in vascular disease, hypertension, coronary heart disease, and myocardial infarction. Altern Ther Health Med. 2007;13:S128–S133. [PubMed] [Google Scholar]
- Huang YL, Cheng SL, Lin TH. Lipid peroxidation in rats administrated with mercuric chloride. Biol Trace Elem Res. 1996;52:193–206. doi: 10.1007/BF02789461. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Oguri T, Tada M. Effect of methylmercury (CH3HgCl) injury on nitric oxide synthase (NOS) activity in cultured human umbilical vascular endothelial cells. Toxicology. 1995;103:1–7. doi: 10.1016/0300-483x(95)99370-r. [DOI] [PubMed] [Google Scholar]
- Langworth S, Sallsten G, Barregard L, Cynkier I, Lind ML, Soderman E. Exposure to mercury vapor and impact on health in the dental profession in Sweden. J Dent Res. 1997;76:1397–1404. doi: 10.1177/00220345970760071001. [DOI] [PubMed] [Google Scholar]
- Mahboob M, Shireen KF, Atkinson A, Khan AT. Lipid peroxidation and antioxidant enzyme activity in different organs of mice exposed to low level of mercury. J Environ Sci Health B. 2001;36:687–697. doi: 10.1081/PFC-100106195. [DOI] [PubMed] [Google Scholar]
- Martín MC, Balfagón G, Minoves N, Blanco-Rivero J, Ferrer M. Androgen deprivation increases neuronal nitric oxide metabolism and its vasodilator effect in rat mesenteric arteries. Nitric Oxide. 2005;12:163–176. doi: 10.1016/j.niox.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Miller DM, Woods JS. Urinary porphyrins as biological indicators of oxidative stress in the kidney. Interaction of mercury and cephaloridine. Biochem Pharmacol. 1993;46:2235–2241. doi: 10.1016/0006-2952(93)90614-3. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ. Small artery remodeling and significance in the development of hypertension. News Physiol Sci. 2002;17:105–109. doi: 10.1152/nips.01366.2001. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Osto E, Coppolino G, Volpe M, Cosentino F. Restoring the dysfunctional endothelium. Curr Pharm Des. 2007;13:1053–1068. doi: 10.2174/138161207780487566. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31:S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- Peçanha FM, Wiggers GA, Briones AM, Pérez-Girón JV, Miguel M, García-Redondo AB, et al. The role of cyclooxygenase (COX)-2 derived prostanoids on vasoconstrictor responses to phenylephrine is increased by exposure to low mercury concentration. J Physiol Pharmacol. 2010;61:29–36. [PubMed] [Google Scholar]
- Reus IS, Bando I, Andrés D, Cascales M. Relationship between expression of HSP70 and metallothionein and oxidative stress during mercury chloride induced acute liver injury in rats. J Biochem Mol Toxicol. 2003;17:161–168. doi: 10.1002/jbt.10074. [DOI] [PubMed] [Google Scholar]
- Riganti C, Costamagna C, Doublier S, Miraglia E, Polimeni M, Bosia A, et al. The NADPH oxidase inhibitor apocynin induces nitric oxide synthesis via oxidative stress. Toxicol Appl Pharmacol. 2008;228:277–285. doi: 10.1016/j.taap.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Salonen J, Seppanen K, Nyyssonen K, Korpela H, Kauhanen J, Kantola M, et al. Intake of mercury from fish, lipid peroxidation, and the risk of myocardial infarction and coronary, cardiovascular, and any death in Eastern Finnish men. Circulation. 1995;91:645–655. doi: 10.1161/01.cir.91.3.645. [DOI] [PubMed] [Google Scholar]
- Salonen JT, Seppanen K, Lakka TA, Salonen R, Kaplan GA. Mercury accumulation and accelerated progression of carotid atherosclerosis: a population-based prospective 4-year follow-up study in men in eastern Finland. Atherosclerosis. 2000;148:265–273. doi: 10.1016/s0021-9150(99)00272-5. [DOI] [PubMed] [Google Scholar]
- Sandborgh-Englund G, Elinder CG, Johanson G, Lind B, Skare I, Ekstrand J. The absorption, blood levels, and excretion of mercury after a single dose of mercury vapor in humans. Toxicol Appl Pharmacol. 1998;150:146–153. doi: 10.1006/taap.1998.8400. [DOI] [PubMed] [Google Scholar]
- Torres AD, Rai AN, Hardiek ML. Mercury intoxication and arterial hypertension: report of two patients and review of the literature. Pediatrics. 2000;105:E34. doi: 10.1542/peds.105.3.e34. [DOI] [PubMed] [Google Scholar]
- Virtanen JK, Voutilainen S, Rissanen TH, Mursu J, Tuomainen TP, Korhonen MJ, et al. Mercury, fish oils, and risk of acute coronary events and cardiovascular disease, coronary heart disease, and all-cause mortality in men in eastern Finland. Arterioscler Thromb Vasc Biol. 2005;25:228–233. doi: 10.1161/01.ATV.0000150040.20950.61. [DOI] [PubMed] [Google Scholar]
- Wei Y, Whaley-Connell AT, Chen K, Habibi J, Uptergrove GM, Clark SE, et al. NADPH oxidase contributes to vascular inflammation, insulin resistance, and remodeling in the transgenic (mRen2) rat. Hypertension. 2007;50:384–391. doi: 10.1161/HYPERTENSIONAHA.107.089284. [DOI] [PubMed] [Google Scholar]
- Wiggers GA, Peçanha FM, Briones AM, Pérez-Girón JV, Miguel M, Vassallo DV, et al. Low mercury concentrations cause oxidative stress and endothelial dysfunction in conductance and resistance arteries. Am J Physiol Heart Circ Physiol. 2008;295:H1033–H1043. doi: 10.1152/ajpheart.00430.2008. [DOI] [PubMed] [Google Scholar]
- Wolf MB, Baynes JW. Cadmium and mercury cause an oxidative stress-induced endothelial dysfunction. Biometals. 2007;20:73–81. doi: 10.1007/s10534-006-9016-0. [DOI] [PubMed] [Google Scholar]
- Xu S, Touyz RM. Reactive oxygen species and vascular remodelling in hypertension: still alive. Can J Cardiol. 2006;22:947–951. doi: 10.1016/s0828-282x(06)70314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.