

Abstract
Systemic mastocytosis is a neoplastic disease of mast cells harboring the activating KIT mutation D816V. In most patients, mast cell infiltration in the bone marrow is accompanied by marked microenvironment alterations, including increased angiogenesis, osteosclerosis, and sometimes fibrosis. Little is known about the mast cell-derived molecules contributing to these bone marrow alterations. We show here that neoplastic mast cells in patients with systemic mastocytosis express oncostatin M (OSM), a profibrogenic and angiogenic modulator. To study the regulation of OSM expression, KIT D816V was inducibly expressed in Ba/F3 cells and was found to up-regulate OSM mRNA and protein levels, suggesting that OSM is a KIT D816V-dependent mediator. Correspondingly, KIT D816V+ HMC-1.2 cells expressed significantly higher amounts of OSM than the KIT D816V− HMC-1.1 subclone. RNA interference-induced knockdown of STAT5, a key transcription factor in KIT D816V+ mast cells, inhibited OSM expression in HMC-1 cells, whereas a constitutively activated STAT5 mutant induced OSM expression. Finally, OSM secreted from KIT D816V+ mast cells stimulated growth of endothelial cells, fibroblasts, and osteoblasts, suggesting that mast cell-derived OSM may serve as a key modulator of the marrow microenvironment and thus contribute to the pathology of systemic mastocytosis.
Systemic mastocytosis is a clonal disease of myelomastocytic progenitors.1 The clinical manifestation of the disease is variable, ranging from an indolent course to highly aggressive variants with short survival time. Based on clinical findings, four major subtypes of systemic mastocytosis have been defined by the World Health Organization: indolent systemic mastocytosis, mastocytosis with an associated clonal hematological non-mast cell lineage disease, aggressive systemic mastocytosis, and mast cell leukemia.2 The pathological hallmark of systemic mastocytosis is the multifocal dense infiltrate of mast cells in the bone marrow. Apart from mast cell infiltrates, alterations in the bone marrow microenvironment, including increased angiogenesis, thickened bone trabeculae, and sometimes bone marrow fibrosis, are found in these patients.3–5 These bone marrow alterations are detectable independent of the category of mastocytosis, presence of eosinophilia, or the mast cell burden. It has also been shown that bone marrow stromal cells support growth and differentiation of mast cells.6–9 So far, however, little is known about the interplay between neoplastic mast cells and stromal cells and its relevance for the pathogenesis of systemic mastocytosis.
The receptor tyrosine kinase KIT and its ligand, stem cell factor (SCF), play a pivotal role in mast cell differentiation and growth. In most patients with systemic mastocytosis, neoplastic cells harbor the KIT mutation D816V, which affects the ATP-binding domain of the KIT receptor and results in a gain of function.10–12 KIT D816V has been shown to promote mast cell differentiation in a ligand-independent manner and to produce an indolent mast cell disease in mice.13 Several studies have suggested that the D816V mutation leads to a quantitative as well as qualitative alteration in KIT-dependent signaling.14–16 In particular, various signal transduction pathways including the phosphoinositide 3-kinase (PI3-kinase)/Akt pathway, the mitogen-activated protein kinases (MAP kinases), and signal transducer and activator of transcription 5 (STAT5) have been shown to be activated by KIT D816V.14,15 Several observations suggest that STAT5 may play a particular role in growth and survival of normal and neoplastic mast cells. Specifically, STAT5-deficient mice have a profound defect in mast cell development.17 It has also been reported that neoplastic mast cells in systemic mastocytosis express phosphorylated STAT5, and that inhibition of STAT5 negatively affects mast cell growth.18,19 So far, however, the pathogenetic significance of STAT5 activation in neoplastic mast cells has not been defined.
Oncostatin M (OSM) is a pleiotropic cytokine belonging to the interleukin-6 (IL-6) family. Produced mainly by activated T cells and monocytes, OSM can elicit different biological effects, depending on the cell type.20 Two types of human OSM receptors have been described. The type I OSM receptor is composed of gp130 and the leukemia inhibitory factor (LIF) receptor β-subunit (LIFR),21,22 and the type II OSM receptor is composed of gp130 and the OSM-specific receptor β-subunit (OSMR).23 Initially, OSM was recognized by its inhibitory activity against A375 melanoma cells.24 Later, OSM was shown to act as a growth factor for mesenchymal cells, including endothelial cells, fibroblasts, and osteoblasts.25–28 Recently, OSM has been implicated as an important cytokine in hematopoiesis. Transplantation experiments with OSM-deficient mice have shown that OSM stimulates stromal cells as well as hematopoietic progenitors and is required for the generation and maintenance of a proper microenvironment in the bone marrow.29–31
In the present study, we examined the expression and potential role of OSM in neoplastic mast cells. We found that neoplastic mast cells express OSM in a constitutive manner and that OSM expression is dependent on KIT D816V and downstream STAT5. We also found that mast cell-derived OSM stimulates the growth of microvascular endothelial cells, osteoblasts, and fibroblasts, suggesting a potential pathogenetic role of this mediator in systemic mastocytosis.
Materials and Methods
Patients
Eighteen patients with systemic mastocytosis and seven control subjects (staging of lymphomas or reactive bone marrow) were examined. Mastocytosis was diagnosed according to World Health Organization criteria.2 In the systemic mastocytosis group, 14 patients had indolent systemic mastocytosis, two had systemic mastocytosis with associated clonal hematological non-mast cell lineage disease, and two had mast cell leukemia (see Supplemental Table S1 at http://ajp.amjpathol.org). Routine staging included physical examination, abdominal ultrasound imaging, complete blood count, serum tryptase measurement, bone marrow histology, IHC (tryptase and CD25), cytologic examination of bone marrow cells on Wright-Giemsa-stained bone marrow smears, flow cytometry for detection of CD2 and CD25 in bone marrow mast cells, and analysis of bone marrow cells for KIT D816V by restriction fragment length polymorphism as described previously.2 Informed consent was obtained in each case before blood examinations and bone marrow puncture.
Reagents
Midostaurin (PKC412) was kindly provided by Johannes Roesel and Paul Manley (Novartis Pharma AG, Basel, Switzerland).32 Recombinant human stem cell factor (SCF) was purchased from Strathmann Biotec (Hannover, Germany). Recombinant murine IL-3 and human IL-6 were from Miltenyi Biotec (Auburn, CA). RPMI 1640 medium, Iscove's modified Dulbecco's medium, Dulbecco's modified Eagle's medium, Dulbecco's modified Eagle's medium/F12(1:1) without Phenol Red, MDCB131 medium, and fetal calf serum (FCS) were from Invitrogen (Carlsbad, CA) and polybrene was from Sigma-Aldrich (St. Louis, MO). The MAP kinase/extracellular signal-regulated kinase (MEK) inhibitor PD98059, the PI3-kinase inhibitor LY294002, and piceatannol were purchased from Calbiochem (La Jolla, CA).
Cells and Cell Culture
Ba/F3 cells with inducible expression of KIT D816V (Ton.Kit.D816V) or wild-type KIT (Ton.Kit.wt)15 were grown in RPMI 1640 medium with 10% FCS and IL-3 (1 ng/mL) at 37°C and 5% CO2. (Ba/F3 is a murine, IL-3-dependent progenitor cell line.) The mast cell line HMC-1 was kindly provided by Joseph H. Butterfield (Mayo Clinic, Rochester, MN).33 Two subclones were used: HMC-1.1, harboring KIT V560G but not KIT D816V34 (referred to as KIT D816V− HMC-1 cells), and HMC-1.2, harboring KIT V560G as well as KIT D816V33,34 (referred to as KIT D816V+ HMC-1 cells). HMC-1 cells were cultured in Iscove's modified Dulbecco's medium with 10% FCS. The human microvascular endothelial cell line HMEC-1 was kindly provided by Edwin Ades (Centers for Disease Control and Prevention, Atlanta, GA).35,36 HMEC-1 cells were cultured in MDCB131 medium with 10% FCS. hFOB 1.19 cells (ATCC, Manassas, VA) were cultured in DMEM/F12 medium without Phenol Red with 10% FCS. After informed consent was obtained, CD34+ progenitor cells were isolated from cord blood by magnetic-activated cell separation using a CD34 MicroBead kit (Miltenyi Biotec) and then were cultured in StemPro-34 serum-free complete medium (Invitrogen) in the presence of SCF and IL-6 (both at 100 ng/mL). After 4 weeks, RPMI 1640 medium containing 10% FCS was used instead of serum-free medium. Cytokines were replaced weekly. After 7 weeks, 70% to 80% of cells were mature mast cells, as evidenced by Wright-Giemsa staining.
Primary lung mast cells were prepared as described previously.37 Lung tissue was obtained from patients undergoing lobectomy for bronchiogenic carcinoma (n = 4), after informed consent was given. Tissue was placed into Ca/Mg-free Tyrode's buffer immediately after resection, then was cut into small pieces and washed in Tyrode's buffer. Tissue fragments were then incubated in collagenase type II (1.5 mg/mL; Worthington, Lakewood, NJ) at 37°C for 60 minutes. Dispersed cells were recovered by filtration through Nitex cloth, washed in RPMI 1640 medium with 10% FCS and then filtered through a cell strainer (70 μm). Mast cells were purified by fluorescence-activated cell sorting on a FACSAria system (Becton Dickinson, Franklin Lakes, NJ) using a phycoerythrin-labeled antibody against CD117 (Becton Dickinson) and then were cultured in RPMI 1640 medium containing 10% FCS and SCF (100 ng/mL) for 3 days.
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (local ethics committee) of the Medical University of Vienna.
Generation of Ba/F3 Cells with Conditional Expression of STAT5 S710F
Ba/F3 cells expressing the reverse tetracycline-transactivator38 were retrovirally transduced with the pRevTRE plasmid (Clontech, Palo Alto, CA) containing murine STAT5A S710F (kindly provided by Richard Moriggl, Ludwig Boltzmann Institute for Oncology Research, Vienna, Austria).39 Cells were selected with puromycin (1 μg/mL) and then cloned by limiting dilution. Expression of STAT5 S710F in the presence of doxycycline (1 μg/mL) was analyzed by immunoblotting.
Knockdown of KIT and STAT5
For knockdown of KIT, a pLKO.1 clone containing a short hairpin RNA (shRNA) targeting human KIT (5′-GCCGGTCGATTCTAAGTTCTAC-3′) was obtained from Open Biosystems (Huntsville, AL). For knockdown of STAT5, an oligonucleotide targeting human and murine STAT5A and STAT5B40 (5′-GCAGCAGACCATCATCCTG-3′) was cloned into a modified pLKO.1 lentiviral vector. Recombinant VSV-G pseudotyped lentiviruses were produced as described previously.41 HMC-1 and Ton.Kit.D816V cells were transduced in the presence of polybrene (7 μg/mL) and were selected with puromycin (2 μg/mL) for 48 hours. Knockdown of STAT5 was confirmed by immunoblotting and knockdown of KIT by fluorescence-activated cell sorting analysis.
IHC, Immunocytochemistry, and Immunofluorescence
IHC was performed on FFPE bone marrow biopsy sections using the indirect immunoperoxidase staining technique as described previously.19 Slides were pretreated in a microwave oven and endogenous peroxidase was blocked by methanol/H2O2 before sections were incubated with anti-tryptase G3 antibody (Ventana Medical Systems, Tucson, AZ) or a polyclonal rabbit anti-OSM antibody (sc-129, dilution 1:500; Santa Cruz Biotechnology, Santa Cruz, CA). In select experiments, the OSM antibody was preincubated with a blocking peptide (Santa Cruz Biotechnology). After washing, slides were incubated with biotinylated horse anti-mouse or goat anti-rabbit IgG for 30 minutes, washed, and exposed to avidin-biotin-peroxidase complex for 30 minutes. 3-Amino-9-ethyl-carbazole was used as chromogen. Slides were counterstained in Mayer's hemalaun and mounted in Aquatex (Merck, Darmstadt, Germany).
Immunocytochemistry was performed on cytospin preparations of HMC-1 and lung mast cells as described previously,19 using a polyclonal rabbit anti-OSM antibody (Santa Cruz Biotechnology). Cytospin slides were incubated with the primary antibody overnight, washed, and thereafter exposed to biotinylated goat anti-rabbit IgG (Biocare Medical, San Diego, CA) for 30 minutes. Streptavidin-alkaline-phosphatase complex (Biocare Medical) was used as chromogen. Antibody reactivity was made visible using neofuchsin. Slides were counterstained in Mayer's hemalaun and mounted. For staining of phosphorylated STAT5, the anti-pSTAT5a/b antibody AX1 from Advantex BioReagents (El Paso, TX) was used as described previously.19 Images were prepared using an Olympus DP11 camera connected to an Olympus BX50F4 microscope equipped with 100×/1.35 UPlan-Apo objective lens (Olympus, Hamburg, Germany) and processed with Adobe Photoshop CS2 software version 9.0 (Adobe Systems, San Jose, CA).
Immunofluorescence double labeling was performed on formalin-fixed, paraffin-embedded bone marrow and lung biopsy sections. Slides were pretreated in a microwave oven, blocked with 10% FCS, and incubated with rabbit anti-OSM antibody (dilution 1:250) overnight. Sections were then incubated with mouse anti-tryptase G3 antibody for 1 hour. After a washing, slides were exposed to Alexa Fluor 555-conjugated anti-rabbit and Alexa Fluor 488-conjugated anti-mouse secondary antibodies (Invitrogen). Nuclei were counterstained with DAPI (Sigma-Aldrich). Images were acquired by confocal laser scanning microscopy using a Zeiss LSM 710 system (Carl Zeiss MicroImaging, Jena, Germany) with a 40× Plan-Apochromat objective (1.4 oil/DICIII) and a pinhole of 0.8 airy units.
Detection of OSM by ELISA
To determine the amount of secreted OSM, an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) was used according to the recommendations of the manufacturer. The detection limit of the ELISA amounted to 30 pg/mL OSM.
Immunoblotting
Whole cell extracts from Ton.Kit.D816V, Ton.Stat5.S710F, and HMC-1 cells were subjected to SDS-polyacrylamide gel electrophoresis analysis and immunoblotting was performed using antibodies against OSM (Santa Cruz Biotechnology), STAT5 (Becton Dickinson), phosphorylated STAT5 (Cell Signaling Technology, Danvers, MA), and β-actin (Sigma-Aldrich) as described previously.15
Reporter Gene Assay
A firefly luciferase reporter plasmid containing 0.7 kb of the 5′-flanking region of the murine OSM gene (pGV-OSM-luc) was kindly provided by Akihiko Yoshimura (Research Institute of Bioregulation, Kyushu University, Fukuoka, Japan).42 The pRL-TK plasmid (Promega, Madison, WI) containing the Renilla luciferase gene driven by a herpes simplex virus thymidine kinase promoter was used as an internal control reporter. pGV-OSM-luc (2 μg) and pRL-TK (20 ng) plasmids were transfected into Ton.Kit.D816V cells by means of Amaxa nucleofection (Lonza, Cologne, Germany) according to the recommendations of the manufacturer. After transfection, cells were split and incubated with or without doxycycline (1 μg/mL) for 24 hours. OSM promoter activity was determined using a dual-luciferase reporter assay system (Promega) and a BioTek Synergy 2 luminometer (BioTek Solutions, Santa Barbara, CA). OSM promoter activity was normalized to Renilla luciferase activity and expressed as percentage of control (absence of doxycycline defined the control).
Real-Time PCR
OSM, OSMR, LIFR, and ABL mRNA copy numbers were quantified on a 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA) using iTaq SYBR Green Supermix with ROX (Bio-Rad Laboratories, Hercules, CA) and plasmid standards. Copy numbers were normalized to ABL copy numbers and expressed as percentage of ABL or as ΔCT values (ΔCT = CT(ABL) − CT(mRNA Sample)). Primer sequences are listed in Table 1.
Table 1.
Primer Sequences
| Gene | Primer | Sequence |
|---|---|---|
| ABL1 | ABL_209_fwd | 5′-TGTATGATTTTGTGGCCAGTGGAG-3′ |
| ABL_279_rev | 5′-GCCTAAGACCCGGAGCTTTTCA-3′ | |
| OSM | OSM_67_fwd | 5′-CACACAGAGGACGCTGCTCA-3′ |
| OSM_126_rev | 5′-ATGCTCGCCATGCTTGGAA-3′ | |
| OSMR | OSMR_1902_fwd | 5′-TGGGTGCTTCTCCTGCTTCTG-3′ |
| OSMR_1992_rev | 5′-CCACCCTCTGTGCCTGCAAT-3′ | |
| LIFR | LIFR_93_fwd | 5′-GCTCAGAAAGGGAGCCTCTGC-3′ |
| LIFR_160_rev | 5′-CCATCATCTGTGCAATGCAGTC-3′ | |
| HDC | HDC_376_fwd | 5′-GGAGACATGCTGGCTGATGC-3′ |
| HDC_443_rev | 5′-TCTGTACACGCAGGGCTGGA-3′ | |
| BCL2L1 | BCL2L1_161_fwd | 5′-CTCCTCTCCCGACCTGTGAT-3′ |
| BCL2L1_260_rev | 5′-AAGATTCTGAAGGGAGAGAAAGAGA-3′ | |
| PIM1 | PIM1_1124_fwd | 5′-TCTTCAGGCAGAGGGTCTCTTCA-3′ |
| PIM1_1184_rev | 5′-CTCAGGGCCAAGCACCATCT-3′ | |
| Abl1 (murine) | Abl_262_fwd | 5′-TCTATGATTTTGTGGCCAGTGGAG-3′ |
| Abl_332_rev | 5′-ACCCAAGACCCGGAGCTTTTCA-3′ | |
| Osm (murine) | Osm_577_fwd | 5′-GCTGCAGAATCAGGCGAACC-3′ |
| Osm_677_rev | 5′-TGCAGGCAGCTCTCAGGTCA-3′ |
[3H]Thymidine Incorporation Assay
To determine effects of mast cell-derived OSM on growth of mesenchymal cells, HMEC-1 cells, hFOB 1.19 cells, and primary bone marrow fibroblasts were cultured in 96-well microtiter plates and incubated with supernatant from KIT D816V+ or KIT D816V− HMC-1 cells in the presence or absence of a neutralizing OSM antibody (R&D Systems) for 72 hours. Next, 1 μCi [3H]thymidine was added to each well. At 12 hours, cells were harvested and bound radioactivity was measured as described previously.41 For production of supernatants, HMC-1 cells (2 × 106 cells/mL) were cultured in the presence of 1% FCS (for stimulation of hFOB 1.19 cells) or 10% FCS (for stimulation of HMEC-1 cells and primary fibroblasts) for 24 hours.
Statistical Analysis
To determine the level of significance, Student's t-test was applied. Results were considered to be significantly different when the P value was <0.05.
Results
Neoplastic Mast Cells Constitutively Express OSM
To investigate OSM expression in neoplastic mast cells, we first examined bone marrow sections obtained from patients with systemic mastocytosis. As assessed by IHC and immunofluorescence, tryptase-positive spindle-shaped neoplastic mast cells reacted with the anti-OSM antibody (Figure 1). OSM was expressed in neoplastic mast cells in all 18 patients investigated (see Supplemental Table S1 at http://ajp.amjpathol.org) and in all variants of systemic mastocytosis (indolent systemic mastocytosis, systemic mastocytosis with associated clonal hematological non-mast cell lineage disease, and mast cell leukemia) with comparable staining intensity (Table 2). Preadsorption with a specific blocking peptide completely abolished immunoreactivity of the anti-OSM antibody, thereby confirming the specificity of the staining reaction (Figure 2, A–D). Apart from neoplastic mast cells, megakaryocytes, endothelial cells, myeloid progenitors, and endosteal cells were also found to react with the anti-OSM antibody (Figure 2, E and F, and Table 2). Erythroid cells and mature granulocytes did not express OSM.
Figure 1.
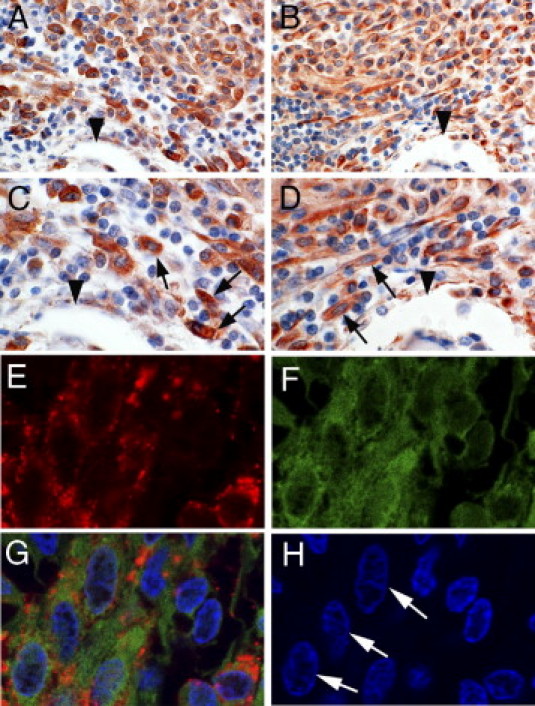
Detection of oncostatin M (OSM) in neoplastic mast cells in the bone marrow of patients with systemic mastocytosis. A–D: Serial sections prepared from paraffin-embedded bone marrow of a patient with systemic mastocytosis were stained with anti-tryptase antibody (A and C) or anti-OSM antibody (B and D) as described under Materials and Methods. Typical spindle-shaped neoplastic mast cells (arrows) and endothelial cells were found to express OSM. Arrowheads indicate the same bone trabecule in two adjacent sections (A and B; C and D). Original magnification: ×400 (A and B); ×1000 (C and D). E–H: Neoplastic mast cells were examined for expression of tryptase (E, red) and OSM (F, green) in bone marrow sections from a patient with systemic mastocytosis by a double immunofluorescence staining technique. The merged image (G) confirms coexpression of OSM and tryptase in single neoplastic mast cells. H: DAPI stain. Arrows indicate spindle-shaped OSM/tryptase-positive mast cells.
Table 2.
Cellular Distribution of Oncostatin M In Bone Marrow Sections in Mastocytosis and Controls
| Cell type | Normal BM | ISM | MCL | SM-AHNMD |
|---|---|---|---|---|
| Megakaryocytes | ++ | ++ | ++ | ++ |
| Myeloid progenitors | + | + | + | + |
| Neutrophil granulocytes | − | − | − | − |
| Eosinophil granulocytes | − | − | − | − |
| Erythroid progenitors | − | − | − | − |
| Mast cells | ND⁎ | + | + | + |
| Endothelial cells | + | + | + | + |
| Endosteal cells | + | + | + | + |
BM, bone marrow; ISM, indolent systemic mastocytosis; MCL, mast cell leukemia; ND, no data; SM-AHNMD, systemic mastocytosis with associated clonal hematologic non-mast cell lineage disease; −, negative; +, positive; ++, strongly reactive.
Bone marrow sections were stained with an antibody against oncostatin M as described under Materials and Methods.
Only a few diffusely spread mast cells were found in normal bone marrow.
Figure 2.
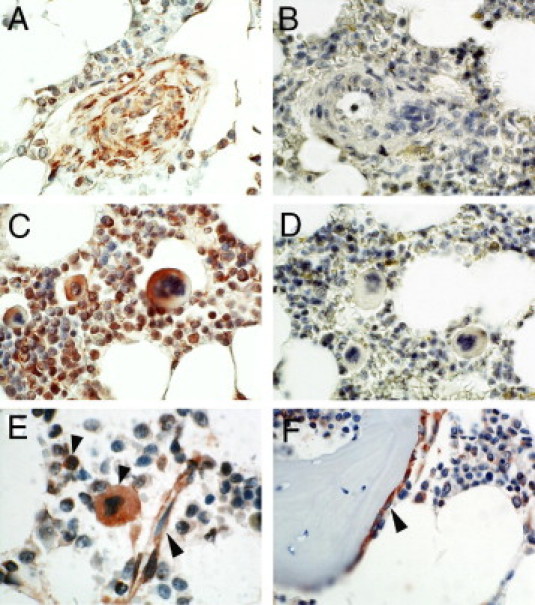
Expression of OSM in the bone marrow. A and C: Bone marrow sections from a patient with systemic mastocytosis were stained with anti-OSM antibody. B and D: Preincubation of anti-OSM antibody with a specific blocking peptide abolished OSM immunoreactivity. E and F: Bone marrow sections from a control patient were stained with anti-OSM antibody. Myeloid progenitors, megakaryocytes, endothelial cells (E, arrowheads), and endosteal cells (F, arrowhead) stained positive for OSM in the normal bone marrow, whereas erythroid cells and mature granulocytes stained negative for OSM.
Next, we asked whether OSM serum levels are increased in patients with systemic mastocytosis. OSM serum levels were determined in 16 mastocytosis patients (see Supplemental Table S2 at http://ajp.amjpathol.org) and in 20 healthy control individuals by ELISA. We found that 6 of the 16 patients with systemic mastocytosis had significantly increased OSM serum levels (median 126.3 pg/mL, range 47.7 pg/mL to 188.2 pg/mL), whereas OSM could not be detected in the serum of healthy individuals. Taken together, our data show that neoplastic mast cells in patients with systemic mastocytosis express OSM and that OSM serum levels are elevated in a subset of these patients.
Role of KIT D816V in Expression of OSM in Neoplastic Mast Cells
Given the high levels of OSM in mast cells of mastocytosis patients and the strong link between systemic mastocytosis and the KIT D816V mutation, we evaluated whether this mutation might be involved in regulating OSM expression. For this purpose, both KIT D816V+ and KIT D816V− HMC-1 cells were analyzed for OSM levels using immunocytochemistry. KIT D816V+ HMC-1 cells were found to express substantial higher amounts of the OSM protein, compared with KIT D816V− HMC-1 cells (Figure 3A). Preincubation of the anti-OSM antibody with a blocking peptide abolished OSM immunoreactivity (Figure 3A). In line with our immunostaining results, KIT D816V+ HMC-1 cells were found to express higher levels of OSM mRNA (Figure 3B) and secreted higher amounts of the OSM protein into culture supernatants (Figure 3C) than KIT D816V− HMC-1 cells. Both normal cord blood-derived mast cells and primary human lung mast cells expressed substantially lower OSM mRNA levels, compared with HMC-1 cells (Figure 3B). In line with these data, double labeling of lung tissue sections for tryptase and OSM by immunofluorescence showed that normal lung mast cells do not express detectable amounts of the OSM protein (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 3.
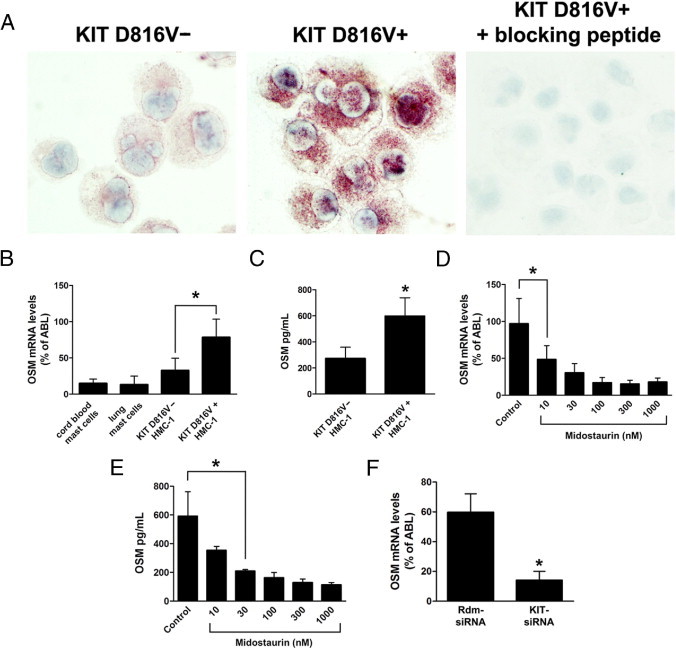
Expression of OSM in the HMC-1 mast cell line. A:KIT D816V− and KIT D816V+ HMC-1 cells were spun onto cytospin slides and expression of OSM was analyzed by immunocytochemistry using an anti-OSM antibody. Preincubation of the anti-OSM antibody with a specific blocking peptide abolished OSM immunoreactivity. B: Cord blood-derived mast cells, primary human lung mast cells, KIT D816V− HMC-1 cells, and KIT D816V+ HMC-1 cells were analyzed for expression of OSM by quantitative real-time PCR. C:KIT D816V− HMC-1 and KIT D816V+ HMC-1 cells were incubated at a density of 4 × 106 cells/mL for 24 hours. Supernatants were then harvested, and the amount of secreted OSM was determined by ELISA. D and E:KIT D816V+ HMC-1 cells (4 × 106 cells/mL) were treated with midostaurin at concentrations from 10 nmol/L to 1000 nmol/L for 24 hours. Cells and supernatants were then harvested, and expression of OSM mRNA (D) and of secreted OSM protein (E) was determined. F:KIT D816V+ HMC-1 cells were lentivirally transduced with a small interfering RNA targeting KIT (KIT-siRNA) or with a random sequence siRNA (Rdm-siRNA). At 24 hours after transduction, cells were selected with puromycin (2 μg/mL) for 48 hours. Expression of OSM was then determined by quantitative real-time PCR. All results represent the mean ± SD of three independent experiments. *P < 0.05.
To determine whether OSM expression in neoplastic mast cells is dependent on KIT D816V kinase activity, KIT D816V+ HMC-1 cells were treated with midostaurin, a multikinase inhibitor reported to block the kinase activity of KIT D816V.32 As expected, inhibition of KIT D816V by midostaurin decreased the expression of OSM mRNA and OSM protein levels in HMC-1 cells in a dose-dependent manner (Figure 3, D and E). Moreover, we found that RNA-interference (RNAi) knockdown of KIT led to a substantial down-regulation of OSM expression in KIT D816V+ HMC-1 cells (Figure 3F). In these experiments, we also noticed that treatment with either midostaurin or RNAi knockdown of KIT led to rapid induction of apoptosis in HMC-1 cells. As determined by a Trypan Blue exclusion test, midostaurin (1 μmol/L) decreased viability of HMC-1 cells from >90% to 60% within 24 hours. RNAi knockdown of KIT reduced viability of HMC-1 cells even to 30% within 4 days after transduction. To exclude the possibility that down-regulation of OSM expression by midostaurin or knockdown of KIT is a nonspecific consequence of decreased viability, we generated HMC-1 cells stably overexpressing BCL-2. As expected, overexpression of BCL-2 effectively counteracted induction of apoptosis by midostaurin or RNAi knockdown of KIT (viability >90% in all experimental settings). Nonetheless, exposure to midostaurin or RNAi knockdown of KIT resulted in decreased expression of OSM in these cells (see Supplemental Figure S2 at http://ajp.amjpathol.org), similar to results obtained in nontransfected HMC-1 cells (Figure 3, D and F). Taken together, these data suggest a regulatory role for KIT D816V in expression of OSM in neoplastic mast cells.
KIT D816V Promotes Expression of OSM in Ba/F3 Cells
To provide further evidence that KIT D816V promotes expression of OSM, Ba/F3 cells with doxycycline-inducible expression of KIT D816V (Ton.Kit.D816V) or of wild-type KIT (Ton.Kit.wt) were used.15 Induction of KIT D816V by addition of doxycycline up-regulated OSM promoter activity as measured by a luciferase reporter assay, and increased OSM mRNA and OSM protein levels in Ba/F3 cells (Figure 4, A–C). The effects of KIT D816V on expression of OSM were more pronounced in the absence of IL-3 (approximately 100-fold up-regulation; Figure 4B) than in the presence of IL-3 (approximately 10-fold up-regulation; Figure 4D), which is best explained by the previously described stimulatory effect of IL-3 on expression of OSM in Ba/F3 cells.43 The KIT D816V-dependent up-regulation of OSM in Ba/F3 cells was inhibited by midostaurin in a dose-dependent manner (Figure 4D) and in the same concentration range as observed with KIT D816V+ HMC-1 cells (Figure 3, D and E). Importantly, wild-type KIT did not affect expression of OSM in Ba/F3 cells, neither in the absence nor in the presence of SCF (Figure 4B). Taken together, these data show that KIT D816V promotes expression of OSM.
Figure 4.
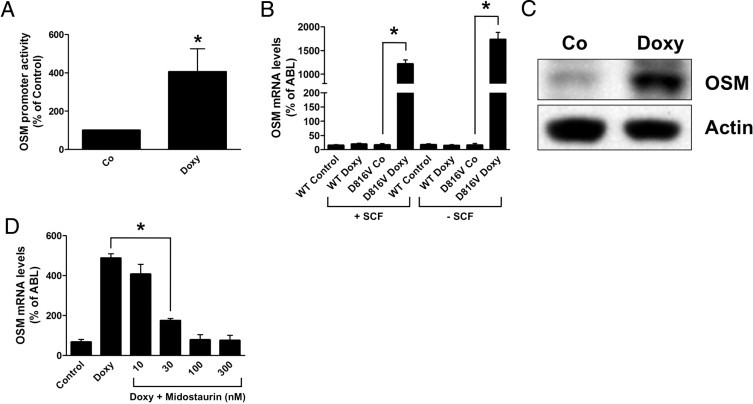
KIT D816V induces expression of OSM in Ba/F3 cells. A: Ton.Kit.D816V cells kept with IL-3 were transfected with plasmids pGV-OSM-luc and pRL-TK and then kept in the absence [control (Co)] or presence (Doxy) of doxycycline for 24 hours. Luciferase activities were then measured. OSM promoter activity was normalized to Renilla luciferase activity and reported as % of control ( = cells kept in the absence of doxycycline). B: Ton.Kit.wt [wild type (WT)] and Ton.Kit.D816V cells (D816V) kept in the absence of IL-3 were cultured without [control (Co)] or with doxycycline for 16 hours. In addition, cells were incubated with or without stem cell factor (SCF). Expression of OSM was then determined by quantitative real-time PCR. C: Ton.Kit.D816V cells kept in the absence of IL-3 were cultured in the absence [control (Co)] or presence of doxycycline. Expression of OSM was then determined by immunoblotting. Actin served as a loading control. D: Ton.Kit.D816V cells kept with IL-3 were cultured in the absence (Control) or presence (Doxy) of doxycycline and were treated with various concentrations of midostaurin as indicated for 16 hours. Expression of OSM was then determined by quantitative real-time PCR. All results represent the mean ± SD of three independent experiments. *P < 0.05.
KIT D816V Induces Expression of OSM through Activation of STAT5
KIT D816V is known to activate several signaling pathways.14,15 To identify the signal transduction pathways involved in KIT D816V-dependent expression of OSM, we first applied pharmacological inhibitors of known downstream effectors of KIT D816V, namely, MEK, PI3-kinase, and STAT5. Inhibition of MEK by PD98059 or PI3-kinase by LY294002 did not affect expression of OSM in KIT D816V+ HMC-1 cells (Figure 5A). In contrast, piceatannol, a compound targeting multiple targets including STAT5,44 inhibited expression of OSM mRNA and OSM protein levels in a dose-dependent manner in KIT D816V+ HMC-1 cells (Figure 5, A and B), as well as in Ba/F3 cells inducibly expressing KIT D816V (Figure 5C). To determine whether STAT5 regulates the expression of OSM, we silenced STAT5 by RNAi (Figure 5D). As assessed by real-time PCR, knockdown of STAT5 significantly decreased expression of OSM in KIT D816V+ HMC-1 cells, as well as in Ba/F3 cells expressing KIT D816V (Figure 5, E and F).
Figure 5.
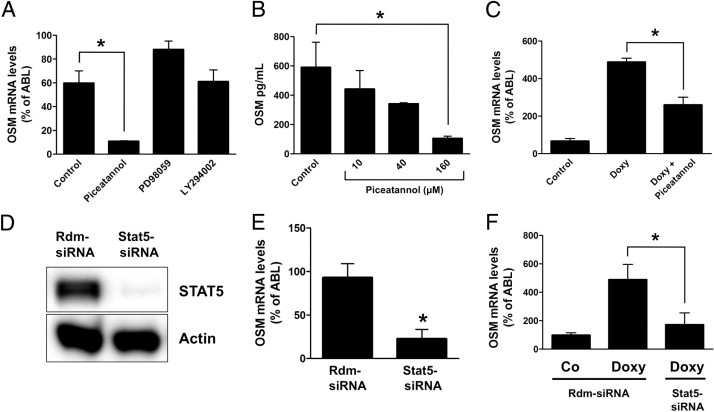
KIT D816V-induced expression of OSM is STAT5-dependent. A:KIT D816V+ HMC-1 cells were incubated with piceatannol (160 μmol/L), PD98059 (50 μmol/L), or LY294002 (20 μmol/L) for 16 hours. Cells were then harvested, and expression of OSM was determined by real-time PCR. Results represent the mean ± SD of three independent experiments. *P < 0.05. B:KIT D816V+ HMC-1 cells (4 × 106 cells/mL) were incubated with piceatannol at concentrations of 10, 40, or 160 μmol/L for 24 hours. Supernatants were then harvested, and the amount of secreted OSM was determined by ELISA. Results represent the mean ± SD of three independent experiments. *P < 0.05. C: Ton.Kit.D816V cells kept with IL-3 were cultured in the absence (Control) or presence (Doxy) of doxycycline (1 μg/mL). Cells cultured with doxycycline were treated with piceatannol (10 μmol/L) for 16 hours. Expression of OSM was then determined by real-time PCR. Results represent the mean ± SD of three independent experiments. *P < 0.05. D:KIT D816V+ HMC-1 cells were lentivirally transduced with a random sequence siRNA (rdm-siRNA) or an siRNA targeting STAT5 (Stat5-siRNA). At 24 hours after infection, transduced cells were selected with puromycin (2 μg/mL) for 48 hours. Cells were then harvested, and expression of STAT5 was determined by immunoblotting. Actin served as a loading control. E:KIT D816V+ HMC-1 cells were transduced with a random sequence siRNA (Rdm-siRNA) or an siRNA targeting STAT5 (Stat5-siRNA) and selected as described above. Expression of OSM was determined by real-time PCR. Results represent the mean ± SD of three independent experiments. *P < 0.05. F: Ton.Kit.D816V cells kept in the presence of IL-3 were transduced with a random sequence siRNA (Rdm-siRNA) or an siRNA targeting STAT5 (Stat5-siRNA) and were selected as described above. At 2 days after infection, cells were split and cultured in the absence [control (Co)] or presence (Doxy) of doxycycline for 16 hours. Expression of OSM was then determined by real-time PCR. Results represent the mean ± SD of three independent experiments. *P < 0.05.
To further analyze the role of STAT5 in expression of OSM, we generated Ba/F3 cells with doxycycline-inducible expression of a constitutively activated STAT5 mutant, STAT5 S710F (Figure 6A). Expression of STAT5 S710F did not substitute for IL-3 as a growth factor, but prolonged survival of Ba/F3 cells in the absence of IL-3 (Figure 6B). As expected, STAT5 S710F led to an increase in expression of OSM in the presence as well as in the absence of IL-3 (Figure 6C). Moreover, overexpression of STAT5 S710F by lentiviral-mediated gene transfer further up-regulated expression of OSM in KIT D816V+ HMC-1 cells (Figure 6D). Taken together, our data show that KIT D816V promotes expression of OSM through activation of STAT5.
Figure 6.
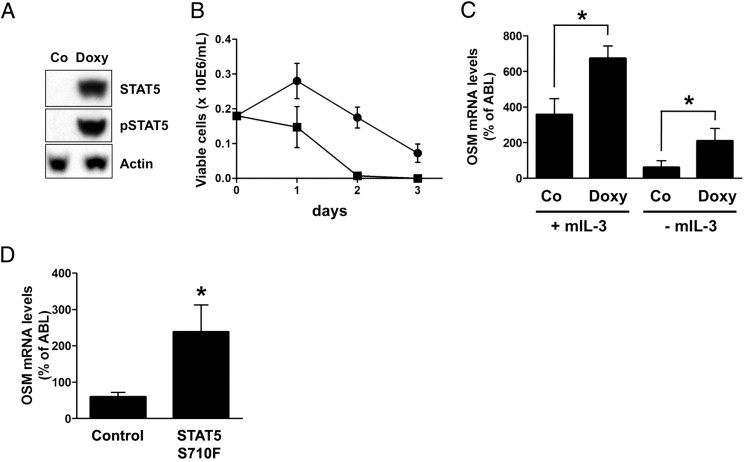
STAT5 S710F promotes expression of OSM. A: Ton.Stat5.S710F cells were incubated in the absence [control (Co)] or presence (Doxy) of doxycycline (1 μg/mL) for 24 hours. Cells were then harvested, and expression of total STAT5 and phosphorylated STAT5 (pSTAT5) was determined. Actin served as a loading control. B: Ton.Stat5.S710F cells kept without IL-3 were cultured in the absence (squares) or presence (circles) of doxycycline (without IL-3), and the number of viable cells was determined every 24 hours. C: Ton.Stat5.S710F cells were cultured in the presence or absence of doxycycline with or without IL-3 for 24 hours. Expression of OSM was then determined by real-time PCR. Results represent the mean ± SD of three independent experiments. D:KIT D816V+ HMC-1 cells were lentivirally transduced with STAT5 S710F. At 24 hours after transduction, cells were selected with puromycin (2 μg/mL) for 48 hours. Expression of OSM was then determined by quantitative real-time PCR. Results represent the mean ± SD of three independent experiments. *P < 0.05.
Effects of Wild-Type KIT on Activation of STAT5 and OSM Expression
Of note, wild-type KIT (in the presence of SCF) did not activate STAT515 or induce expression of OSM in Ba/F3 cells (Figure 4). To investigate the effects of wild-type KIT and SCF on expression of OSM in greater detail, CD34+ cord blood cells were analyzed. CD34+ progenitor cells cultured in the presence of SCF for 24 hours expressed substantial amounts of OSM (Figure 7A); however, differentiation of these cells into normal mast cells was associated with down-regulation of OSM expression (Figure 7A). As a control, we determined expression of the mast cell marker histidine decarboxylase. As expected, SCF-induced mast cell differentiation led to up-regulation of histidine decarboxylase (Figure 7B). In a next step, expression of phosphorylated STAT5 and OSM was investigated in primary human lung mast cells. As assessed by immunocytochemistry, lung mast cells were found to stain negative for phosphorylated STAT5 and expressed only low levels of the OSM protein, compared with KIT D816V+ HMC-1 cells (Figure 7, C and D). Moreover, lung mast cells expressed significantly lower mRNA levels of other STAT5-target genes such as PIM1 and BCL2L1 (alias BCL-XL), compared with KIT D816V+ HMC-1 cells (Figure 7, E and F), whereas no significant difference was found when comparing histidine decarboxylase mRNA levels (not shown). These data suggest that wild-type KIT (in the presence of SCF) can promote expression of STAT5-target genes in CD34+ progenitor cells, but not in a more mature mast cell context such as cord blood-derived mast cells or primary lung mast cells.
Figure 7.
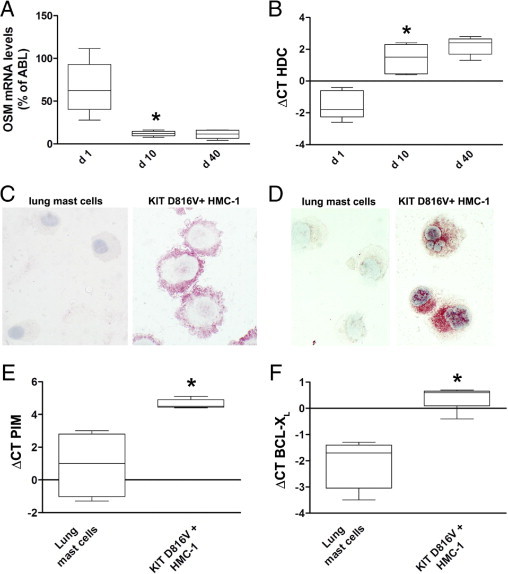
Effects of wild-type KIT on activation of STAT5 and expression of OSM. (A, B) CD34+ progenitor cells isolated from cord blood (n = 5) were cultured in the presence of SCF and IL-6 (both 100 ng/mL). Box plots show expression of OSM (A) or histidine decarboxylase (HDC; B) mRNA levels as determined by real-time PCR on day 1, 10, and 40. *P < 0.05. C: Primary human lung mast cells cultured in the presence of SCF were spun onto cytospin slides and expression of phosphorylated STAT5 was analyzed by immunocytochemistry using the anti-pSTAT5a/b antibody AX1. KIT D816V+ HMC-1 cells were included as a positive control. D: Primary human lung mast cells and KIT D816V+ HMC-1 cells were analyzed for expression of the OSM protein by immunocytochemistry as described above. E and F: Primary lung mast cells obtained from four different patients and KIT D816V+ HMC-1 cells (five independent experiments) were analyzed for expression of PIM1 (E) or BCL-XL (F) mRNA levels by real-time PCR. Results are shown as box plots. *P < 0.05.
OSM Derived from Neoplastic Mast Cells Promotes Growth of Microvascular Endothelial Cells, Osteoblasts, and Fibroblasts
OSM has been reported to promote proliferation of mesenchymal cells25,26 and thus may be important for the bone marrow alterations associated with systemic mastocytosis. Therefore, we next wished to determine whether OSM derived from KIT D816V+ neoplastic cells can stimulate growth of mesenchymal cells. For this purpose, human microvascular endothelial cells (HMEC-1 cells), human immortalized osteoblasts (hFOB 1.19 cells), and primary human bone marrow fibroblasts were incubated with cell culture supernatants from HMC-1 cells. As assessed by [3H]thymidine incorporation assay, supernatants from KIT D816V+ HMC-1 cells were found to stimulate growth of these cells, whereas supernatants from KIT D816V− HMC-1 cells had no effect on cell growth (Figure 8, A–C). This growth-stimulatory effect was inhibited by preincubation of supernatants with a neutralizing anti-OSM antibody (Figure 8, A–C). Preincubation of supernatants with a control antibody had no effect (data not shown). In a final step, we were able to show that HMEC-1 cells, hFOB 1.19 cells, and primary bone marrow fibroblasts all expressed transcripts for the OSM receptor (Table 3). By contrast, neoplastic or normal mast cells did not express mRNA for OSM receptors, excluding an autocrine growth regulation of neoplastic mast cells involving OSM. Taken together, our data suggest that neoplastic mast cells produce functionally active OSM, which may play a role as a mediator of growth of various stromal cells.
Figure 8.
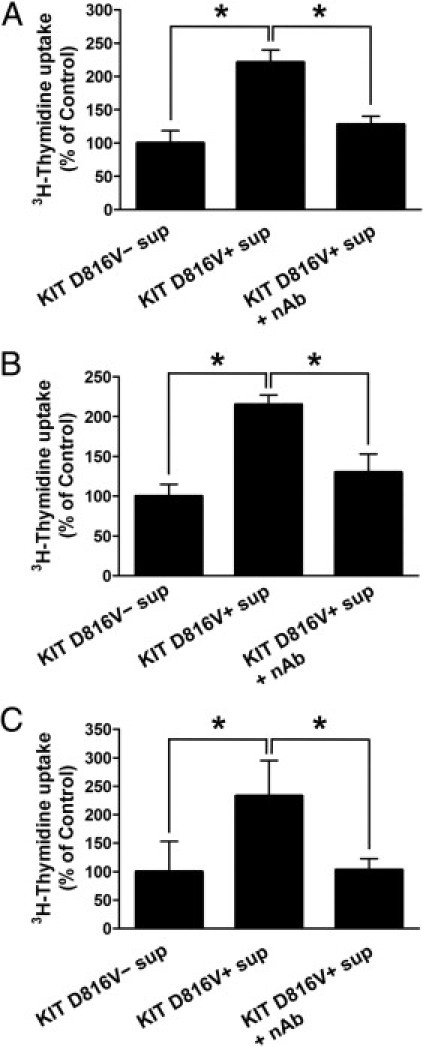
OSM derived from KIT D816V+ cells stimulates proliferation of mesenchymal cells. A–C: Human microvascular endothelial cells (HMEC-1 cells; A), human immortalized osteoblasts (B) and primary human bone marrow fibroblasts (C) were incubated with supernatant (sup) from KIT D816V− HMC-1 or KIT D816V+ HMC-1 cells. In addition, cells were treated with supernatant from KIT D816V+ HMC-1 cells preincubated with a neutralizing antibody against OSM (nAb) at a concentration of 10 μg/mL. Cells were incubated with supernatants for 72 hours. Proliferation of cells was then determined by [3H]thymidine incorporation assay. Results represent the mean ± SD of triplicates and represent one of three almost identical experiments. *P < 0.05.
Table 3.
Expression of Oncostatin M Receptors as Determined by Real-Time PCR
| Cell line | OSM receptor | LIF receptor |
|---|---|---|
| HMC-1 cells | − | − |
| CB-derived mast cells | − | − |
| CB-derived CD34+ progenitor cells | − | − |
| HMEC-1 cells | + | − |
| hFOB 1.19 cells | + | − |
| Primary bone marrow fibroblasts | + | − |
CB, cord blood; LIF, leukemia inhibitory factor; OSM, oncostatin M; −, negative; +, positive.
Discussion
Numerous studies have implicated KIT D816V in the pathogenesis of systemic mastocytosis; however, the mechanisms underlying the biological effects of the mutated KIT receptor are not well understood, and little is known about KIT D816V-specific gene expression leading to the typical changes in the bone marrow microenvironment found in patients with systemic mastocytosis. The present results show that KIT D816V promotes expression of OSM through activation of STAT5. Neoplastic mast cells in patients with mastocytosis showed constitutive expression of OSM and mast cell-derived OSM promoted growth of various mesenchymal cells. Thus, our data suggest that OSM could be an important KIT D816V-dependent modulator of the bone marrow microenvironment involved in the pathogenesis of systemic mastocytosis.
OSM is a well-established regulator of mesenchymal cells and has been implicated in the pathogenesis of various inflammatory and neoplastic disorders.20 In the present study, we found that neoplastic mast cells in patients with systemic mastocytosis show constitutive expression of OSM. Recently, it has been reported that cord blood-derived mast cells and primary human lung mast cells, as well as the human LAD 2 mast cell line, are negative for OSM but show substantial up-regulation of OSM expression when incubated with activated T-cells.45 This is in good agreement with the present data showing that unstimulated cord blood-derived mast cells and normal lung mast cells express only baseline levels of OSM, compared with KIT D816V+ neoplastic mast cells. Finally, the observation that wild-type KIT (in the presence of SCF) does not promote expression of OSM in Ba/F3 cells also supports the notion that normal human mast cells do not express OSM at a high level.
So far, little is known about expression of OSM in the various cell types of the human bone marrow. We found that megakaryocytes, endothelial cells, and myeloid progenitors stain positive for OSM, whereas erythroid cells and mature granulocytes were OSM-negative. Remarkably, this expression pattern is identical to the cellular distribution described for phosphorylated STAT5.18,19 The finding that megakaryocytes express substantial amounts of OSM is noteworthy, given that OSM has been reported to act as a maturation factor for these cells.46 In addition, mice deficient for the OSM receptor have been reported to have marked thrombocytopenia.30 It has also been reported that OSM is expressed in murine erythroblasts and supports survival of erythroid progenitors in mice under anemic conditions.47 In the present study, however, we did not detect expression of OSM in erythroid progenitors in human bone marrow. This finding was somewhat unexpected, but fits with the observation that human erythroid progenitors also did not express phosphorylated STAT5. The most likely explanation for this discrepancy is that phosphorylated STAT5 and OSM are not expressed in steady-state hematopoiesis but only under anemic conditions, as has been reported for phenylhydrazine-induced anemia in mice.47
The majority of patients with systemic mastocytosis harbor the KIT mutation D816V. In this study, we found that a KIT D816V− clone of the HMC-1 cell line displayed significantly lower OSM expression levels than KIT D816V+ HMC-1 cells. In addition, midostaurin, an inhibitor of KIT D816V inhibited expression of OSM in a dose-dependent manner. Finally, we showed that doxycycline-inducible expression of KIT D816V directly promoted expression of OSM in Ba/F3 cells. Taken together, these in vitro data suggest that KIT D816V plays an important role in expression of OSM in neoplastic mast cells. Notably, when analyzing expression of OSM in neoplastic mast cells in patients with systemic mastocytosis, we did not find major differences between KIT D816V+ and KIT D816V− patients. This finding was not completely unexpected, however, because KIT D816V− patients express equal levels of phosphorylated STAT5, compared with KIT D816V+ patients.18,19 Therefore, we conclude that activation of STAT5 is a general phenomenon in neoplastic mast cells and that KIT D816V− systemic mastocytosis patients probably carry other STAT5-activating hits.
STAT5 plays a decisive role in mast cell biology.48 STAT5 is critical for normal mast cell development and survival, and also for IgE-mediated mast cell function.17,49 More recently, we and others have shown that neoplastic mast cells display constitutive expression of phosphorylated STAT5.18,19 Moreover, we have previously shown that growth of neoplastic mast cells is inhibited by expression of a dominant negative-acting STAT5 mutant.19 Nonetheless, little is known about effector molecules in neoplastic mast cells downstream of STAT5 and their role in the pathogenesis of systemic mastocytosis. The present findings show that KIT D816V-dependent expression of OSM is mediated by STAT5. RNAi-induced knockdown of STAT5 inhibited expression of OSM, whereas expression of a constitutively activated STAT5 mutant (STAT5 S710F) promoted OSM expression. Interestingly, Salamon et al45 showed that up-regulation of OSM in T-cell-stimulated mast cells is dependent on the MAP kinase pathway. This observation is in contrast to our data, because inhibition of MEK did not affect expression of OSM in neoplastic mast cells. This could indicate that different mechanisms underlie T-cell-dependent and KIT D816V-dependent expression of OSM.
Another noteworthy aspect of the present findings was the observation that wild-type KIT (in the presence of SCF) did not up-regulate expression of OSM in Ba/F3 cells. This result is explained by the fact that wild-type KIT cannot activate STAT5 in the same way as KIT D816V in these cells.15 At first glance, it seems that these data are in contrast to the well-established role of STAT5 in mast cell development17,48 and to a recent report showing that SCF leads to phosphorylation of STAT5 in human CD34+ progenitor cells isolated from cord blood.50 This discrepancy is most likely explained by differences in the cellular context. Han et al51 recently showed that KIT-dependent STAT5 activation is highly variable in different cell types and can be triggered by SCF only in distinct subsets of progenitor cells. These findings match well to our observation that CD34+ cord blood cells down-regulate expression of OSM when induced to differentiate into mast cells and are also in good agreement with our data showing that mature lung mast cells do not express phosphorylated STAT5. Finally, the study of Salomon et al45 also supports the notion that mature mast cells do not express high levels of STAT5-target genes such as OSM. All in all, our data suggest that KIT D816V leads to aberrant activation of STAT5 in neoplastic mast cells.
The pathology of systemic mastocytosis is characterized by an accumulation of neoplastic mast cells. Apart from mast cell infiltrates, changes in the bone marrow microenvironment are frequently noticed. These alterations include neoangiogenesis, thickened bone trabeculae, and sometimes massive bone marrow fibrosis.3–5 In the present study, we show that HMC-1 cell-derived OSM stimulates in vitro growth of microvascular endothelial cells, osteoblasts, and fibroblasts. This finding indicates that neoplastic mast cells secrete sufficient amounts of active OSM to stimulate proliferation of mesenchymal cells. Therefore, it is likely that mast cell-derived OSM contributes to the bone marrow pathologies observed in systemic mastocytosis. The bone marrow microenvironment is supposed to play a key role in the pathogenesis of systemic mastocytosis as it supports growth and differentiation of neoplastic mast cells by generation of cytokines, as well as by direct interactions (cell-cell adhesion). Recently, it has been reported that OSM promotes growth of murine bone marrow mast cells through a fibroblast-dependent pathway.52 Thus, it is tempting to speculate that mast cell-derived OSM stimulates growth of neoplastic cells in a paracrine way. An autocrine growth stimulation by OSM is unlikely, because mature (neoplastic) mast cells did not express OSM receptors. Nonetheless, it cannot be excluded that OSM directly affects myelomastocytic progenitors, as has been suggested for erythrocytic and megakaryocytic progenitors.30
Together, our data show that neoplastic mast cells express functionally active OSM in a KIT D816V-dependent and STAT5-dependent manner. Thus, OSM could represent a novel key molecule and potential target in the pathogenesis of systemic mastocytosis.
Acknowledgments
We thank Sabine Rauscher (Core Facility Imaging, Medical University of Vienna, Vienna, Austria) for help with confocal microscopy, Hans-Peter Horny, M.D. (Institute of Pathology Ansbach, Ansbach, Germany), for reviewing IHC stainings and data, and Helen Pickersgill, Ph.D. (independent scientific editor), for critical reading of the manuscript.
Footnotes
Supported by the Medical Scientific Fund of the Mayor of the City of Vienna, by a grant from the Austrian Society of Laboratory Medicine and Clinical Chemistry, by the Hans and Blanca Moser Foundation, and by the Funds for Scientific Research in Austria [Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich (FWF)] grant no. P21173-B13.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.020.
Supplementary data
Expression of OSM in normal (non-neoplastic) lung mast cells. Lung sections were double stained for tryptase (red) and OSM (green) by immunofluorescence. The merged image shows that tryptase-positive lung mast cells do not express detectable amounts of OSM. DAPI (blue) identifies all nucleated cells. Two representative mast cells are shown (#1 and #2).
Expression of OSM in the HMC-1 mast cell line. A:KIT D816V+ HMC-1 cells stably overexpressing BCL-2 were treated with midostaurin at concentrations from 10 nmol/L to 1000 nmol/L for 24 hours. Cells were then harvested, and expression of OSM mRNA was determined by quantitative RT-PCR. B:KIT D816V+ HMC-1 stably overexpressing BCL-2 cells were lentivirally transduced with an siRNA targeting KIT (KIT-siRNA), with an siRNA targeting STAT5 (STAT5-siRNA), or with a random sequence siRNA (Rdm-siRNA). At 24 hours after transduction, cells were selected with puromycin (2 μg/mL) for 48 hours. Expression of OSM was then determined by quantitative RT-PCR. All results represent the mean ± SD of three independent experiments. ⁎P < 0.05. Viability was >90% in all experimental settings, as determined by a Trypan Blue exclusion test.
References
- 1.Akin C., Metcalfe D.D. Systemic mastocytosis. Annu Rev Med. 2004;55:419–432. doi: 10.1146/annurev.med.55.091902.103822. [DOI] [PubMed] [Google Scholar]
- 2.Valent P., Horny H.P., Li C.Y., Longley B.J., Metcalfe D.D., Parwaresch M.R., Bennett J.M. Mastocytosis (mast cell disease): Pathology and genetics of tumours of haematopoietic and lymphoid tissues. In: Jaffe E.S., Harris N.L., Stein H., Vardiman J.W., editors. World Health Organization Classification of Tumours. IARC Press; Lyon: 2001. pp. 291–302. [Google Scholar]
- 3.Horny H.P., Valent P. Diagnosis of mastocytosis: general histopathological aspects, morphological criteria, and immunohistochemical findings. Leuk Res. 2001;25:543–551. doi: 10.1016/s0145-2126(01)00021-2. [DOI] [PubMed] [Google Scholar]
- 4.Li C.Y., Baek J.Y. Mastocytosis and fibrosis: role of cytokines. Int Arch Allergy Immunol. 2002;127:123–126. doi: 10.1159/000048182. [DOI] [PubMed] [Google Scholar]
- 5.Wimazal F., Jordan J.H., Sperr W.R., Chott A., Dabbass S., Lechner K., Horny H.P., Valent P. Increased angiogenesis in the bone marrow of patients with systemic mastocytosis. Am J Pathol. 2002;160:1639–1645. doi: 10.1016/S0002-9440(10)61111-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furitsu T., Saito H., Dvorak A.M., Schwartz L.B., Irani A.M., Burdick J.F., Ishizaka K., Ishizaka T. Development of human mast cells in vitro. Proc Natl Acad Sci USA. 1989;86:10039–10043. doi: 10.1073/pnas.86.24.10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irani A.A., Craig S.S., Nilsson G., Ishizaka T., Schwartz L.B. Characterization of human mast cells developed in vitro from fetal liver cells cocultured with murine 3T3 fibroblasts. Immunology. 1992;77:136–143. [PMC free article] [PubMed] [Google Scholar]
- 8.Levi-Schaffer F., Kelav-Appelbaum R., Rubinchik E. Human foreskin mast cell viability and functional activity is maintained ex vivo by coculture with fibroblasts. Cell Immunol. 1995;162:211–216. doi: 10.1006/cimm.1995.1071. [DOI] [PubMed] [Google Scholar]
- 9.Sellge G., Lorentz A., Gebhardt T., Levi-Schaffer F., Bektas H., Manns M.P., Schuppan D., Bischoff S.C. Human intestinal fibroblasts prevent apoptosis in human intestinal mast cells by a mechanism independent of stem cell factor, IL-3, IL-4, and nerve growth factor. J Immunol. 2004;172:260–267. doi: 10.4049/jimmunol.172.1.260. [DOI] [PubMed] [Google Scholar]
- 10.Nagata H., Worobec A.S., Oh C.K., Chowdhury B.A., Tannenbaum S., Suzuki Y., Metcalfe D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longley B.J., Jr, Metcalfe D.D., Tharp M., Wang X., Tyrrell L., Lu S.Z., Heitjan D., Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feger F., Ribadeau Dumas A., Leriche L., Valent P., Arock M. Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int Arch Allergy Immunol. 2002;127:110–114. doi: 10.1159/000048179. [DOI] [PubMed] [Google Scholar]
- 13.Zappulla J.P., Dubreuil P., Desbois S., Létard S., Hamouda N.B., Daëron M., Delsol G., Arock M., Liblau R.S. Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation. J Exp Med. 2005;202:1635–1641. doi: 10.1084/jem.20050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chian R., Young S., Danilkovitch-Miagkova A., Ronnstrand L., Leonard E., Ferrao P., Ashman L., Linnekin D. Phosphatidylinositol 3 kinase contributes to the transformation of hematopoietic cells by the D816V c-Kit mutant. Blood. 2001;98:1365–1373. doi: 10.1182/blood.v98.5.1365. [DOI] [PubMed] [Google Scholar]
- 15.Mayerhofer M., Gleixner K.V., Hoelbl A., Florian S., Hoermann G., Aichberger K.J., Bilban M., Esterbauer H., Krauth M.T., Sperr W.R., Longley J.B., Kralovics R., Moriggl R., Zappulla J., Liblau R.S., Schwarzinger I., Sexl V., Sillaber C., Valent P. Unique effects of KIT D816V in BaF3 cells: induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J Immunol. 2008;180:5466–5476. doi: 10.4049/jimmunol.180.8.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piao X., Paulson R., van der Geer P., Pawson T., Bernstein A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc Natl Acad Sci USA. 1996;93:14665–14669. doi: 10.1073/pnas.93.25.14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shelburne C.P., McCoy M.E., Piekorz R., Sexl V., Roh K.H., Jacobs-Helber S.M., Gillespie S.R., Bailey D.P., Mirmonsef P., Mann M.N., Kashyap M., Wright H.V., Chong H.J., Bouton L.A., Barnstein B., Ramirez C.D., Bunting K.D., Sawyer S., Lantz C.S., Ryan J.J. Stat5 expression is critical for mast cell development and survival. Blood. 2003;102:1290–1297. doi: 10.1182/blood-2002-11-3490. [DOI] [PubMed] [Google Scholar]
- 18.Zuluaga Toro T., Hsieh F.H., Bodo J., Dong H.Y., Hsi E.D. Detection of phospho-STAT5 in mast cells: a reliable phenotypic marker of systemic mast cell disease that reflects constitutive tyrosine kinase activation. Br J Haematol. 2007;139:31–40. doi: 10.1111/j.1365-2141.2007.06708.x. [DOI] [PubMed] [Google Scholar]
- 19.Baumgartner C., Cerny-Reiterer S., Sonneck K., Mayerhofer M., Gleixner K.V., Fritz R., Kerenyi M., Boudot C., Gouilleux F., Kornfeld J.W., Sillaber C., Moriggl R., Valent P. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: subcellular distribution and role of the transforming oncoprotein KIT D816V. Am J Pathol. 2009;175:2416–2419. doi: 10.2353/ajpath.2009.080953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez-Lechón M.J. Oncostatin M: signal transduction and biological activity. Life Sci. 1999;65:2019–2030. doi: 10.1016/s0024-3205(99)00296-9. [DOI] [PubMed] [Google Scholar]
- 21.Gearing D.P., Comeau M.R., Friend D.J., Gimpel S.D., Thut C.J., McGourty J., Brasher K.K., King J.A., Gillis S., Mosley B. The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science. 1992;255:1434–1437. doi: 10.1126/science.1542794. [DOI] [PubMed] [Google Scholar]
- 22.Gearing D.P., Bruce A.G. Oncostatin M binds the high-affinity leukemia inhibitory factor receptor. New Biol. 1992;4:61–65. [PubMed] [Google Scholar]
- 23.Mosley B., De Imus C., Friend D., Boiani N., Thoma B., Park L.S., Cosman D. Dual oncostatin M (OSM) receptors: cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J Biol Chem. 1996;271:32635–32643. doi: 10.1074/jbc.271.51.32635. [DOI] [PubMed] [Google Scholar]
- 24.Zarling J.M., Shoyab M., Marquardt H., Hanson M.B., Lioubin M.N., Todaro G.J. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci USA. 1986;83:9739–9743. doi: 10.1073/pnas.83.24.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasse M., Pourtau J., Trochon V., Muraine M., Vannier J.P., Lu H., Soria J., Soria C. Oncostatin M induces angiogenesis in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1999;19:1835–1842. doi: 10.1161/01.atv.19.8.1835. [DOI] [PubMed] [Google Scholar]
- 26.Scaffidi A.K., Mutsaers S.E., Moodley Y.P., McAnulty R.J., Laurent G.J., Thompson P.J., Knight D.A. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br J Pharmacol. 2002;136:793–801. doi: 10.1038/sj.bjp.0704769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jilka R.L., Weinstein R.S., Bellido T., Parfitt A.M., Manolagas S.C. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- 28.Heymann D., Rousselle A.V. gp130 Cytokine family and bone cells. Cytokine. 2000;12:1455–1468. doi: 10.1006/cyto.2000.0747. [DOI] [PubMed] [Google Scholar]
- 29.Minehata K., Takeuchi M., Hirabayashi Y., Inoue T., Donovan P.J., Tanaka M., Miyajima A. Oncostatin M maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int J Hematol. 2006;84:319–327. doi: 10.1532/IJH97.06090. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka M., Hirabayashi Y., Sekiguchi T., Inoue T., Katsuki M., Miyajima A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood. 2003;102:3154–3162. doi: 10.1182/blood-2003-02-0367. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka M., Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52. doi: 10.1007/s10254-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 32.Fabbro D., Ruetz S., Bodis S., Pruschy M., Csermak K., Man A., Campochiaro P., Wood J., O'Reilly T., Meyer T. PKC412—a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15:17–28. [PubMed] [Google Scholar]
- 33.Butterfield J.H., Weiler D., Dewald G., Gleich G.J. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345–355. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- 34.Akin C., Brockow K., D'Ambrosio C., Kirshenbaum A.S., Ma Y., Longley B.J., Metcalfe D.D. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol. 2003;31:686–692. doi: 10.1016/s0301-472x(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 35.Foster C.A., Dreyfuss M., Mandak B., Meingassner J.G., Naegeli H.U., Nussbaumer A., Oberer L., Scheel G., Swoboda E.M. Pharmacological modulation of endothelial cell-associated adhesion molecule expression: implications for future treatment of dermatological diseases. J Dermatol. 1994;21:847–854. doi: 10.1111/j.1346-8138.1994.tb03300.x. [DOI] [PubMed] [Google Scholar]
- 36.Ades E.W., Candal F.J., Swerlick R.A., George V.G., Summers S., Bosse D.C., Lawley T.J. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 37.Valent P., Ashman L.K., Hinterberger W., Eckersberger F., Majdic O., Lechner K., Bettelheim P. Mast cell typing: demonstration of a distinct hematopoietic cell type and evidence for immunophenotypic relationship to mononuclear phagocytes. Blood. 1989;73:1778–1785. [PubMed] [Google Scholar]
- 38.Daley G.Q., Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci USA. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grebien F., Kerenyi M.A., Kovacic B., Kolbe T., Becker V., Dolznig H., Pfeffer K., Klingmuller U., Muller M., Beug H., Mullner E.W., Moriggl R. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008;111:4511–4522. doi: 10.1182/blood-2007-07-102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheeren F.A., Naspetti M., Diehl S., Schotte R., Nagasawa M., Wijnands E., Gimeno R., Vyth-Dreese F.A., Blom B., Spits H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat Immunol. 2005;6:303–313. doi: 10.1038/ni1172. [DOI] [PubMed] [Google Scholar]
- 41.Mayerhofer M., Gleixner K.V., Mayerhofer J., Hoermann G., Jaeger E., Aichberger K.J., Ott R.G., Greish K., Nakamura H., Derdak S., Samorapoompichit P., Pickl W.F., Sexl V., Esterbauer H., Schwarzinger I., Sillaber C., Maeda H., Valent P. Targeting of heat shock protein 32 (Hsp32)/heme oxygenase-1 (HO-1) in leukemic cells in chronic myeloid leukemia: a novel approach to overcome resistance against imatinib. Blood. 2008;111:2200–2210. doi: 10.1182/blood-2006-11-055723. [DOI] [PubMed] [Google Scholar]
- 42.Yoshimura A., Ichihara M., Kinjyo I., Moriyama M., Copeland N.G., Gilbert D.J., Jenkins N.A., Hara T., Miyajima A. Mouse oncostatin M: an immediate early gene induced by multiple cytokines through the JAK-STAT5 pathway. EMBO J. 1996;15:1055–1063. [PMC free article] [PubMed] [Google Scholar]
- 43.Jaster R., Tschirch E., Bittorf T., Brock J. Role of STAT5 in interferon-alpha signal transduction in Ba/F3 cells. Cell Signal. 1999;11:331–335. doi: 10.1016/s0898-6568(98)00049-7. [DOI] [PubMed] [Google Scholar]
- 44.Su L., David M. Distinct mechanisms of STAT phosphorylation via the interferon-alpha/beta receptor: selective inhibition of STAT3 and STAT5 by piceatannol. J Biol Chem. 2000;275:12661–12666. doi: 10.1074/jbc.275.17.12661. [DOI] [PubMed] [Google Scholar]
- 45.Salamon P., Shoham N.G., Puxeddu I., Paitan Y., Levi-Schaffer F., Mekori Y.A. Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J Allergy Clin Immunol. 2008;121 doi: 10.1016/j.jaci.2007.08.054. 448–455.e5. [DOI] [PubMed] [Google Scholar]
- 46.Wallace P.M., MacMaster J.F., Rillema J.R., Peng J., Burstein S.A., Shoyab M. Thrombocytopoietic properties of oncostatin M. Blood. 1995;86:1310–1315. [PubMed] [Google Scholar]
- 47.Menon M.P., Karur V., Bogacheva O., Bogachev O., Cuetara B., Wojchowski D.M. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006;116:683–694. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shelburne C.P., McCoy M.E., Piekorz R., Sexl V.V., Gillespie S.R., Bailey D.P., Gharse A., Mirmonsef P., Mann M.N., Kashyap M., Wright H.V., Chong H.J., Bouton L.A., Ramirez C.D., Lantz C.S., Ryan J.J. Stat5: an essential regulator of mast cell biology. Mol Immunol. 2002;38:1187–1191. doi: 10.1016/s0161-5890(02)00061-5. [DOI] [PubMed] [Google Scholar]
- 49.Barnstein B.O., Li G., Wang Z., Kennedy S., Chalfant C., Nakajima H., Bunting K.D., Ryan J.J. Stat5 expression is required for IgE-mediated mast cell function. J Immunol. 2006;177:3421–3426. doi: 10.4049/jimmunol.177.5.3421. [DOI] [PubMed] [Google Scholar]
- 50.Harir N., Boudot C., Friedbichler K., Sonneck K., Kondo R., Martin-Lannerée S., Kenner L., Kerenyi M., Yahiaoui S., Gouilleux-Gruart V., Gondry J., Bénit L., Dusanter-Fourt I., Lassoued K., Valent P., Moriggl R., Gouilleux F. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood. 2008;112:2463–2473. doi: 10.1182/blood-2007-09-115477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han L., Wierenga A.T., Rozenveld-Geugien M., van de Lande K., Vellenga E., Schuringa J.J. Single-cell STAT5 signal transduction profiling in normal and leukemic stem and progenitor cell populations reveals highly distinct cytokine responses. PLoS One. 2009;4:e7989. doi: 10.1371/journal.pone.0007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gyotoku E., Morita E., Kameyoshi Y., Hiragun T., Yamamoto S., Hide M. The IL-6 family cytokines, interleukin-6, interleukin-11, oncostatin M, and leukemia inhibitory factor, enhance mast cell growth through fibroblast-dependent pathway in mice. Arch Dermatol Res. 2001;293:508–514. doi: 10.1007/pl00007465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of OSM in normal (non-neoplastic) lung mast cells. Lung sections were double stained for tryptase (red) and OSM (green) by immunofluorescence. The merged image shows that tryptase-positive lung mast cells do not express detectable amounts of OSM. DAPI (blue) identifies all nucleated cells. Two representative mast cells are shown (#1 and #2).
Expression of OSM in the HMC-1 mast cell line. A:KIT D816V+ HMC-1 cells stably overexpressing BCL-2 were treated with midostaurin at concentrations from 10 nmol/L to 1000 nmol/L for 24 hours. Cells were then harvested, and expression of OSM mRNA was determined by quantitative RT-PCR. B:KIT D816V+ HMC-1 stably overexpressing BCL-2 cells were lentivirally transduced with an siRNA targeting KIT (KIT-siRNA), with an siRNA targeting STAT5 (STAT5-siRNA), or with a random sequence siRNA (Rdm-siRNA). At 24 hours after transduction, cells were selected with puromycin (2 μg/mL) for 48 hours. Expression of OSM was then determined by quantitative RT-PCR. All results represent the mean ± SD of three independent experiments. ⁎P < 0.05. Viability was >90% in all experimental settings, as determined by a Trypan Blue exclusion test.