

Abstract
Dynamin 2 (DNM2) is a large GTPase implicated in many cellular functions, including cytoskeleton regulation and endocytosis. Although ubiquitously expressed, DNM2 was found mutated in two genetic disorders affecting different tissues: autosomal dominant centronuclear myopathy (ADCNM; skeletal muscle) and peripheral Charcot-Marie-Tooth neuropathy (peripheral nerve). To gain insight into the function of DNM2 in skeletal muscle and the pathological mechanisms leading to ADCNM, we introduced wild-type DNM2 (WT-DNM2) or R465W DNM2 (RW-DNM2), the most common ADCNM mutation, into adult wild-type mouse skeletal muscle by intramuscular adeno-associated virus injections. We detected altered localization of RW-DNM2 in mouse muscle. Several ADCNM features were present in RW-DNM2 mice: fiber atrophy, nuclear mislocalization, and altered mitochondrial staining, with a corresponding reduction in specific maximal muscle force. The sarcomere and triad structures were also altered. We report similar findings in muscle biopsy specimens from an ADCNM patient with the R465W mutation. In addition, expression of wild-type DNM2 induced some muscle defects, albeit to a lesser extent than RW-DNM2, suggesting that the R465W mutation has enhanced activity in vivo. In conclusion, we show the RW-DNM2 mutation acts in a dominant manner to cause ADCNM in adult muscle, and the disease arises from a primary defect in skeletal muscle rather than secondary to peripheral nerve involvement. Therefore, DNM2 plays important roles in the maintenance of adult muscle fibers.
Dynamins are large, multifunctional GTPase proteins that were initially identified as microtubule-binding proteins1 and more recently were recognized to play an important role in actin cytoskeleton assembly2 and membrane trafficking and endocytosis.3,4 Dynamins can form polymerized rings around membrane tubules,5 and the mechanism by which dynamins regulate membrane fission has been intensively studied.6,7
Dynamins contain an N-terminal GTPase domain, middle domain, Pleckstrin homology (PH) domain (phosphoinositide binding), GTPase effector domain (GED), and a Proline-rich domain (PRD) for protein-protein interactions. Dynamin 1 is expressed specifically in neurons, dynamin 3 is expressed mainly in the brain and testis, and dynamin 2 (DNM2) is ubiquitously expressed. Despite its ubiquitous expression, mutations in DNM2 induce tissue-specific diseases; missense mutations in the middle and C-terminal part of the PH domain lead to autosomal dominant centronuclear myopathy (ADCNM; OMIM 160150), which affects skeletal muscle,8 whereas mutations in the N-terminal part of the PH domain are linked to dominant Charcot-Marie-Tooth (OMIM 606482) peripheral neuropathy.9 Although studies on DNM2 have been performed mainly in cultured cells, the function of DNM2 in skeletal muscle has barely been studied. Moreover, mechanisms of the tissue-specific impact of DNM2 mutations leading to myopathy are not characterized, and it is not clear whether DNM2 mutations have an impact on muscle development or adult muscle maintenance.
Centronuclear myopathies (CNMs) are congenital myopathies characterized by muscle weakness associated with fiber atrophy, predominance of type I fibers, and increased centralization of nuclei not secondary to muscle regeneration.10 DNM2-related CNM commonly presents as an adult-onset, mild form of CNM8; however, cases of severe neonatal onset have been identified.11 In addition to the autosomal dominant form, the X-linked form or myotubular myopathy (OMIM 310400) is due to mutations in the phosphoinositides phosphatase myotubularin,12 and several autosomal recessive CNM patients (OMIM 160150) are linked to mutations in the membrane remodeling protein amphiphysin 2 (BIN1).13 Rare sporadic cases of CNM have also been identified with variants in the ryanodine receptor (RyR1)14,15 or in the myotubularin-related protein hJUMPY/MTMR14.16
In this study, we expressed DNM2 containing the most common ADCNM mutation, R465W, in adult wild-type skeletal muscle by intramuscular adeno-associated virus (AAV) injections. We induced several features of CNM, and we have compared these findings with muscle biopsy specimens from a patient with the same mutation. We show that DNM2 is important in maintenance of adult muscle fibers and that this DNM2 mutation acts in a dominant manner to perturb DNM2 function.
Materials and Methods
Materials
Full-length human isoform DNM2 cDNA was purchased from Geneservice (DNAFORM, Kanagawa, Japan) (IMAGE clone 5722134, GenBank accession number NM BC039596) and was cloned into pENTR1A (Invitrogen, Carlsbad, CA) and then recombined into a pAAV-MCS vector using the Gateway system, with or without a C-terminal green fluorescent protein (GFP) fusion tag. The R465W mutation was introduced by primer-directed PCR mutagenesis. All constructs were verified by sequencing. pXR1 (AAV1) plasmid was a gift from Jude Samulski at the Gene Therapy Center, the University of North Carolina at Chapel Hill. Informed consent was obtained from human subjects. The patient with a DNM2 Arg465Trp mutation will be reported in more detail elsewhere. Primary antibodies used were mouse anti–α-actinin (EA-53; Sigma-Aldrich, St. Louis, MO), DHPRα1 (Cav1.1) subunit (MA3-920; Affinity Bioreagents, Golden, CO), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; MAB374; Chemicon, Temecula, CA), and desmin (Y-20; Santa Cruz Biotechnology, Santa Cruz, CA). Rabbit anti-DNM2 antibodies (2680 and 2865) were made onsite at the polyclonal antibody facility of the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) (described below). Rabbit anti-dystrophin antibody was a gift from Karim Hnia and Dominique Mornet, ERI25, Montpellier, France. Alexa-conjugated secondary antibodies were from Invitrogen. Secondary antibodies against mouse and rabbit IgG, conjugated with horseradish peroxidase (HRP), were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). An ECL chemiluminescent reaction kit was purchased from Pierce (Rockford, IL). Hoechst was purchased from Sigma-Aldrich (B2883). The AAV Helper-Free system was purchased from Stratagene (La Jolla, CA) (catalog number 240071).
Antibody Production
Two DNM2 rabbit antibodies were generated: R2680, raised against the linker between the PH and GED domains of DNM2 (peptide EKDQAENEDGAQENTF), and R2865, raised against the PRD of DNM2 (peptide HSPTPQRRPVSSVHPPGRPPAVR). Serum samples were purified on peptide-coupled SulfoLink columns (Pierce) (R2680) or with the Imject Maleimide Activated Mariculture KLH kit (INTERCHIM SA, Montlucon, France) (R2865). DNM2 antibodies were validated for immunoblotting in COS-1 cells transfected with human DNM2 using peptide competition and validated for immunofluorescence in mouse muscle sections using peptide competition and showed similar results.
Production and Purification of AAV
AAV2/1 vectors were generated by a triple transfection of AAV-293 cell line with pAAV2 insert containing the insert under the control of the CMV promoter and flanked by serotype 2 inverted terminal repeats, pXR1 containing rep and cap genes of AAV serotype 1, and pHelper encoding the adenovirus helper functions. Cell lysates were subjected to 3 freeze/thaw cycles, then treated with 50 U/mL of Benzonase (Sigma) for 30 minutes at 37°C, and clarified by centrifugation. Viral vectors were purified by Iodixanol gradient ultracentrifugation followed by dialysis and concentration against Dulbecco's Phosphate Buffered Saline using centrifugal filters (Amicon Ultra-15 Centrifugal Filter Devices 30K, Millipore, Bedford). Physical particles were quantified by real-time PCR using a plasmid standard pAAV-eGFP, and titers are expressed as viral genomes per milliliter (vg/mL). rAAV titers used in these experiments were 5 to 7 × 1011 vg/mL.
AAV Transduction of Wild-Type Tibialis Anterior Muscle of Mice
Five- to 6-week-old, male, wild-type, 129PAS mice were anesthetized by i.p. injection of 5 μL/g of ketamine (20 mg/mL; Virbac, Carros, France) and xylazine (0.4%, Rompun; Bayer, Wuppertal, Germany). Tibialis anterior (TA) muscles were injected with 25 μL of AAV2/1 preparations or sterile PBS solution. Animals were housed in a temperature-controlled room (19°C to 22°C) with a 12:12-hour light/dark cycle. Mice were sacrificed by CO2 inhalation followed by cervical dislocation, according to national and European legislations on animal experimentation. The TA muscles were dissected [with the mice under anesthesia when required for transmission electron microscopy (TEM)] 2 to 4 weeks post injection (PI) and frozen in nitrogen-cooled isopentane and liquid nitrogen for histologic and immunoblot assays, respectively. In mice injected i.m. with GFP-AAV2/1, we observed a transduction efficiency of 75% to 100%, which is comparable to previously published results.17
Histologic and Immunofluorescence Analysis of Skeletal Muscle
Longitudinal and transverse cryosections (8 μm) or semithin (500 nm) sections of mouse TA skeletal muscles and human muscle were prepared, fixed, and stained with antibodies to DHPRα1 (1:500), desmin (1:500), α-actinin (1:1000), R2680-DNM2 (1:500), R2865-DNM2 (1:500), and dystrophin (1:400). Nuclei were detected by costaining with Hoechst (Sigma-Aldrich) for 10 minutes. Sections were viewed using a laser scanning confocal microscope (TCS SP2; Leica Microsystems, Sunnyvale, CA). Intensity of staining (Figure 1B) was measured using the plot profile function in ImageJ analysis software (W.S. Rasband, ImageJ, National Institutes of Health, Bethesda, MD, 1998–2009; http://rsb.info.nih.gov/ij). Alternatively, air-dried transverse sections were fixed and stained with hematoxylin-eosin (HE), succinate dehydrogenase (SDH) or NADH-tetrazolium reductase (TR) activity and viewed with a fluorescence microscope (DM4000; Leica Microsystems). The cross-sectional area was analyzed in HE sections from TA mouse skeletal muscle, using the software MetaMorph (Molecular Devices). The cross-sectional area was calculated (>500 fibers per mouse) from three to five mice per group. The percentage of TA muscle fibers with centralized or internalized nuclei was counted in more than 400 fibers in at least three mice using the cell counter plugin in ImageJ image analysis software.
Figure 1.
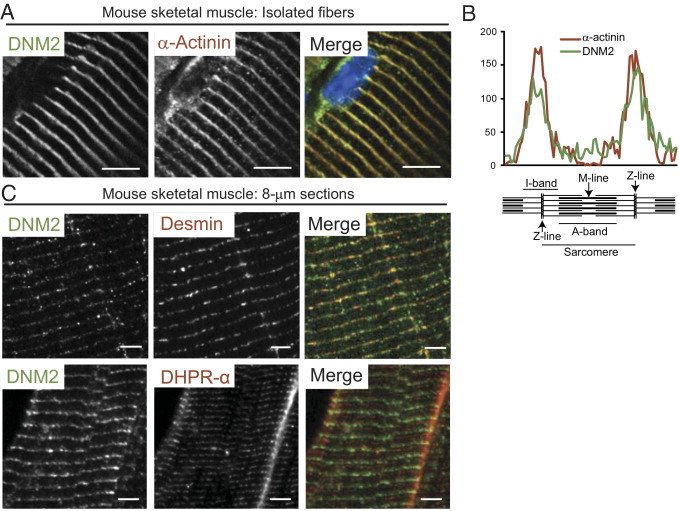
DNM2 localization in wild-type mouse muscle. A: Wild-type mouse isolated muscle fibers costained with DNM2 (green) and α-actinin (red) antibodies and Hoechst nuclear stain (blue) and viewed by confocal microscopy. Scale bar = 5 μm. B: Intensity of sarcomeric staining shown in A, spanning one complete sarcomere. Similar peaks are seen in both red and green channels, corresponding to the Z-line. C: Longitudinal mouse semithin (500 nm) skeletal muscle sections costained with DNM2 (green) and desmin (red, top) or DHPRα (red, bottom) antibodies and viewed by fluorescence microscopy. Scale bar = 5 μm.
Preparation of Isolated Muscle Fibers
The TA muscle was dissected from the hind limb and fixed in 4% paraformaldehyde in PBS for 30 minutes. Muscles were then incubated with 0.1 mol/L glycine in PBS for 30 minutes, followed by 30% sucrose/PBS solution overnight at 4°C. Muscle were washed with PBS, and fibers were then isolated under a binocular microscope. Fibers were then permeabilized and stained with DNM2 and α-actinin antibodies and viewed by confocal microscopy.
Transmission Electron Microscopy
Mice were anesthetized by intraperitoneal injection of 10 μL/g of ketamine (20 mg/mL; Virbac) and xylazine (0.4%, Rompun; Bayer). Muscle biopsy specimens from hind limbs were fixed with 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.2) and processed as described.18 For T-tubule analysis, the T-tubule shape factor was measured by manually outlining the shape of the T-tubule and using the circularity measurement in ImageJ image analysis software. On average, 50 T-tubules per mouse for two mice per group were measured. Potassium ferrocyanide staining was performed as described previously.19
Muscle Contractile Properties
Muscle force measurements were evaluated by measuring in situ muscle contraction in response to nerve and muscle stimulation, as described previously.20,21 Briefly, animals were anesthetized (i.p., pentobarbital sodium, 50 mg × kg−1). The distal tendon of the TA was detached and tied with a silk ligature to an isometric transducer (Harvard Bioscience, Holliston, MA). The sciatic nerve was distally stimulated, response to tetanic stimulation (pulse frequency of 50 to 143 Hz) was recorded, and absolute maximal force was determined. After contractile measurements, the animals were sacrificed by cervical dislocation. To determine specific maximal force, TA muscles were dissected and weighted. Muscles were then stored as described for further analysis.
Preparation of Samples for Western Blotting
Total, soluble, and insoluble proteins were extracted from the skeletal muscle of mice. Mouse TA muscle (stored at −80°C before use) was minced and homogenized on ice for 3 × 30 seconds (Ultra Thurax homogenizer) in 10 times the w/v of 1% NP-40 Tris-Cl buffer (pH 8) then extracted for 30 minutes at 4°C, and used for Western blotting. For preparation of soluble and insoluble fractions, lysates were then centrifuged at 8000 × g for 5 minutes, and the soluble fraction (supernatant) and insoluble fraction (pellet solubilized in 8 mol/L urea) were collected. Protein concentration was determined using a DC protein assay kit (Bio-Rad Laboratories, Hercules, CA) and lysates analyzed by SDS–polyacrylamide gel electrophoresis and Western blotting (nitrocellulose membrane). Primary antibodies used were R2865-DNM2 (1:1000) and GAPDH (1:10,000); secondary antibodies were anti-rabbit HRP or anti-mouse HRP.
Densitometry Analysis
Western blot films were scanned and band signal intensities were determined using ImageJ software. Densitometry values were expressed as a fold difference relative to the control, standardized to corresponding total GAPDH values.
Microscopy and Statistical Analysis
All microscopy was performed at the Imaging Centre of the IGBMC. All samples for microscopy were mounted in Fluorsave reagent (Merck, Summit, NJ) and viewed at room temperature. Confocal microscopy was performed using a confocal laser scanning microscope (TCS SP2; Leica Microsystems) on a DMRXA2 upright microscope. Fluorescence and light microscopy was performed using a fluorescence microscope (DM4000; Leica Microsystems) fitted with a color CCD camera (Coolsnap cf color; Photometrics, Tucson, AZ) camera. Metamorph software (Molecular Devices) and ImageJ analysis software were used for image analysis. Statistical analysis was performed using the unpaired Student's t-test unless stated otherwise. P < 0.05 was considered significant.
Results
DNM2 Localizes Close to the Z-line in Striated Muscle
DNM2 is ubiquitously expressed in human tissues22,23; however, the localization of DNM2 in skeletal muscle has not been well characterized. In mouse isolated muscle fibers, DNM2 colocalized with the Z-line marker α-actinin, as observed by immunofluorescence (Figure 1A) and confirmed by analyzing the intensity of staining of DNM2 and α-actinin, which displayed a consistent overlapping profile at the Z-line (Figure 1B). In mouse skeletal muscle sections, desmin, a different Z-line marker, colocalized with DNM2, although this was slightly more discontinuous compared with the colocalization observed with α-actinin. In contrast, DHPRα, which labels T-tubules, formed a doublet band that did not colocalize but aligned in close proximity with DNM2 staining (Figure 1C). Therefore, in mouse striated muscle DNM2 appears to localize to the Z-line, in close proximity to the T-tubules. DNM2 localization in human skeletal muscle was consistent with that observed in mouse muscle. In neonatal human muscle (1.5 months old), DNM2 formed a transverse banding pattern consistent with Z-line staining, whereas DHPRα was located in a longitudinal arrangement (Figure 2A), indicating that DNM2 localizes to the sarcomeric unit before the final maturation of T-tubules to a transverse position. In adult human muscle the DNM2 localization was consistent with Z-line striations on longitudinal sections (Figure 2C, top left), whereas on transverse sections an intricate network of DNM2 staining was observed within the fiber, with a more dense localization at the subsarcolemmal region (Figure 2C, bottom left). Therefore, DNM2 is recruited to the sarcomeric unit before the maturation of T-tubules is complete and localizes to the Z-line in mature muscle.
Figure 2.
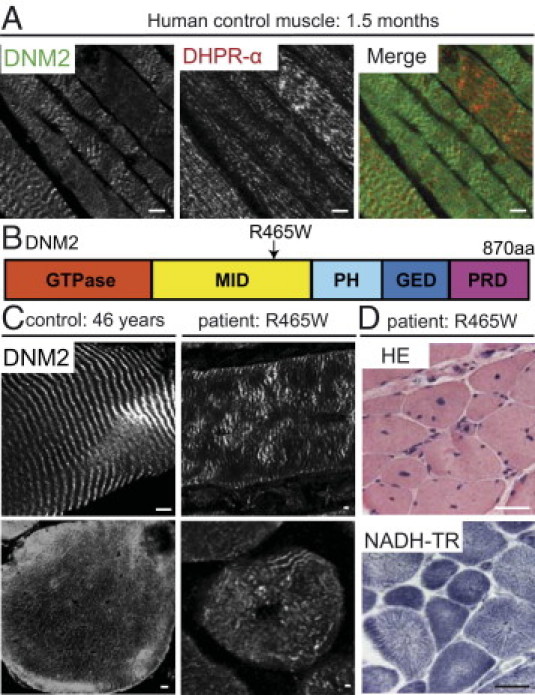
DNM2 in normal and pathologic human muscles. A: Longitudinal, human, 10-μm sections (1.5-month-old control muscle) costained with R2680-DNM2 (green) and DHPRα (red) antibodies and viewed by confocal microscopy. Scale bar = 5 μm. B: DNM2 protein consists of GTPase, middle (MID), PH, GED, and PRD. The most common DNM2 mutation, R465W, is indicated. C: Human muscle sections (left: control muscle; right: R465W patient muscle) stained with R2680-DNM2 antibody and viewed by confocal microscopy. Scale = bar 5 μm. D: Transverse R465W patient muscle sections stained with HE or for NADH-TR and viewed by fluorescence microscopy. Scale bar = 50 μm.
Histopathology in a Patient with the Common R465W DNM2 Mutation
The most common mutation found in patients with ADCNM is an arginine to tryptophan missense at position 465 (R465W), located within the middle domain of DNM2 (RW-DNM2) (Figure 2B).8,24 Patients with this mutation exhibit the three typical CNM features: atrophic fibers, centralized and internalized nuclei, and abnormal oxidative staining with the appearance of radial sarcoplasmic strands (RSSs) on NADH-TR staining (Figure 2D).25 In muscle sections from an RW-DNM2 patient, DNM2 localized as a striated pattern on longitudinal sections, although this pattern appeared disturbed and more irregular (Figure 2C, top right). On transversal sections, DNM2 was present but the intricate network of staining appeared perturbed (Figure 2C, bottom right). Altogether, this suggests that the RW-DNM2 mutation is linked to abnormal protein localization and muscle intracellular organization.
AAV Expression of Mutated DNM2 in Wild-Type Muscle
Complete deletion of DNM2 in mouse resulted in embryonic lethality,26 and RW-DNM2 heterozygous knock-in mice exhibit a mild impairment in skeletal muscle structure, with skeletal muscle atrophy and a corresponding reduction in force detected.27 However, a strong CNM phenotype was not observed in these mice, and no increase in central or internalized nuclei was detected. It is currently unknown whether the CNM phenotype is due to a haploinsufficiency of DNM2 expression or function or whether the mutant protein acts in a dominant manner. In addition, whether the disease results from the altered function of DNM2 in muscle development or in adult muscle maintenance is not clear. Moreover, it was not clear whether the muscle phenotype is due to a primary impairment of peripheral nerves because some DNM2 mutations cause a peripheral neuropathy. To decipher these three points, we used an original in vivo approach to attempt to recreate ADCNM in wild-type adult mouse skeletal muscle and to investigate the normal function of DNM2 in wild-type muscle. Expression of DNM2 containing the R465W mutation was achieved by intramuscular injections of an AAV cognate vector into the TA muscle of wild-type 5- to 6-week-old mice, an age where the muscle mass is nearly fully developed. This allowed the role of DNM2 to be studied specifically in adult skeletal muscle. Mice were injected with WT-DNM2 or RW-DNM2 with a C-terminal GFP tag or GFP alone as a control. The injection of WT-DNM2, while acting as a control for RW-DNM2–injected muscle, also allowed us to investigate the function of WT-DNM2 in muscle. The addition of a GFP tag allowed the differentiation between AAV-transduced and endogenous DNM2 expression. Untagged DNM2 constructs were also used and gave comparable results as described below.
The level of DNM2 protein expression was determined using Western blot analysis and densitometry. In both WT-DNM2– and RW-DNM2–transduced muscles a fivefold increase in total DNM2 expression was observed compared with GFP-transduced and noninjected muscles (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org). No significant difference in protein solubility was observed between exogenous WT-DNM2 or RW-DNM2 and endogenous DNM2 (see Supplemental Figure S1, C and D, at http://ajp.amjpathol.org), indicating AAV-DNM2 expression did not alter DNM2 association to membranes. Therefore, full-length AAV-DNM2 is expressed in mouse skeletal muscle.
RW-DNM2 Expression Induces a CNM-like Phenotype
Because full-length RW-DNM2 was efficiently expressed, we next determined whether any features of ADCNM were exhibited in this model. At 2 weeks PI, mice transduced with RW-DNM2 exhibited a slight but significant reduction in TA muscle mass, and by 4 weeks PI the muscles were reduced to almost 50% the weight of GFP control and PBS-injected muscles (Figure 3A). To confirm that the decrease in muscle mass was due to muscle fiber atrophy, the cross-sectional area of muscle fibers was determined from transverse sections stained with HE (Figure 3, D and F). A decrease in fiber area was observed in RW-DNM2 transduced TA muscle both 2 weeks (Figure 3, B and D) and 4 weeks PI (Figure 3, B and F), corresponding with the decrease in muscle mass. The decrease in average muscle fiber area was due to an increase in atrophic fibers compared with WT-DNM2 and GFP-transduced muscle (Figure 3C), confirming RW-DNM2 transduction into wild-type muscle induced muscle fiber atrophy. WT-DNM2 also exhibited a slight but significant decrease in muscle weight and fiber size, however to a significantly lesser extent than RW-DNM2 muscle. It is possible that the level of endogenous DNM2 expression needs to be tightly regulated and that overexpression of WT-DNM2 causes some perturbations in muscle size.
Figure 3.
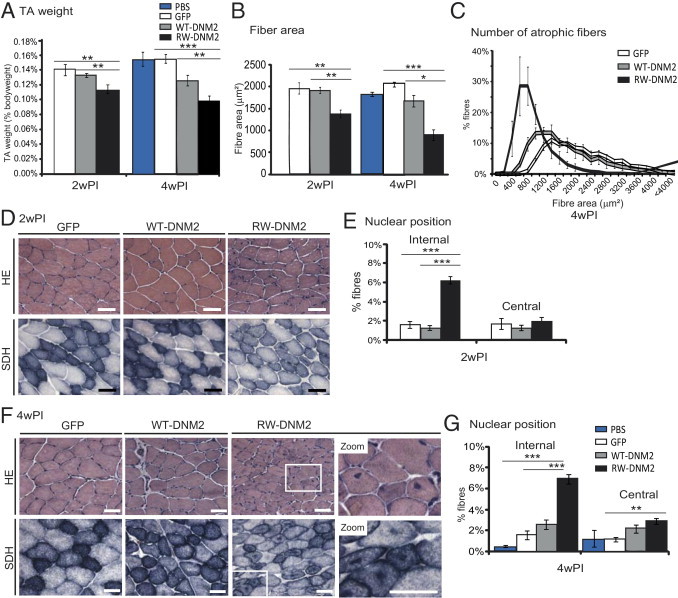
CNM features observed in RW-DNM2 mice. A: Immediately after dissection TA muscles were weighed from mice 2 and 4 weeks PI. Graph represents TA weight as a percentage of total body weight (n = 3 to 6 mice). B: HE-stained transversal sections from D and F were analyzed for fiber area (n = 3 to 5 mice). C: Transverse muscle sections were analyzed for fiber area. Fiber size is grouped into 200-μm intervals and represented as the percentage of total fibers in each group (n = 5 mice, 4 weeks PI). D: Transverse TA sections from AAV-transduced GFP, WT-DNM2, or RW-DNM2 mice 2 weeks PI (2wPI) stained with HE (top) or SDH (bottom) and viewed by light microscopy. Scale bar = 50 μm. E: The frequency of fibers with internal or central nuclei 2wPI were scored (n = 3 to 5 mice). Internal nuclei are defined as neither subsarcolemmal nor central. F: Transverse muscle sections 4 weeks PI (4wPI). Higher magnification (zoom) images are shown. Scale bar = 50 μm. G: Nuclear position; 4wPI (n = 5 to 6 mice). All graphs depict mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
Healthy muscle fibers contain nuclei beneath the sarcolemma, and a hallmark of DNM2-related CNM is the large increase in fibers containing internal or central nuclei (Figure 2D).10,25,28 To determine whether wild-type TA muscle expressing RW-DNM2 also exhibited this feature, fibers with central or internal nuclei were quantified from HE images. At 2 weeks PI, a threefold increase of fibers with internal nuclei was observed in mice transduced with RW-DNM2 (Figure 3E). By 4 weeks PI, this difference increased slightly, and in addition a twofold increase in centralized nuclei was observed in comparison to both GFP-transduced and PBS-injected muscles (Figure 3G). No significant difference in myonuclei positioning between WT-DNM2 and GFP-transduced or PBS-injected muscle was noted. Therefore, RW-DNM2 expression induces mislocalization of muscle nuclei, as observed in patients with DNM2-related CNM.
Additional characteristics of ADCNM patient muscle include a high percentage of fibers with RSSs and unevenly dispersed mitochondria, which tend to stain more densely toward the center and/or periphery of the fiber (Figure 2D).25,29 These features can be observed with oxidative staining methods (NADH/SDH) and electron microscopy. In GFP- and WT-DNM2–transduced muscle, an even distribution of SDH staining was observed within each fiber; however, in RW-DNM2 muscle a clear redistribution occurred, with an increase in staining at the center and periphery of many fibers (Figure 3, D and F). In some cases this gave the appearance of radial-like strands; however, by TEM radial strands were not observed, indicating that this specific feature is not reproducible in our mouse model. CNM patients also exhibit an increase in type 1 fibers; however, we did not detect an increase in type 1 fibers in our model (not shown).
To confirm that the C-terminal GFP tag had no influence on our results, we repeated the above experiments with untagged WT-DNM2 and RW-DNM2. AAV injection of untagged RW-DNM2 induced an increased number of atrophic fibers (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org), increased internal and centralized nuclei (see Supplemental Figure S2, A and C, at http://ajp.amjpathol.org), and altered oxidative (SDH) staining, similar to GFP-tagged RW-DNM2. Therefore, mice transduced with GFP-tagged or untagged RW-DNM2 exhibit most but not all features seen in ADCNM patients.
RW-DNM2 Affects Muscle Strength
We next determined whether the histologic features observed in our model corresponded to a decrease in force by measuring the force of AAV-transduced TA muscles 4 weeks PI. The muscle was stimulated by the sciatic nerve, and the maximal force produced was recorded. RW-DNM2–transduced muscle displayed a severe decrease in absolute maximal force compared with both GFP- and WT-DNM2–transduced muscle (Figure 4A). Interestingly, WT-DNM2–transduced muscle also exhibited a strong reduction in absolute maximal force. To determine whether reduced force was due to a reduction in muscle fiber “quality” in addition to the atrophy observed, the specific maximal force was calculated: absolute maximal force was normalized to muscle mass. A large reduction in specific maximal force was observed in RW-DNM2 muscle compared with GFP and WT-DNM2 muscle (Figure 4B), indicating that expression of RW-DNM2 impairs the muscle's ability to produce maximal force, independently of muscle size. WT-DNM2 expression also decreased the specific maximal force produced, although to a significantly lesser extent than RW-DNM2. For all experiments no difference in maximal force was observed between sciatic nerve stimulation and direct muscle stimulation, indicating the nerve-muscle communication is unaffected. Therefore, expression of WT-DNM2 and RW-DNM2 causes a reduction in maximal force due to both a reduction in fiber size and the reduced contractile abilities of transduced fibers.
Figure 4.
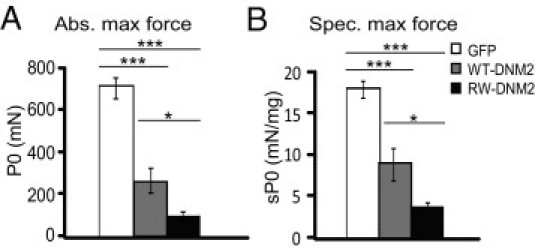
Altered muscle force in RW-DNM2 mice. A: The absolute (Abs.) maximal force of the TA muscle was measured 4 weeks PI (n = 6 to 8 mice). B: The specific (Spec.) maximal force of TA muscle represents the absolute maximal force related to muscle weight (n = 6 to 8 mice). GFP-transduced muscle produced specific maximal force measurements that were statistically similar to PBS control mice. All graphs depict mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001). P0, maximum isometric force; sP0, specific isometric force.
Defects in Sarcomere Structure in RW-DNM2–Transduced Muscle
Because our model recapitulates most features of ADCNM, we examined the effect of AAV-DNM2 expression on muscle fiber structure and protein localization to gain insight to the cellular function of DNM2. To determine the localization of DNM2 in AAV-transduced mouse muscle compared with human muscle, we labeled longitudinal sections from AAV-transduced mice with a DNM2 antibody, which detects both endogenous and exogenous DNM2 localization. DNM2 localized in a transverse banding pattern on longitudinal sections from GFP-transduced and WT-DNM2–transduced muscle (Figure 5B, lower panel ), reminiscent of Z-line staining observed in wild-type mouse muscle (Figure 1)27 and human muscle (Figure 2C). In longitudinal sections of RW-DNM2–transduced muscle, DNM2 exhibited transverse localization akin to WT-DNM2 staining (Figure 5B, high-exposure image); however, in some fibers a punctate staining pattern in the subsarcolemmal region and center of the fiber was more prominent (Figure 5B, lower panel). This variation in RW-DNM2 localization between fibers was also observed in untagged RW-DNM2 transduced fibers (see Supplemental Figure S3, M–U, at http://ajp.amjpathol.org), indicating the GFP tag is not causing misfolding of the RW-DNM2 protein. To confirm the transverse banding pattern was Z-line localization, 8-μm sections from muscle transduced with untagged DNM2 constructs were costained with DNM2 and α-actinin. DNM2 colocalized with α-actinin at the Z-line in both muscles injected with PBS (see Supplemental Figure S3, A–F, at http://ajp.amjpathol.org) and WT-DNM2 (see Supplemental Figure S3, G–L, at http://ajp.amjpathol.org) using two DNM2 antibodies derived to different sequences within DNM2 (R2680 and R2865, see Materials and Methods for details). In muscle transduced with RW-DNM2, DNM2 localized to the Z-line in some fibers (see Supplemental Figure S3, M–R, at http://ajp.amjpathol.org); however, in other fibers DNM2 appeared mislocalized, gathering in the subsarcolemmal region of the fiber (see Supplemental Figure S3, S–U, at http://ajp.amjpathol.org) consistent with RW-DNM2-GFP localization (Figure 5B). Therefore, mutant DNM2 exhibits altered localization in RW-DNM2–transduced mouse muscle.
Figure 5.
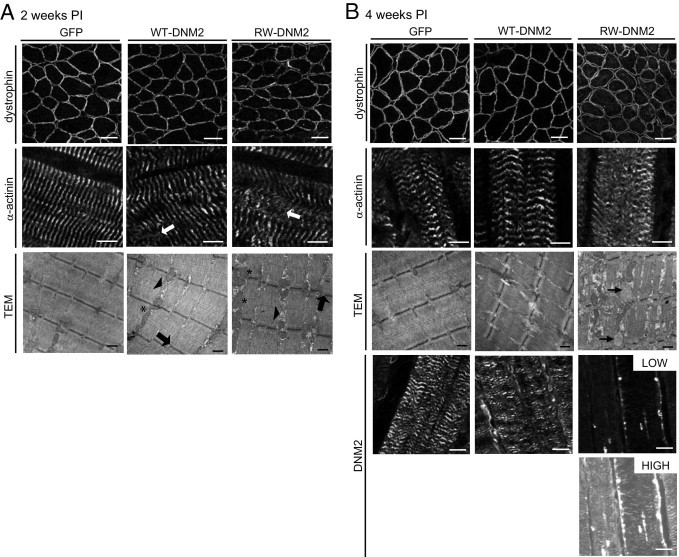
Altered sarcomeric structure in RW-DNM2 mice. TA sections from GFP, WT-DNM2, and RW-DNM2 muscles were stained for dystrophin (top, scale bar = 50 μm), α-actinin (middle, scale bar = 5 μm) or DNM2 (lower in B, scale bar = 20 μm) and imaged by confocal microscopy. Arrows, misaligned Z-lines. For RW-DNM2 staining, low-exposure (labeled low) and high-exposure (high) images are shown to observe striated staining pattern of DNM2. Alternatively, AAV-transduced TA muscles were imaged by TEM. Arrows, misaligned Z-lines; arrowheads, membrane accumulation; asterisks indicate elongated mitochondria (A). Arrows in B indicate abnormal vesicles. In GFP-transduced TA, the α–actinin staining appears slightly disorganized in some fibers expressing GFP, although the Z-line and sarcomeric arrangements were not altered as seen by TEM. Scale bar = 0.5 μm. All images are from mice 2 weeks PI (A) or 4 weeks PI (B).
Sarcolemmal dystrophin staining appeared undisturbed in RW-DNM2 mice compared with GFP and WT-DNM2 muscle at 2 weeks PI (Figure 5A). At 4 weeks PI there was a slight increase in the cytoplasmic pool of dystrophin in RW-DNM2 muscle; however, the levels of dystrophin at the sarcolemma did not appear significantly reduced (Figure 5B), indicating that there is no severe disruption of the dystrophin complex at the sarcolemma. Staining with α-actinin antibodies was performed on longitudinal sections to assess the integrity of sarcomeres. In GFP control muscle 2 weeks PI, α-actinin appeared as a transverse banding pattern (Figure 5A), indicating the Z-line is correctly aligned. In WT-DNM2 muscle the staining appeared relatively normal; however, some disruptions were observed (Figure 5A). In contrast, RW-DNM2–transduced muscle exhibited many areas with misalignment of the Z-line (Figure 5A). This defect was more severe 4 weeks PI (Figure 5B).
To confirm that this was due to a disruption of the Z-line and not α-actinin alone, TEM was performed. Two weeks PI, GFP-transduced muscle appeared normal, with intact aligned Z-lines (Figure 5A). WT-DNM2 muscle sarcomeres also seemed normal, with a small increase in membrane accumulations between myofibrils and a slight misalignment of the Z-line. In contrast, RW-DNM2 muscle exhibited striking defects, with numerous Z-lines misaligned, increased accumulations of membrane structures between myofibrils, and abnormally shaped and orientated mitochondria. By 4 weeks PI, the muscle structure had further deteriorated, with vesicles proliferation (Figure 5B). The sarcomeric structure, which is responsible for enabling muscle fiber contraction, is thus severely affected in RW-DNM2 transduced muscle 4 weeks PI, which may explain at least in part the reduced specific maximal force.
Triad Defects in RW-DNM2–Transduced Muscle
Triads enable excitation-contraction coupling and are composed of a membrane invagination (T-tubule), which associates with two sarcoplasmic reticuli. Defects in the triad structure and orientation have been reported in zebra fish and mouse models for X-linked CNM deficient in myotubularin.19,30 BIN1, a binding partner of DNM2, localizes to T-tubules and plays a role in T-tubule biogenesis,28,31,32 and mutations in BIN1 are responsible for autosomal recessive CNM.13 Furthermore, mutations in RyR1, which is responsible for coupling the sarcoplasmic reticulum to the T-tubule and calcium release into the fiber on excitation, were suggested to cause CNM,14,15 indicating that there may be a common molecular mechanism between the different forms of CNM affecting triad structure and function.28
To determine whether our model also exhibits triad defects, triad structures were examined with TEM in mice 4 weeks PI. In GFP-transduced muscle, T-tubules were observed correctly aligned between two sarcoplasmic reticuli (Figure 6B). In WT-DNM2–transduced muscle, the T-tubules also looked healthy; however, in comparison, many T-tubules appeared rounder in RW-DNM2 muscles (Figure 6B, lower panel). The shape factor of T-tubules was measured to determine whether there were significant irregularities. A circle has a shape factor of one, whereas an object approaching a straight line has a shape factor near zero. Indeed, no difference was seen between the shape factor for GFP- or WT-DNM2–transduced muscles; however, the shape factor increased significantly in RW-DNM2–transduced muscle (Figure 6D), indicating T-tubules were rounder in our model. This was measured also at 2 weeks PI, and a significant but less severe difference was observed (Figure 6, A and C), indicating the abnormalities in T-tubule morphology increase over time.
Figure 6.
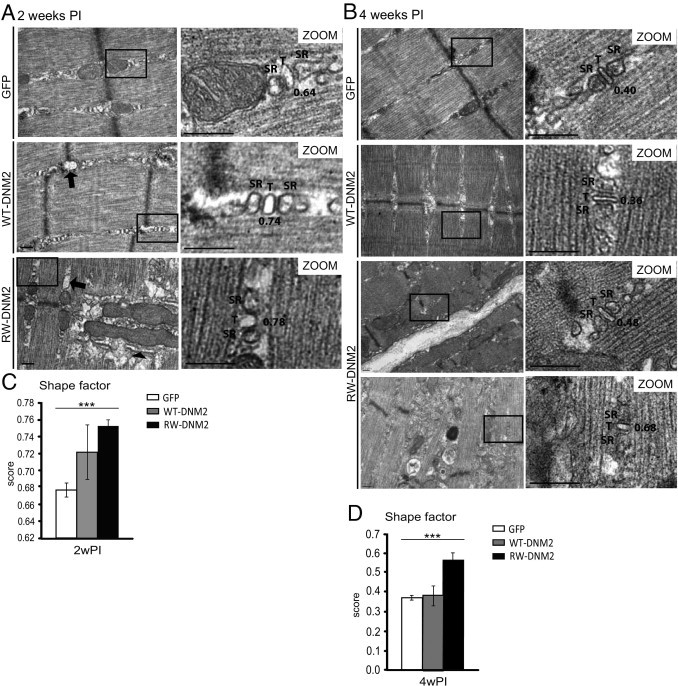
Altered T-tubule structures in RW-DNM2 mice. TA muscles from AAV-transduced GFP, WT-DNM2, or RW-DNM2 mice 2 (A) and 4 (B) weeks PI imaged by TEM. Magnified (zoom) images shown on the right. SR, sarcoplasmic reticulum; T, T-tubule. Arrows: swollen sarcoplasmic reticulum; arrowhead: abnormal membrane structures near mitochondria. Scale bar = 0.2 μm. The shape factor was calculated from T-tubules shown in A and B. C: 2 weeks PI (2wPI). D: 4 weeks PI (4wPI). A shape factor of 1 = circle and 0 = straight line (shape factors for T-tubules in images in A and B are indicated). Graph represents mean ± SEM (***P < 0.001) (n = approximately 50 T-tubules per mouse for two mice per group).
In addition to T-tubule abnormalities, structures that appeared to be swollen sarcoplasmic reticulum were observed in both WT-DNM2 and RW-DNM2 muscle (Figure 6A, arrow). In RW-DNM2 muscle, triads were not easily detected near enlarged mitochondria; however, the presence of abnormal membrane structures around the mitochondria was common. Whether these membranes are remnants of sarcoplasmic reticulum or longitudinal T-tubule structures cannot be determined from standard TEM. An additional drawback of TEM is that T-tubules must be recognizable within a triad structure to be measured. Therefore, to determine the extent of damage to triad structures in RW-DNM2 mice 4 weeks PI, the morphology of triads was analyzed by electron microscopy using potassium ferrocyanide, which selectively stains plasma membrane invaginations such as T-tubules. In both GFP- and WT-DNM2–transduced muscle, T-tubules appeared normally shaped and organized between sarcoplasmic reticuli, in an orientation matching the Z-line (Figure 7, A–D). Occasionally misoriented T-tubules were observed in WT-DNM2–transduced muscle (Figure 7C). In contrast, T-tubules in muscle transduced with RW-DNM2 were severely affected (Figure 7, E and F). Some T-tubules were observed, which, although rounder than normal (confirming results seen in Figure 6), were distinguishable as triads (Figure 7F). However, many areas of enlarged, misoriented T-tubules were observed (Figure 7F), often associated near sarcoplasmic reticulum (Figure 7F). Therefore, triad structures and mitochondria are profoundly altered in muscle transduced with RW-DNM2.
Figure 7.
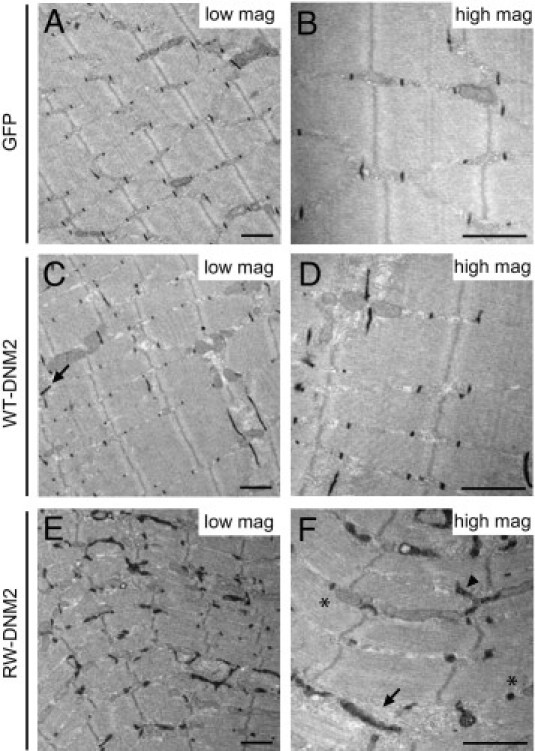
Triad disorganization in RW-DNM2 mice. Electron microscopy images from muscles transduced with GFP (A and B), WT-DNM2 (C and D), and RW-DNM2 (E and F), labeled with potassium ferrocyanide [K3Fe(CN)6] staining. High-magnification (high mag) images are shown on the right and low-magnification (low mag) images on the left. C: Arrow indicates misoriented T-tubule. F: Round T-tubules (asterisk), enlarged, misoriented T-tubules (arrowhead), and sarcoplasmic reticulum (arrow). Scale bar = 1 μm.
Discussion
In this study, we examined the function of DNM2 in skeletal muscle and the pathophysiology of ADCNM using AAV to express WT-DNM2 or RW-DNM2 mutation, the most common mutation observed in patients. We characterized a mammalian model with a DNM2 defect and showed that RW-DNM2 expression in adult wild-type muscle can induce most features of CNM compared with muscle biopsy specimens from a patient with the same mutation.
DNM2 in CNM
Our RW-DNM2 mice muscles recapitulate the increased internal and centralized nuclei, abnormal oxidative (SDH) staining, and fiber atrophy. It also shows that the muscle weakness is due to a reduction in specific maximal force and not only to muscle atrophy. However, we did not observe an increase in either type 1 fibers or the radial strand organization of oxidative staining, the latter being a diagnosis marker to differentiate with the X-linked form of CNM. Because we injected the TA muscle, which contains <1% of type 1 fibers, it is possible that a difference was not detectable. Alternatively, R465W mutant expression could affect fiber-type determination during muscle differentiation; however, we injected adult muscle. The RSS appearance is caused by a difference in myofibril width between myofibrils in the center and periphery. This abnormality could appear with time because such a phenotype was rarely observed in neonatal ADCNM cases.11 Importantly, the appearance of most CNM features after injection of adult muscles supports a defect in muscle structure maintenance in the disease.
While our original manuscript was submitted, a RW-DNM2 knock-in mouse was described, in whom RW-DNM2 was expressed throughout muscle development.27 Heterozygous knock-in mice exhibited mild skeletal muscle atrophy and muscle weakness, with minor alterations in mitochondrial and reticular organization. Similar to our model, no difference in fiber-type expression or RSSs was apparent, indicating that these features are most likely not dependent on expression of RW-DNM2 during early stages of muscle development. One important CNM feature, which was not observed in heterozygous knock-in mice, was an alteration in nuclei position27 compared with our AAV expression system in which an increase in central and internally placed nuclei was observed, a hallmark feature in CNM patients. It may be that a higher level of overexpression of RW-DNM2 is required to induce this feature in mice. Interestingly, in surviving homozygous knock-in mice an increase in internal and central nuclei was observed,27 akin to that observed in mice 4 weeks PI of RW-DNM2-AAV, supporting the belief that a higher level of RW-DNM2 expression/activity is required to induce CNM-like features in mice.
Distinct from the work of Durieux et al,27 this study indicates that the most common DNM2 mutation is acting in a dominant manner, as wild-type mice were injected. It rules out haploinsufficiency as a cause of the muscle disease and parallels recent findings showing that the R465W missense mutation increases the dynamin oligomer stability and GTPase activity.33,34 Interestingly, in our study we also observed an effect by expression of WT-DNM2 in wild-type mouse muscle, mainly on fiber size, nuclei centralization, and maximal force, but to a lesser extent than with RW-DNM2. Because this is the first study to overexpress DNM2 in a system that already has normal DNM2 levels, we can begin to hypothesize the mechanism(s) by which this effect was induced. Increased expression of WT-DNM2 increased the pool of DNM2 within muscle, which may result in an overall increase in GTPase activity. It may also promote DNM2 dimerization.35,36 Together with biochemical characterization of DNM2 mutants,33,34 our results strongly support that the CNM muscle phenotype observed is due to an increase in DNM2 GTPase activity and that the R465W mutation acts in a dominant manner by increasing the GTPase activity or DNM2 oligomerization in vivo.
Although DNM2 is mutated in both CNM and peripheral neuropathy, our data show that a primary defect of skeletal muscle is at the basis of CNM as muscles were injected. Moreover, maximal force measurement indicated the nerve-muscle communication appeared unaffected.
Roles of DNM2 in Skeletal Muscle
DNM2 is implicated in ADCNM; however, its function in muscle is not understood. Because adult mice were transduced, we can begin to discern the specific functions of DNM2 in muscle development compared with muscle maintenance. DNM2 appears to localize to the Z-line, and because this occurs before the transverse alignment of T-tubules, DNM2 may participate in T-tubule reorientation during maturation. Indeed, DNM2 interacts directly with BIN1 in vitro, and BIN1 is localized at T-tubules and is implicated in T-tubule biogenesis.13,31 T-tubules appeared rounder, enlarged, and misoriented in RW-DNM2 muscle, suggesting that DNM2 might also be important for the maintenance of these structures. Of note, T-tubules in skeletal muscle are approximately 20 to 40 nm in diameter,37 wider than the internal diameter of the dynamin helix (15 to 20 nm),38 suggesting that DNM2 is not oligomerizing around T-tubules. Whether and when a possible interaction between DNM2 and BIN1 exists in skeletal muscle remains to be determined.
In RW-DNM2 mice we also observed swelling of the sarcoplasmic reticulum and abnormally shaped mitochondria. In muscle fibers, the sarcoplasmic reticulum and mitochondria are tightly linked,39 and a defect in both triads and mitochondrial/sarcoplasmic reticulum signaling can lead to defects in Ca2+ handling.40 Therefore, in RW-DNM2 mice there may be a defect in excitation-contraction coupling during muscle contraction. The decrease in specific maximal force in these mice could also be due to the strong disruption of sarcomere structure. The involvement of DNM2 in excitation-contraction coupling is supported by the severe decrease in specific maximal force observed in WT-DNM2–transduced mice, despite the sarcomere structure appearing similar to control muscle. It also suggests that the level and/or activity of DNM2 needs to be tightly regulated in skeletal muscle. Defects in these functions (sarcomere maintenance and triad organization) may be at the basis of the histologic and clinical phenotypes of ADCNM patients.
In conclusion, using AAV transduction we developed an animal model for ADCNM that recapitulates several key histologic features of the disease together with altered mitochondrial and triad structures, resulting in muscle weakness. Although AAV vectors are mostly used for preclinical or clinical gene therapy approaches, exogenous expression of mutant proteins represents an original approach, as an alternative to engineering of germline mouse mutations, to test the impact of different patient mutations or artificial DNM2 mutations to dissect the molecular regulation of DNM2 function in vivo. Data obtained with this approach point to distinctive roles of DNM2 in maintenance of adult muscle structure.
Acknowledgments
We thank Christine Kretz, Johann Boehm, Marc Koch, Yannick Schwab, Nadia Messaddeq, and Jean-Marie Garnier for excellent technical assistance, the animal house and imaging center of the IGBMC for support, and Jude Samulski (Gene Therapy Center, the University of North Carolina at Chapel Hill) for providing the pXR1-AAV1 plasmid.
Footnotes
Supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, University of Strasbourg, Collège de France, Association Française contre les Myopathies, Fondation pour la Recherche Médicale (DEQ20071210538), Agence Nationale de la Recherche (ANR 07-0065 and 08-005), E-rare program, and a College de France postdoctoral fellowship (B.S.C.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.054.
Supplemental data
DNM2 expression in AAV-DNM2 mice. A: Skeletal muscle lysates 4 weeks after injection were immunoblotted for DNM2 and GAPDH in untransduced (control) muscle and AAV-transduced GFP, WT-DNM2, or RW-DNM2 muscles. The numbers 1 and 2 indicates different mice. Endogenous DNM2 appeared as a doublet. No degradation products were observed for the exogenous GFP-DNM2 proteins. B: Relative expression of total DNM2 expression determined by densitometry of DNM2-immunoreactive polypeptides, standardized relative to GAPDH loading. DNM2 expression is represented as the fold difference from control lysate. C: Soluble and insoluble muscle lysates were immunoblotted for DNM2 and GAPDH. D: The ratio of soluble to insoluble DNM2 was quantified by densitometry of DNM2-immunoreactive polypeptides in C, standardized relative to GAPDH loading. All graphs depict mean ± SEM (n = 3 to 4 AAV-transduced mice, *P < 0.05).
Mice injected with untagged RW-DNM2 exhibit CNM-like features. A: Transverse TA sections from mice were stained with HE (top panel) or SDH (bottom panel) and viewed by light microscopy. Scale bar = 50 μm. B: Transverse HE muscle sections were analyzed for fiber area, then the fiber area was grouped into 200-μm intervals and represented as the percentage of total fibers in each group (n = 3 to 4 mice). C: The frequency of fibers with internal or central nuclei was scored in transverse sections stained with HE. Internal nuclei are defined as not subsarcolemmal nor central (n = 3 to 5 mice). All graphs depict mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001). All mice were sacrificed 4 weeks PI for analysis.
DNM2 localization in AAV-DNM2 mice. Longitudinal muscle sections from PBS-injected (A-F), untagged WT-DNM2–transduced (G-L), and RW-DNM2–transduced (M-U) mice were stained with either R2680 or R2865 DNM2 antibodies (green) and costained with an α-actinin antibody (red) and imaged by confocal microscopy. Scale bar = 20 μm. Note the different staining patterns for RW-DNM2–transduced fibers (P-R vs S-U).
References
- 1.Shpetner H.S., Vallee R.B. Identification of dynamin, a novel mechanochemical enzyme that mediates interactions between microtubules. Cell. 1989;59:421–432. doi: 10.1016/0092-8674(89)90027-5. [DOI] [PubMed] [Google Scholar]
- 2.Unsworth K.E., Mazurkiewicz P., Senf F., Zettl M., McNiven M., Way M., Holden D.W. Dynamin is required for F-actin assembly and pedestal formation by enteropathogenic Escherichia coli (EPEC) Cell Microbiol. 2007;9:438–449. doi: 10.1111/j.1462-5822.2006.00801.x. [DOI] [PubMed] [Google Scholar]
- 3.Jones S.M., Howell K.E., Henley J.R., Cao H., McNiven M.A. Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science. 1998;279:573–577. doi: 10.1126/science.279.5350.573. [DOI] [PubMed] [Google Scholar]
- 4.van der Bliek A.M., Meyerowitz E.M. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351:411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- 5.Takei K., McPherson P.S., Schmid S.L., De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 6.Roux A., Uyhazi K., Frost A., De Camilli P. GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature. 2006;441:528–531. doi: 10.1038/nature04718. [DOI] [PubMed] [Google Scholar]
- 7.Pucadyil T.J., Schmid S.L. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell. 2008;135:1263–1275. doi: 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitoun M., Maugenre S., Jeannet P.Y., Lacene E., Ferrer X., Laforet P., Martin J.J., Laporte J., Lochmuller H., Beggs A.H., Fardeau M., Eymard B., Romero N.B., Guicheney P. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 9.Zuchner S., Noureddine M., Kennerson M., Verhoeven K., Claeys K., De Jonghe P., Merory J., Oliveira S.A., Speer M.C., Stenger J.E., Walizada G., Zhu D., Pericak-Vance M.A., Nicholson G., Timmerman V., Vance J.M. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- 10.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bitoun M., Bevilacqua J.A., Prudhon B., Maugenre S., Taratuto A.L., Monges S., Lubieniecki F., Cances C., Uro-Coste E., Mayer M., Fardeau M., Romero N.B., Guicheney P. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. 2007;62:666–670. doi: 10.1002/ana.21235. [DOI] [PubMed] [Google Scholar]
- 12.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 13.Nicot A.S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.M., Biancalana V., Oldfors A., Mandel J.L., Laporte J. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 14.Jungbluth H., Zhou H., Sewry C.A., Robb S., Treves S., Bitoun M., Guicheney P., Buj-Bello A., Bonnemann C., Muntoni F. Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2007;17:338–345. doi: 10.1016/j.nmd.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Wilmshurst J.M., Lillis S., Zhou H., Pillay K., Henderson H., Kress W., Muller C.R., Ndondo A., Cloke V., Cullup T., Bertini E., Boennemann C., Straub V., Quinlivan R., Dowling J.J., Al-Sarraj S., Treves S., Abbs S., Manzur A.Y., Sewry C.A., Muntoni F., Jungbluth H. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. 2010;68:717–726. doi: 10.1002/ana.22119. [DOI] [PubMed] [Google Scholar]
- 16.Tosch V., Rohde H.M., Tronchere H., Zanoteli E., Monroy N., Kretz C., Dondaine N., Payrastre B., Mandel J.L., Laporte J. A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. 2006;15:3098–3106. doi: 10.1093/hmg/ddl250. [DOI] [PubMed] [Google Scholar]
- 17.Louboutin J.P., Wang L., Wilson J.M. Gene transfer into skeletal muscle using novel AAV serotypes. J Gene Med. 2005;7:442–451. doi: 10.1002/jgm.686. [DOI] [PubMed] [Google Scholar]
- 18.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A.H., Allard B., Mandel J.L., Laporte J., Jacquemond V., Buj-Bello A. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci U S A. 2009;106:18763–18768. doi: 10.1073/pnas.0900705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vignaud A., Cebrian J., Martelly I., Caruelle J.P., Ferry A. Effect of anti-inflammatory and antioxidant drugs on the long-term repair of severely injured mouse skeletal muscle. Exp Physiol. 2005;90:487–495. doi: 10.1113/expphysiol.2005.029835. [DOI] [PubMed] [Google Scholar]
- 21.Vignaud A., Hourde C., Medja F., Agbulut O., Butler-Browne G., Ferry A. Impaired skeletal muscle repair after ischemia-reperfusion injury in mice. J Biomed Biotechnol. 2010;2010:724914. doi: 10.1155/2010/724914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook T.A., Urrutia R., McNiven M.A. Identification of dynamin 2, an isoform ubiquitously expressed in rat tissues. Proc Natl Acad Sci U S A. 1994;91:644–648. doi: 10.1073/pnas.91.2.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diatloff-Zito C., Gordon A.J., Duchaud E., Merlin G. Isolation of an ubiquitously expressed cDNA encoding human dynamin II, a member of the large GTP-binding protein family. Gene. 1995;163:301–306. doi: 10.1016/0378-1119(95)00275-b. [DOI] [PubMed] [Google Scholar]
- 24.Susman R.D., Quijano-Roy S., Yang N., Webster R., Clarke N.F., Dowling J., Kennerson M., Nicholson G., Biancalana V., Ilkovski B., Flanigan K.M., Arbuckle S., Malladi C., Robinson P., Vucic S., Mayer M., Romero N.B., Urtizberea J.A., Garcia-Bragado F., Guicheney P., Bitoun M., Carlier R.Y., North K.N. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul Disord. 2010;20:229–237. doi: 10.1016/j.nmd.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Romero N.B. Centronuclear myopathies: a widening concept. Neuromuscul Disord. 2010;20:223–228. doi: 10.1016/j.nmd.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 26.Ferguson S.M., Raimondi A., Paradise S., Shen H., Mesaki K., Ferguson A., Destaing O., Ko G., Takasaki J., Cremona O., O'Toole E., De Camilli P. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell. 2009;17:811–822. doi: 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durieux A.C., Vignaud A., Prudhon B., Viou M.T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Laine J., Romero N.B., Guicheney P., Bitoun M. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet. 2010;19:4820–4836. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- 28.Toussaint A., Cowling B.S., Hnia K., Mohr M., Oldfors A., Schwab Y., Yis U., Maisonobe T., Stojkovic T., Wallgren-Pettersson C., Laugel V., Echaniz-Laguna A., Mandel J.L., Nishino I., Laporte J. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2010;121:253–266. doi: 10.1007/s00401-010-0754-2. [DOI] [PubMed] [Google Scholar]
- 29.Zanoteli E., Vergani N., Campos Y., Vainzof M., Oliveira A.S., d'Azzo A. Mitochondrial alterations in dynamin 2-related centronuclear myopathy. Arq Neuropsiquiatr. 2009;67:102–104. doi: 10.1590/s0004-282x2009000100023. [DOI] [PubMed] [Google Scholar]
- 30.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee E., Marcucci M., Daniell L., Pypaert M., Weisz O.A., Ochoa G.C., Farsad K., Wenk M.R., De Camilli P. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 32.Razzaq A., Robinson I.M., McMahon H.T., Skepper J.N., Su Y., Zelhof A.C., Jackson A.P., Gay N.J., O'Kane C.J. Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev. 2001;15:2967–2979. doi: 10.1101/gad.207801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L., Barylko B., Byers C., Ross J.A., Jameson D.M., Albanesi J.P. Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J Biol Chem. 2010;285:22753–22757. doi: 10.1074/jbc.C110.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenniston J.A., Lemmon M.A. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010;29:3054–3067. doi: 10.1038/emboj.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol. 2009;10:423–429. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- 36.Chappie J.S., Acharya S., Leonard M., Schmid S.L., Dyda F. G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature. 2010;465:435–440. doi: 10.1038/nature09032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franzini-Armstrong C., Landmesser L., Pilar G. Size and shape of transverse tubule openings in frog twitch muscle fibers. J Cell Biol. 1975;64:493–497. doi: 10.1083/jcb.64.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mears J.A., Ray P., Hinshaw J.E. A corkscrew model for dynamin constriction. Structure. 2007;15:1190–1202. doi: 10.1016/j.str.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossi A.E., Boncompagni S., Dirksen R.T. Sarcoplasmic reticulum-mitochondrial symbiosis: bidirectional signaling in skeletal muscle. Exerc Sport Sci Rev. 2009;37:29–35. doi: 10.1097/JES.0b013e3181911fa4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durham W.J., Aracena-Parks P., Long C., Rossi A.E., Goonasekera S.A., Boncompagni S., Galvan D.L., Gilman C.P., Baker M.R., Shirokova N., Protasi F., Dirksen R., Hamilton S.L. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNM2 expression in AAV-DNM2 mice. A: Skeletal muscle lysates 4 weeks after injection were immunoblotted for DNM2 and GAPDH in untransduced (control) muscle and AAV-transduced GFP, WT-DNM2, or RW-DNM2 muscles. The numbers 1 and 2 indicates different mice. Endogenous DNM2 appeared as a doublet. No degradation products were observed for the exogenous GFP-DNM2 proteins. B: Relative expression of total DNM2 expression determined by densitometry of DNM2-immunoreactive polypeptides, standardized relative to GAPDH loading. DNM2 expression is represented as the fold difference from control lysate. C: Soluble and insoluble muscle lysates were immunoblotted for DNM2 and GAPDH. D: The ratio of soluble to insoluble DNM2 was quantified by densitometry of DNM2-immunoreactive polypeptides in C, standardized relative to GAPDH loading. All graphs depict mean ± SEM (n = 3 to 4 AAV-transduced mice, *P < 0.05).
Mice injected with untagged RW-DNM2 exhibit CNM-like features. A: Transverse TA sections from mice were stained with HE (top panel) or SDH (bottom panel) and viewed by light microscopy. Scale bar = 50 μm. B: Transverse HE muscle sections were analyzed for fiber area, then the fiber area was grouped into 200-μm intervals and represented as the percentage of total fibers in each group (n = 3 to 4 mice). C: The frequency of fibers with internal or central nuclei was scored in transverse sections stained with HE. Internal nuclei are defined as not subsarcolemmal nor central (n = 3 to 5 mice). All graphs depict mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001). All mice were sacrificed 4 weeks PI for analysis.
DNM2 localization in AAV-DNM2 mice. Longitudinal muscle sections from PBS-injected (A-F), untagged WT-DNM2–transduced (G-L), and RW-DNM2–transduced (M-U) mice were stained with either R2680 or R2865 DNM2 antibodies (green) and costained with an α-actinin antibody (red) and imaged by confocal microscopy. Scale bar = 20 μm. Note the different staining patterns for RW-DNM2–transduced fibers (P-R vs S-U).