

Abstract
A heterotelechelic biotin-maleimide polymer containing a cleavable disulfide bond was synthesized by RAFT polymerization and used to reversibly modify surfaces with proteins.
Conjugation of proteins and other biomolecules to surfaces is of interest for applications in proteomics, biomaterials, and nanotechnology.1, 2 Reactive polymer scaffolds are well suited for this purpose.3, 4 Orientated immobilization is essential for retaining bioactivity and for studying protein function, protein-ligand and protein-protein interactions.3, 5, 6 Chemospecific reaction at a defined location on the protein ensures correct orientation and further improves homogeneity of the surface.3, 7, 8 Incorporation of a cleavable handle provides a method for capture and release.9, 10
In particular polymers with a surface anchoring group at one end and a moiety for site-specific attachment of biomolecules at the other end are convenient for protein immobilization. Compared to small molecule linkers, polymers may have certain advantages for applications where conformational flexibility of the protein on the surface is required. Recently, we described the synthesis of a biotin-maleimide functional polymer (P3, Scheme 1).11 The streptavidin (SAv)-, avidin- or neutravidin-biotin interaction is often exploited for surface immobilization, and the maleimide functional group chemospecifically reacts with cysteines in the presence of amine residues at mild pH.12 In this report, we disclose for the first time the synthesis of poly(N-isopropylacrylamide) (pNIPAAm) containing a disulfide linkage between the biotin functional group and the maleimide-terminated polymer (P4, Scheme 1) for use in reversible modification of surfaces with proteins. A key feature of the polymer synthesis is that the disulfide did not participate in the polymerization or radical coupling reaction used to install the maleimide end group; this is similar to what was observed in previous studies involving activated or internal disulfides.10, 13–22
Scheme 1.

Synthesis of biotin-maleimide pNIPAAm with (P2 and P4) and without (P1 and P3) a reversible disulfide bond.
Reversible addition-fragmentation chain transfer (RAFT), mediated by a chain transfer agent (CTA), is a robust polymerization method.23 CTAs containing protein reactive R- or Z- groups result in the direct synthesis of α- or ω functional polymers, respectively.11, 14, 24–27
Alternatively, the thiocarbonylthio functional group, which is located on the chain end of the polymer, can be transformed post-polymerization into a protein-reactive functional group.11, 28–31 We chose to incorporate a biotin with a reversible disulfide bond into the R-moiety of the CTA (2). After polymerization a maleimide was installed via radical coupling, which concurrently removed the trithiocarbonate (Scheme 1).
RAFT polymerization of NIPAAm was explored in N,N-dimethylformamide (DMF). pNIPAAm is a thermoresponsive polymer that has been utilized for capture and release of biomolecules,27, 32 as well as for cell sheet formation,33 which makes it an attractive choice for surface modification. 2,2′-Azobisisobutyronitrile (AIBN) initiated polymerization of NIPAAm was conducted at 70 °C using molar ratios of 1:250:0.1 of 2:NIPAAm:AIBN. The resultant biotinylated pNIPAAm had a number average molecular weight (M n) of 34,200 Da and a polydispersity index (PDI) of 1.15 by gel permeation chromatography (GPC) analysis (ESI Fig. S2).
Radical cross-coupling between dithioester chain-ends and azo-initiators has been employed to remove the thiocarbonylthio moiety.34 We have recently shown that protein-reactive functionality can be incorporated into the azo-initiator to produce telechelic polymers.11 In a similar manner, the maleimide chain-end was installed via a radical cross coupling with oxo norbornene azo-initiator 3 in a 1:1 mixture of DMF:dioxane at 70 °C, affording P2. A protected maleimide derivative was employed to avoid radical polymerization of maleimide. Subsequent deprotection via a retro Diels-Alder to provide the biotin-maleimide P4 was performed. 1H NMR analysis confirmed incorporation of the maleimide group (ESI Fig. S1) while GPC analysis (ESI Fig. S2) demonstrated that the molecular weight distribution remained narrow (PDI = 1.17, Mn = 35,700 Da). These results showed that the disulfide linkage was tolerant to each of these processes. For comparative purposes, the nonreversible telechelic pNIPAAm (P3) was also synthesized (Scheme 1) and utilized for surface studies (Mn = 18,400 Da, PDI 1.10, see ESI).
We confirmed the ability of the end groups to bind proteins. To this end a mutant V131C T4 lysozyme (T4L) was expressed and purified from E. coli. The protein was incubated with an excess of either P4 or P2 in phosphate buffer (PB) pH 7.0 at 4 °C for 24 h. The latter was used as a control. Formation of the crude conjugates was verified by SDS PAGE (ESI, Fig. S3). When the maleimide was available, a conjugate was observed at ~37 kDa; however, when the furan-protected maleimide polymer was incubated with T4L, no conjugate was visible. This confirmed that the disulfide bond did not participate in conjugate formation. The crude T4L-P4 was then incubated with SAv to ensure that the biotin was available. The molecular weight of the conjugate (~125 kDa) was significantly higher (Fig. 1, lane 5) than either T4L (lane 3), SAv (lane 4) or T4L-P4 conjugate (~37 kDa, Fig. S3), which demonstrated that the biotin was available for conjugation. The residual T4L at ~ 12 kDa carried through from the maleimide conjugation was likely a result of partial hydrolysis of the maleimide or oxidation of the protein in that step.
Figure 1.

SDS-PAGE of conjugates. Lane 1: protein marker, lane 2: blank, lane 3: T4L, lane 4: SAv, lane 5: T4L + SAv + P4, lane 6: T4L-pNIPAAm isolated from SAv-resin, lane 7: BSA, lane 8: BSA + P4. Protein crystal structures obtained from PDB: 1SWA, 1P56, and 1E7H. Lanes 3–8 under non-reducing conditions.
We next explored reversible protein immobilization on SAv surfaces (Scheme 2). Modification of T4L with P4 was repeated. In this experiment, however, the protein was in excess. Crude T4L-P4 was then loaded onto a SAv-agarose resin in order to bind the conjugate. Prior to addition of 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), the resin was washed extensively to remove unconjugated protein until no absorbance was observed at 280 nm. After incubating with the reducing agent for 2 h at 22 °C, all fractions that contained an absorbance at 280 nm were pooled, concentrated, and analyzed by gel electrophoresis under nonreducing conditions. Lane 6 (Fig. 1) showed the presence of protein-polymer conjugate. The residual unconjugated T4L that was also observed may have been due to electrostatic interaction of the basic T4L with the positively charged SAv that was not effectively screened by the low salt eluent. Alternatively, it is possible that the free thiol on the T4L, which was used in excess, bound to the cysteines of streptavidin. However, the results clearly demonstrate that the conjugate was immobilized to the SAv by a reversible bond and that the conjugate could be cleaved from the resin using a reducing agent. This overall strategy may be useful for synthesis and purification of protein-polymer conjugates using resins.
Scheme 2.
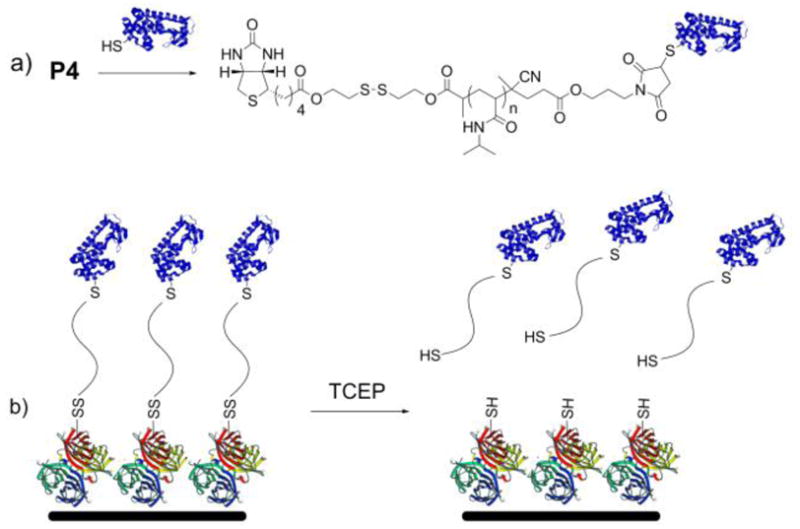
(a) Conjugation of biotin-maleimide pNIPAAm to a free cysteine on a protein; (b) immobilization of the conjugate via the biotin-SAv interaction and removal from the surface by reduction (protein structures from the PDB: 1SWA and 1P56).
To further demonstrate reversible conjugation, immobilization of bovine serum albumin (BSA), a commercially available protein with a surface accessible free cysteine, was attempted. First, BSA was conjugated to P3 and P4 in phosphate buffered saline forming conjugate 1 (biotin-pNIPAAm-BSA) and conjugate 2 (biotin-SS-pNIPAAm-BSA), respectively. The excess polymer was removed by gel filtration. In both cases, the reactions were successful; shifts to higher molecular weights for the conjugates compared to BSA were observed in the SDS PAGE (for P3, data not shown; for P4 Fig. 1, lane 8 compared to lane 7).
BSA immobilization onto surfaces was then evaluated using an enzyme linked immunosorbant assay (ELISA). Neutravidin-coated 96-well plates were first blocked with BSA-free blocking buffer to minimize non-specific binding of the conjugate, and then incubated with solutions of conjugates 1 or 2 with BSA as a control. Anti-BSA-horse radish peroxidase (HRP) was then incubated in each well; the absorbance of the HRP-catalyzed oxidation of the o-phenylenediamine (OPD) substrate to 2,3-diaminophenazine was measured at 450 nm. Shown in Fig. 2, the absorbances at 450 nm of conjugate 1 and 2 were significantly higher (p <0.05) than that of the BSA control and no difference in binding was observed between the two conjugates (p > 0.10).
Figure 2.

ELISA of BSA-pNIPAAm conjugates. Each sample was incubated with 1 mg/mL of OPD substrate; after 30 min the absorbance at 450 nm was measured to determine the relative amount of oxidized substrate. From left to right: BSA, conjugate 1, conjugate 1 (pre-blocked with biotin); conjugate 2; conjugate 2 with addition of 10 mM GSH. Error bars indicate standard deviation from a minimum of 6 measurements. *Denotes statistical deviation from the BSA control (p < 0.05). **Denotes statistical deviation from the BSA control and conjugate 2 (p < 0.05).
In order to confirm that the conjugate was bound to the surface through the biotin binding site of neutravidin, a control study was performed in which the wells were first incubated with a solution of biotin, to pre-block the binding sites of neutravidin. After addition of the substrate, no statistical difference in production of 2,3-diaminophenazine between BSA and biotin-blocked wells was observed (p > 0.68) (Fig. 2). An additional control was performed where the polymer without BSA was conjugated to the surface. Again, binding of the BSA antibody was not observed (ESI, Fig. S4).
To determine if BSA-P4 could be removed from the surface via reduction of the disulfide bond, a biologically relevant concentration of 10 mM glutathione (GSH) was incubated with the immobilized conjugate. GSH contributes to the reducing environment of cells in vivo and was therefore chosen in order to mimic biological conditions.35 ELISA analysis determined that after reduction with GSH, a significant amount of conjugate 2 was cleaved from the surface, as evident by the decrease in production of 2,3-diaminophenazine (Fig. 2, p < 0.05). Taken together, these data demonstrate: (1) The polymers effectively immobilize proteins to surfaces; (2) the conjugates are linked to the biotin binding sites of the neutravidin; (3) the conjugation is reversible when the disulfide linkage between the biotin and polymer is present; and (4) the protein was still able to bind to its antibody while anchored to the surface by the polymer linker.
In conclusion, we have demonstrated the synthesis of a biotinylated CTA containing a disulfide bond. It was shown that this CTA mediates the polymerization of NIPAAm to generate well-defined α-biotin disulfide, ω-trithiocarbonate-pNIPAAm. The trithiocarbonate end group was efficiently transformed via a radical coupling reaction with a furan-protected maleimide azo initiator, resulting in a heterotelechelic α-biotin disulfide, ω-maleimide-pNIPAAm after a retro-Diels Alder reaction. The disulfide bond did not participate in the polymerization or radical coupling reaction. The polymer was conjugated to either V131C T4L or BSA and then anchored to SAv- or neutravidin-functionalized surfaces. The conjugate was liberated from the surfaces under mild reducing conditions demonstrating the reversibility. Furthermore, immobilization of the complimentary antibody revealed that the surface-anchored protein maintained activity towards antibodies. We anticipate that this approach will be generally useful for reversibly modifying surfaces with proteins for a wide variety of applications.
Supplementary Material
Acknowledgments
This work was supported by the NSF (CAREER CHE-0645793). KLH thanks the NIH-sponsored Chemistry-Biology Interface Training Program, the Christopher S. Foote Organic Division Research Fellowship and the UCLA Dissertation Year Fellowship for funding. GNG thanks the Christopher S. Foote Organic Division Research Fellowship and the NIH Biotechnology Training Grant. HDM thanks the Alfred P. Sloan Foundation for additional support.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, data not shown in the text. See DOI: 10.1039/b000000x/
References and Notes
- 1.Angenendt P. Drug Discov Today. 2005;10:503–511. doi: 10.1016/S1359-6446(05)03392-1. [DOI] [PubMed] [Google Scholar]
- 2.Rusmini F, Zhong ZY, Feijen J. Biomacromolecules. 2007;8:1775–1789. doi: 10.1021/bm061197b. [DOI] [PubMed] [Google Scholar]
- 3.Christman KL, Enriquez-Rios VD, Maynard HD. Soft Matter. 2006;2:928–939. doi: 10.1039/b611000b. [DOI] [PubMed] [Google Scholar]
- 4.Tugulu S, Arnold A, Sielaff I, Johnsson K, Klok HA. Biomacromolecules. 2005;6:1602–1607. doi: 10.1021/bm050016n. [DOI] [PubMed] [Google Scholar]
- 5.Chen SF, Liu LY, Zhou J, Jiang SY. Langmuir. 2003;19:2859–2864. [Google Scholar]
- 6.Rao SV, Anderson KW, Bachas LG. Mikrochim Acta. 1998;128:127–143. [Google Scholar]
- 7.Asuri P, Karajanagi SS, Yang HC, Yim TJ, Kane RS, Dordick JS. Langmuir. 2006;22:5833–5836. doi: 10.1021/la0528450. [DOI] [PubMed] [Google Scholar]
- 8.Camarero JA, Kwon Y, Coleman MA. J Am Chem Soc. 2004;126:14730–14731. doi: 10.1021/ja0456611. [DOI] [PubMed] [Google Scholar]
- 9.Carlsson J, Axen R, Unge T. Eur J Biochem. 1975;59:567–572. doi: 10.1111/j.1432-1033.1975.tb02483.x. [DOI] [PubMed] [Google Scholar]
- 10.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2002;35:9009–9014. [Google Scholar]
- 11.Heredia KL, Grover GN, Tao L, Maynard HD. Macromolecules. 2008;42:2360–2367. doi: 10.1021/ma8022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermanson GT. Bioconjugate Techniques. Academic Press; New York: 1996. [Google Scholar]
- 13.Boyer C, Liu J, Wong L, Tippett M, Bulmus V, Davis TP. J Polym Sci, Part A: Polym Chem. 2008;46:7207–7224. [Google Scholar]
- 14.Heredia KL, Nguyen TH, Chang CW, Bulmus V, Davis TP, Maynard HD. Chem Commun. 2008:3245–3247. doi: 10.1039/b804812f. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Macromol Rapid Commun. 2007;28:305–314. [Google Scholar]
- 16.Setijadi E, Tao L, Liu JQ, Jia ZF, Boyer C, Davis TP. Biomacromolecules. 2009;10:2699–2707. doi: 10.1021/bm900646g. [DOI] [PubMed] [Google Scholar]
- 17.Tao L, Liu JQ, Davis TP. Biomacromolecules. 2009;10:2847–2851. doi: 10.1021/bm900678r. [DOI] [PubMed] [Google Scholar]
- 18.Tao L, Liu JQ, Xu JT, Davis TP. Org Biomol Chem. 2009;7:3481–3485. doi: 10.1039/b907061c. [DOI] [PubMed] [Google Scholar]
- 19.Vazquez-Dorbatt V, Tolstyka ZP, Chang CW, Maynard HD. Biomacromolecules. 2009;10:2207–2212. doi: 10.1021/bm900395h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu JT, Boyer C, Bulmus V, Davis TP. J Polym Sci, Part A: Polym Chem. 2009;47:4302–4313. [Google Scholar]
- 21.Xu JT, Tao L, Liu JQ, Bulmus V, Davis TP. Macromolecules. 2009;42:6893–6901. [Google Scholar]
- 22.Bontempo D, Heredia KL, Fish BA, Maynard HD. J Am Chem Soc. 2004;126:15372–15373. doi: 10.1021/ja045063m. [DOI] [PubMed] [Google Scholar]
- 23.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2005;58:379–410. [Google Scholar]
- 24.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, Stenzel MH. Macromolecules. 2008;41:5641–5650. [Google Scholar]
- 25.Hentschel J, Bleek K, Ernst O, Lutz JF, Borner HG. Macromolecules. 2008;41:1073–1075. [Google Scholar]
- 26.Heredia KL, Maynard HD. Org Biomol Chem. 2007;5:45–53. doi: 10.1039/b612355d. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman AS, Stayton PS. Prog Polym Sci. 2007;32:922–932. [Google Scholar]
- 28.Pound G, McKenzie JM, Lange RFM, Klumperman B. Chem Commun. 2008:3193–3195. doi: 10.1039/b803952f. [DOI] [PubMed] [Google Scholar]
- 29.Alconcel SNS, Grover GN, Matsumoto NM, Maynard HD. Aust J Chem. 2009;62:1496–1500. doi: 10.1071/CH09398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grover GN, Alconcel SNS, Matsumoto NM, Maynard HD. Macromolecules. 2009;42:7657–7663. doi: 10.1021/ma901036x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyer C, Bulmus V, Davis TP. Macromol Rapid Commun. 2009;30:493–497. doi: 10.1002/marc.200800708. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman AS, Stayton PS. Macromol Symp. 2004;207:139–151. [Google Scholar]
- 33.Matsuda N, Shimizu T, Yamato M, Okano T. Adv Mater. 2007;19:3089–3099. [Google Scholar]
- 34.Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38:2033–2036. [Google Scholar]
- 35.Wang W, Ballatori N. Pharmacol Rev. 1998;50:335–355. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.