

Abstract
Peroxisome proliferator-activated receptor-γ (PPARγ) is an anti-inflammatory molecule. To assess its biological function in lung alveolar epithelial cells, a CCSP-rtTA/(tetO)7-dnPPARγ bitransgenic mouse model was generated. In this model, a dominant negative (dn) PPARγ-Flag fusion protein was overexpressed in lung alveolar type II (AT II) epithelial cells in an inducible manner to suppress the endogenous PPARγ function. Overexpression of dnPPARγ induces up-regulation of proinflammatory cytokines and chemokines at both mRNA and protein levels in AT II epithelial cells. This up-regulation was due to dnPPARγ directly DNA binding on the promoter regions. Up-regulation of proinflammatory molecules activated multiple intracellular signaling pathways in AT II epithelial cells. In addition, inflammatory CD11b+Gr-1+ myeloid-derived suppressor cells were significantly accumulated but T cells were decreased in the lung and circulation system of doxycycline-treated mice. In vitro, myeloid-derived suppressor cells from the lung suppressed T-cell proliferation and function. As a pathogenic consequence, emphysema was observed in the doxycycline-treated lung in association with up-regulation of matrix metalloproteases. Chronic inflammation and lung injury also induced conversion of bone marrow mesenchymal stem cells into AT II epithelial cells in bitransgenic mice. These findings support that PPARγ and its negatively regulated downstream genes in AT II epithelial cells possess multiple functions to control alveolar homeostasis through inflammatory and noninflammatory mechanisms.
In previous work, we demonstrated that neutral lipids are essential to maintaining alveolar homeostasis. Blockage of cholesteryl ester and triglyceride metabolism in the lysosomal acid lipase (LAL) knockout mouse model resulted in heterogeneous phenotypes in the lung, including hypercellularity and emphysema.1,2 An Affymetrix GeneChip microarray study showed abnormal expression of multiple genes in the lung; in particular, proinflammatory cytokines and chemokines were highly up-regulated in lung alveolar type II (AT II) epithelial cells.2 In this mouse model, myeloid-derived lineage cells (especially immature CD11b+Gr-1+ cells, also called myeloid-derived suppressor cells, or MDSCs) were significantly expanded and infiltrated into the lung and other organs,1–4 whereas T cells were dramatically reduced as a result of MDSC suppression and intrinsic defect.4,5
LAL hydrolyzes cholesteryl esters and triglycerides to generate free fatty acids and cholesterol in the lysosomes of cells. The downstream metabolites of these compounds (eg, HODEs, HETEs, and 15d-PGJ2) serve as hormonal ligands for nuclear receptors, such as peroxisome proliferator-activated receptor-γ (PPARγ). Both PPARγ and retinoid X receptor (RXR) form heterodimer on ligand binding. PPARγ is an anti-inflammatory transcriptional factor by reducing proinflammatory cytokine expression. Various experiments demonstrated that PPARγ ligands blocked induction of interleukins IL-1β, IL-2, and IL-6, as well as of tumor necrosis factor α (TNF-α).6–8 At the transcriptional level, activated PPARγ effectively blocked the IL-6 downstream signal transduction pathway by inhibiting DNA binding and transcriptional activity of activation of signal transducer and activator of transcription 3 (STAT3), a potent proinflammatory transcription factor.9 PPARγ also negatively regulates the expression of proinflammatory and oncogenic apoptosis inhibitor 6 (Api6) and matrix metalloprotease 12 (MMP12), which contribute to emphysema and tumorigenesis in the lung.2,10,11
We have previously reported that PPARγ is expressed in AT II cells and Clara cells.12 We therefore hypothesized that PPARγ in AT II epithelial cells plays important roles in controlling pulmonary inflammation and associated pathogenesis. To test this hypothesis, a doxycycline-controlled CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mouse model was generated. In this model, the dominant-negative PPARγ was overexpressed to inactivate endogenous PPARγ in AT II epithelial cells.
Materials and Methods
Animal Care
All scientific protocols involving the use of animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University School of Medicine and followed guidelines established by the Panel on Euthanasia of the American Veterinary Medical Association. Protocols involving the use of recombinant DNA or biohazardous materials were approved by Biosafety Committee of Indiana University School of Medicine and followed guidelines established by the National Institutes of Health (NIH). Animals were housed under IACUC-approved conditions in a secured animal facility at Indiana University School of Medicine and were regularly screened for common pathogens. Experiments involving animal sacrifice used CO2 narcosis to minimize animal discomfort.
Generation of Doxycycline-Controlled dnPPARγ Transgenic Mice
To generate the (tetO)7-CMV-dnPPARγ transgenic mouse line, murine dnPPARγ cDNA was amplified by PCR using a downstream primer [5′-AAGGAAAAAAGCGGCCGCTTATCACTTGCTATCGTCGTCCTTGTATCATACAAGTCCTTGTA-3′, which contains the NotI site (underlined) and the Flag sequence] and an upstream primer [5′-CTAGACGCGTGCCACCATGGTTGACACACAGATG-3′, which contains the Mlu1 site (underlined) and the Kozak sequence]. The PCR product was digested with Mlu1/NotI and subcloned downstream of the cytomegalovirus (CMV) minimal promoter linked to seven Tet-responsive elements at the Mlu1 and NotI sites in the pTRE2 vector (Clontech, Mountain View, CA). The expression cassette containing the CMV minimal promoter, the dnPPARγcDNA, and the human globin polyadenylation signaling sequence was dissected out and purified for microinjection into FVB/N mice. Founder lines were identified by a pair of primers corresponding to a pTRE2 plasmid sequence (5′-ACGCTGTTTTGACCTCCATAGAAG-3′) and to a dnPPARγ cDNA coding region sequence (5′-AACTGTGGTAAAGGGCTTGATGTC-3′). The CCSP-rtTA transgenic mice were provided by Dr. Jay Tichelaar and Jeffery A. Whitsett (Cincinnati Children's Hospital Medical Center, Cincinnati, OH).13 CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were obtained by crossbreeding CCSP-rtTA transgenic mice and (tetO)7-CMV-dnPPARγ transgenic mice. As reported originally,13 the rat 2.3-kb CCSP (Clara cell secondary promoter) promoter region showed AT II cell specificity, instead of Clara cell specificity. We had confirmed this previously in the CCSP-rtTA/(tetO)7-CMV-Stat3C bitransgenic mouse model14 and in the CCSP-rtTA/(tetO)7-CMV-MMP12 bitransgenic mouse model.11 In general, animals were treated with doxycycline water (0.5 mg/mL) twice a week starting at 1 month of age, after genotyping.
FACS
Fluorescence-activated cell sorting (FACS) was performed as described previously.4 Briefly, peripheral blood mononuclear cells were obtained after red blood cell lysis with distilled water plus 10× PBS. To prepare cells from the lung, the mouse chest cavity was opened using surgical dissection, and the inferior vena cava and abdominal aorta were clamped. The left atrium was opened by incision, and the right ventricle was infused with 1× PBS to remove any residual blood in the pulmonary vasculature. The lung was cut into small pieces and placed in RPMI 1640 medium containing 5% fetal bovine serum, collagenase, and DNase (Sigma-Aldrich, St. Louis, MO). After 40 minutes of digestion at 37°C, lungs were further disrupted by aspiration through an 18-gauge needle and were suspended in FACS buffer. Approximately 1 × 106 to 2 × 106 cells from various organs in FACS buffer were stained with the cell surface markers anti-CD4, anti-CD8, anti-CD3, anti-B220, anti-CD11c, anti-CD11b, and anti-Gr-1 (eBiosciences, San Diego, CA).
For measurement of intracellular signaling molecules in AT II epithelial cells, cell suspensions from the lungs of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were fixed with 2% formaldehyde. After 20 minutes at room temperature, fixation samples were resuspended in methanol at a final 90% concentration for 2 hours at 4°C, and then washed and resuspended in 200 μL 1× permeabilization buffer. Cell suspensions were labeled with SP-C antibody and the primary phospho-specific antibodies at 4°C overnight, then washed in 1× PBS containing 4% fetal bovine serum and labeled using the secondary antibody. Anti-phospho-P44/42 (Erk1/2), anti-phospho-P38, anti-phospho-NF-κB, anti-phospho-Stat1, anti-phospho-Stat2, anti-phospho-Stat3, anti-phospho-Stat4, anti-phospho-Stat5, and anti-phospho-Stat6 antibodies were purchased from Cell Signaling Technology (Danvers, MA). SP-C antibody (FL-197) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Flag polyclonal antibody (F7425) was purchased from Sigma-Aldrich.
For measurement of intracellular signaling molecules in MDSCs, cell suspensions from bone marrow, spleen, blood, and lung of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were stained with CD11b and GR-1 antibodies, and then fixed with 2% formaldehyde. After 10 minutes at 37°C, fixation samples were resuspended in methanol at a final 90% concentration for 30 minutes at 4°C. Cell suspensions were labeled with phospho-specific antibodies at 4°C overnight and then analyzed by flow cytometry. Anti-phospho-P44/42 (Erk1/2), anti-phospho-P38, anti-phospho-NF-κB, anti-phospho-Akt, and anti-phospho-Stat3 antibodies were purchased from Cell Signaling Technology.
Cells were analyzed on an LSR II flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using BD FACStation software (BD CellQuest Pro version 5.2.1; BD Biosciences). The total number of positive cells was calculated as gated viable cells. Isotype controls IgG1, IgG2a, and IgG2b were included in all experiments. Quadrants were assigned using isotype controls.
Western Blot
Cells from the lung of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-dnPPARγ bitransgenic mice were prepared as described above. Protein extracts were prepared in radioimmunoprecipitation assay (RIPA) buffer and fractionated on a polyacrylamide gel (Invitrogen, Carlsbad, CA). After transferring to the immunoblot polyvinylidene difluoride membrane, anti-PPARγ antibody was used to hybridize with the membrane. Protein bands were visualized with a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). Anti-PPARγ antibody (SC-7196) was purchased from Santa Cruz Biotechnology.
Isolation of CD4+ T Cells and MDSCs
To purify CD4+ T cells, erythrocyte-depleted splenocytes were stained with anti-CD4 microbeads and passed over a mass spectrometry column according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). To purify CD11b+Gr-1+ MDSCs, single cells from the bone marrow, spleen, and lung were stained with anti-Gr-1+-biotin antibodies, followed by positive magnetic selection using anti-biotin microbeads according to the manufacturer's instructions (Miltenyi Biotec). The purity of the MDSC population was typically >90%.
In Vitro MDSC Suppression Assay
For the proliferation study, the sorted CD4+ T cells from wild-type spleen were labeled with 1 μmol/L carboxyfluorescein diacetate succinimidyl diester (CFSE; Molecular Probes, Eugene, Oregon) at 37°C for 10 minutes. Cells were pelleted and resuspended in complete medium for 40 minutes, followed by washing twice in PBS. CFSE-labeled CD4+ T cells were cultured in 96-well flat-bottom plates coated with anti-CD3 monoclonal antibody (mAb) (2 μg/mL) and anti-CD28 mAb (5 μg/mL). Purified MDSCs from bone marrow, spleen, and lung of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were added into wells. The ratio of MDSCs to CD4+ T cells was 1:5. After 4 days, cells were harvested, stained with APC-labeled anti-CD4 mAb (eBiosciences), and analyzed by flow cytometry.
For the CD69 expression study, MDSCs from the bone marrow, spleen, and lung of doxycycline-treated or untreated bitransgenic mice and wild-type T splenocytes were cocultured in 96-well plates coated with anti-CD3 mAb (2 μg/mL) and anti-CD28 mAb (5 μg/mL) for 48 hours. Cells were harvested, stained with anti-CD4-APC mAb and anti-CD69-PEcy7 mAb (eBiosciences), and analyzed by flow cytometry. The expression levels of IL-2, IL-4, and IFN-γ in the culturing supernatants were measured using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's instructions (BD Biosciences).
Alveolar Type II Epithelial Cell Purification
AT II epithelial cells were purified from wild-type mice and doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, as described previously.15 After purification, AT II epithelial cells were stained with anti-CD11b and Gr-1 antibodies to make sure there was no contamination by macrophages or neutrophils.
Reverse Transcription and Real-Time PCR
For reverse transcription reactions, total RNAs were extracted from whole lungs or AT II epithelial cells of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice using a Qiagen RNeasy mini kit according to the manufacturer's instruction (Qiagen, Valencia, CA). RNAs (2 μg) were reversely transcribed using Taqman Reverse Transcription Reagent (Applied Biosystems, Branchburg, NJ) in a GeneAmp 9700 thermocycler (Applied Biosystems, Foster City, CA) with cycling conditions as recommended by the manufacturer: 25°C for 10 minutes, 48°C for 30 minutes, and 95°C for 5 minutes. All reactions were performed using a TaqMan reverse transcription kit (Applied Biosystems).
For real-time PCR, 2 μL of cDNA was amplified by a pair of sequence-specific DNA oligonucleotide primers for each molecule in a 50-μL reaction mixture containing SYBR Green PCR master mix (Applied Biosystems). The housekeeping gene GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was an endogenous control in parallel PCR reactions. The reactions were analyzed using a relative quantification assay and a 7500 system sequence detection software package (7500 real-time PCR system version 1.3.1; Applied Biosystems). The default cycling protocol for this software was 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. Relative expression was determined by the 2ΔΔCt method, in which ΔΔCt = ΔCt(+Dox) − ΔCt(−Dox). ΔCt represents the PCR cycle threshold (Ct) of a testing molecule normalized by the Ct of GAPDH, as ΔCt = Ct(testing molecule) – Ct(GAPDH). Primers for real-time PCR are listed in Table 1.
Table 1.
Primers for Real-Time PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| mIFN-α | 5′-AAGGACAGGAAGGATTTTGGATT-3′ | 5′-GAGCCTTCTGGATCTGTTGGTT-3′ |
| mIFN-γ | 5′-CAAGGCGAAAAAGGATGCAT-3′ | 5′-CTGGACCTGTGGGTTGTTGAC-3′ |
| mIL-1β | 5′-TTGACGGACCCCAAAAGATG-3′ | 5′-CAGGACAGCCCAGGTCAAA-3′ |
| mIL-6 | 5′-GAGGCTTAATTACACATGTTC-3′ | 5′-TGCCATTGCACAACTCTTTTCT-3′ |
| mIL-13 | 5′-AGCAACATCACACAAGACCAGACT-3′ | 5′-CCAGGTCCACACTCCATACCA-3′ |
| mMMP-2 | 5′-GAGGCTGACATCATGATCAACTTT-3′ | 5′-GCCATCAAATGGGTATCCATCT-3′ |
| mMMP-7 | 5′-TGAGGACGCAGGAGTGAACTT-3′ | 5′-CCCAGAGAGTGGCCAAATTC-3′ |
| mMMP-8 | 5′-CTCAAGAGACCATGGTGACAATTC-3′ | 5′-AAGGCATGGGCAAGGATTC-3′ |
| mMMP-9 | 5′-GAGGCTGACATCATGATCAACTTT-3′ | 5′-GCCATCAAATGGGTATCCATCT-3′ |
| mMMP-12 | 5′-TGGTATTCAAGGAGATGCACATTT-3′ | 5′-GGTTTGTGCCTTGAAAACTTTTAGT-3′ |
| mTNF-α | 5′-CCCCAAAGGGATGAGAAGTTC-3′ | 5′-TGAGGGTCTGGGCCATAGAA-3′ |
| GAPDH | 5′-GGCCATCAAGCCAGAGCTT-3′ | 5′-CCAAACCATCACTGACACTCAGA-3′ |
Bronchioalveolar Lavage Fluid Collection and Kwik-Diff Staining
Bronchioalveolar lavage fluid (BALF) was collected by 1-mL 1× PBS wash and repeated three times. Cells were centrifuged at 1000 rpm on a Thermo CR4i Centrifuge (Asheville, NC) for 5 minutes and resuspended into 0.5 mL 1× PBS. Total cell number in BALF was determined by hemocytometry. The same volume of cell suspension was spun onto slides using a centrifuge and stained with a Kwik-Diff stain kit (Thermo Shandon, Pittsburgh, PA).
Cytokine Measurement by ELISA
BALF was obtained from the lungs of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. The expression levels of IL-1β, IL-6, and TNF-α in BALF were measured using DuoSet ELISA kits for mouse IL-1β, IL-6, and TNF-α according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
ChIP Assay
Following a procedure described previously,16 single cells were isolated from the lungs of 2-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Proteins were cross-linked to DNA by adding formaldehyde directly to the RPMI 1640 culture medium to a final concentration of 1%, followed by incubation for 10 minutes at room temperature. The cells were washed with 1× PBS three times and then lysed with SDS lysis buffer (0.1% SDS, 3 mmol/L EDTA, 25 mmol/L Tris pH 8.1, 1× protease inhibitor cocktail). Cells were sonicated for 10 seconds three times at 30-second intervals. A fraction of 1% sonicated chromatin was saved from each sample for input normalization of real-time PCR. The remaining chromatin samples were incubated with 20 μg of anti-Flag polyclonal antibody (Sigma-Aldrich) overnight at 4°C. Protein A agarose beads were added to adsorb the immunocomplexes, with rotating at 4°C for 2 hours. DNA-protein immunocomplexes were eluted from the beads with chromatin immunoprecipitation (ChIP) elution buffer (1% SDS, 0.1 mol/L NaHCO3) and were de-crosslinked at 65°C for 4 hours. DNA was isolated by chloroform-phenol extraction. For real-time PCR, 2 μL of DNA as described above was amplified by a pair of sequence-specific DNA oligonucleotide primers for each gene promoter (as listed below) in a 50-μL reaction mixture containing SYBR Green PCR master mix (Applied Biosystems). The reactions were analyzed using a relative quantification assay and 7500 system software. All PCR reactions were normalized by input DNA fraction according to ΔCt = Ct(testing) − (Ct(input) − log2(input dilution factor) as described by the manufacturer (ChampionChIP qPCR Primers User Manual; SABiosciences, Frederick, MD).
Based on the DNA binding sequence (5′-(A/G)GG(T/G)CA(A/G)AGG(T/G)CA-3′) for PPARγ, the putative PPARγ binding sites were identified on the promoters of the Api6 (ENSMUSG00000015854), IL-1β (ENSMUSG00000027398), IL-6 (ENSMUSG00000025746), MMP12 (ENSMUSG00000049723), and TNF-α (ENSMUSG00000024401) genes according to the Ensembl database (http://www.ensembl.org/index.html), using MacVector 8.1.2 software (MacVector, Cary, NC).
Primers for ChIP-real-time PCR were as follows: mApi-6: 5′-GGAAGCTGGTTGAAGGTAGGAA-3′ (upstream) and 5′-GTAAGTCACGATTGTGGCATCTATCT-3′ (downstream); mIL-1β: 5′-CACTATCTGCCACCCCTTGAC-3′ (upstream) and 5′-GAAGAGGCTATTGCTACCCTGAAA-3′ (downstream); mIL-6: 5′-TGGGATCAGCACTAACAGATAAGG-3′ (upstream) and 5′-TGGTCTCTTGGCTATCTTCTTAGTTAAG-3′ (downstream); mMMP12: 5′-GCAGAAAAATTGAAATGGGTAAAGA-3′ (upstream) and 5′-TGGGTTGCTTTGGGAGGTATT-3′ (downstream); mTNF-α: 5′-CCCAGATTGCCACAGAATCC-3′ (upstream) and 5′-CCTACACCTCTGTCTCGGTTTCTT-3′ (downstream).
Lung Histology and Morphometry
The trachea was cannulated and the cannula was tied firmly in place. The lungs were filled through the trachea with 4% paraformaldehyde in PBS at 20 cm H2O pressure by gravity and were maintained overnight at 4°C. The lungs were monitored for leakage during this procedure at 4°C for approximately 24 hours. Only lungs that did not leak were used for further study. The lungs were washed with PBS and dehydrated through an ethanol series, followed by paraffin embedding. Sections (5 μm) were mounted on glass slides and deparaffinized. The adult lung slides were washed in a series of xylene and ethanol to remove paraffin from the tissues. Multiple sections from each lung were stained with H&E. For quantitative characterization, images at ×20 original magnification were transferred by video camera to a computer for viewing on-screen. The measurements of alveolar number, alveolar mean cord length (Lm), alveolar area, alveolar sphere surface area, and alveolar volume were determined by MetaMorph imaging software version 7.1.2.0 (Molecular Devices, Sunnyvale, CA) by selecting appropriate functions. Twenty randomly selected microscopic fields of respiratory alveoli were analyzed.
MMP Activity
BALF was obtained from the lungs of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ mice for MMP activity assay. MMP9 and MMP12 specific activities were measured by fluorescence quenched substrate cleaving using a SensoLyte 490 MMP assay kit (AnaSpec, San Jose, CA). This kit was optimized to detect the activities of MMPs in biological samples using a fluorescence quenched substrate (EDANS/DabcylPlus FRET peptide; AnaSpec) at excitation and emission wavelengths of 340 nm and 490 nm, respectively. Each BALF sample was incubated with 5 μL 1 mmol/L 4-aminophenylmercuric acetate (APMA) for 2 hours at 37°C to activate MMPs. Samples (50 μL) were then added to a 96-well plate, and a standard curve was set up using a serial dilution of purified MMP9 and MMP12 protein (AnaSpec) at concentrations of 1.6 ng/μL, 0.8 ng/μL, 0.4 ng/μL, 0.2 ng/μL, 0.1 ng/μL, 0.05 ng/μL, and 0.0125 ng/μL. MMP12 substrate solution (50 μL per well, 1×) was added to the samples and the standards. The plate was mixed gently and incubated at room temperature for 1 hour and protected from light. Stop solution from AnaSpec (50 μL) was added to each well, and the fluorescence intensity was measured using a VICTOR3 fluorescence plate reader (Perkin Elmer, Waltham, MA) at 490 nm after excitation at 340 nm. MMP9 or MMP12 purified enzyme (Sigma-Aldrich) was used as positive control.
Bone Marrow Stem Cell Injection and Double-Immunofluorescence Staining
For bone marrow mesenchymal stem cells (BMSCs), we followed a procedure described previously.17 BMSCs were isolated from femur and tibia of hSP-B 1.5kb lacZ transgenic mice, 2 to 3 months old,18 and then were cultured in Mesencult MSC basal medium (StemCell Technologies, Vancouver, BC, Canada) at 37°C with 5% CO2. After 1 day, the unattached cells were discarded and, for attached BMSCs, the medium was completely replaced. Thereafter, half the medium was changed every 3 days until confluence was reached. BMSCs were passaged five to six times with split, to amplify the cell population. Each passage took 1 week to reach confluence.
Cultured BMSCs were harvested and resuspended in 1× PBS (1 × 106 BMSCs/100 μL). Each mouse was infused with 200 μL BMSCs through the tail vein. The same procedure was repeated 2 weeks later. After 8 weeks of initial injection, AT II epithelial cells were isolated from recipient mice and double-stained with rabbit anti-proSP-C antibody and mouse anti-β-galactosidase antibody. A Cy2-conjugated donkey anti-rabbit IgG and a Cy3-conjugated donkey anti-mouse IgG were used as the secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Labeled cells were analyzed by fluorescence microscopy or flow analysis. DAPI was used for nuclear staining.
Statistical Analysis
Data are reported as means ± SD of at least three independent experiments. A paired Student's t-test or analysis of variance was used to evaluate the significance of the differences. Statistical significance was set at P < 0.05.
Results
Generation of CCSP-rtTA/(tetO)7-CMV-dnPPARγ Bitransgenic Mice
A doxycycline-inducible bitransgenic mouse model was generated to specifically direct dnPPARγ expression in lung epithelial cells. In this system, a previously established CCSP-rtTA transgenic mouse line13 was crossbred with a newly generated (tetO)7-CMV-dnPPARγ transgenic mouse line. The PPARγ mutant form contains two substitutions (L468A and E471A) in the AF-2 domain and acts as a dominant negative (dn) form.19 A Flag sequence was added at the C terminus of the dnPPARγ cDNA to distinguish it from the endogenous PPARγ molecule. CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were successfully obtained, as detected by PCR using specific primers and mouse tail DNA (Figure 1A). Induction of dnPPARγ mRNA expression was achieved by doxycycline treatment for 1 month, monitored by real-time PCR assay in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, compared with untreated mice (Figure 1B).
Figure 1.
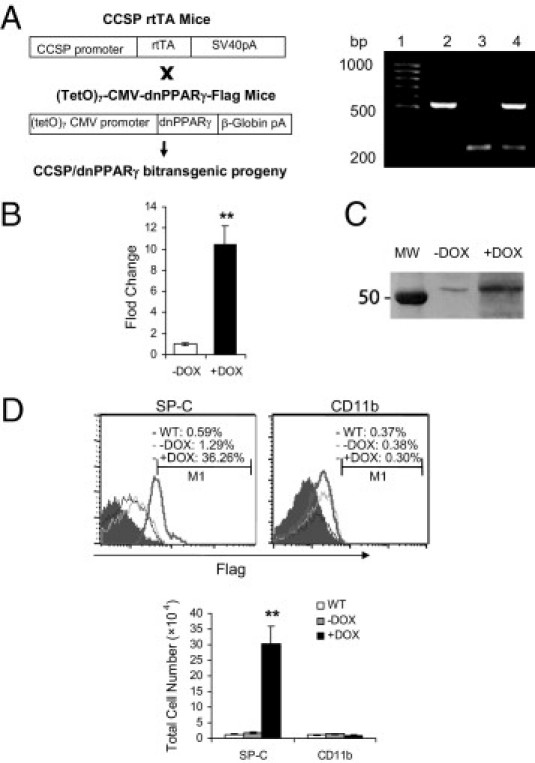
Generation of doxycycline-inducible CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: Construct maps and genotyping for CCSP-rtTA transgenic mice, (TetO)7-CMV-dnPPARγ transgenic mice, and CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Lane 1, DNA molecular marker; lane 2, CCSP-rtTA transgenic mouse; lane 3, (tetO)7-CMV-dnPPARγ transgenic mouse; lane 4, CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mouse. B: Real-time PCR analysis of the dnPPARγ mRNA expression level in the lungs of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice that were treated with doxycycline (+DOX) or untreated (−DOX) for 4 months (n = 4 to 6). Data are reported as means ± SD. **P < 0.01. C: Western blot analysis of PPARγ and dnPPARγ protein expression levels in the lungs of doxycycline-treated (+DOX) or untreated (−DOX) CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. MW, molecular weight marker. D: Expression of the dnPPARγ-Flag fusion protein in the AT II epithelial cells from the lungs of wild-type mice (WT), untreated bitransgenic mice (−DOX), or doxycycline-treated (+DOX) bitransgenic mice (n = 4 to 6) assessed by flow cytometry. Treatment duration: 4 months. The cells were double-labeled with anti-Flag and anti-SP-C antibodies (AT II marker). CD11b is a macrophage marker and served as the negative control. Data are reported as means ± SD. **P < 0.01.
Western blot analysis with PPARγ antibody showed induced expression of dnPPARγ over PPARγ in the lung of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Figure 1C). A basal level of endogenous PPARγ expression was also detected in the lung of doxycycline-untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. To further assess whether dnPPARγ-Flag fusion protein was induced in AT II epithelial cells, flow cytometry was performed to double-label lung single cells using anti-SP-C (AT II epithelial cell marker) and anti-Flag antibodies. Expression of the dnPPARγ-Flag fusion protein was readily detected in SP-C+ AT II epithelial cells using an anti-Flag antibody in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, but not in alveolar CD11b+ macrophages (Figure 1D). No dnPPARγ-Flag fusion protein was detected in SP-C+ AT II epithelial cells from doxycycline-untreated bitransgenic mice. The same negative observation was also made in CCSP-rtTA single transgenic mice (see Supplemental Figure S1A at http://ajp.amjpathol.org).
Overexpression of dnPPARγ Induces Emphysema in the Lung of Bitransgenic Mice
To assess pathogenic phenotypes in the lung as a result of dnPPARγ overexpression in AT II epithelial cells, CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice with 4 months of doxycycline treatment were investigated by histological analysis. Histopathological analysis revealed emphysema in the lung of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Figure 2A). Quantitative analysis with MetaMorph imaging software showed that alveolar numbers of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ mice (65 ± 9) were much less than those of untreated bitransgenic mice (122 ± 13) (Figure 2B). The mean cord length (Lm), alveolar area, alveolar sphere surface area, and alveolar volume were all significantly increased in doxycycline-treated bitransgenic mice, compared with untreated bitransgenic mice (Figure 2C).
Figure 2.
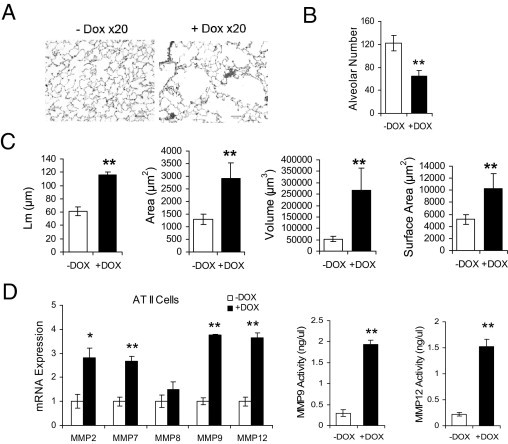
Histopathology and up-regulation of MMPs in the lung of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: H&E staining of lung tissue sections from representative doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice with emphysema. Treatment duration: 4 months. B: Quantitative measurement of the alveolar numbers by MetaMorph imaging software. Data are reported as means ± SD (n = 6). **P < 0.01. C: Quantitative measurement of mean cord length (Lm), alveolar area, alveolar volume, and alveolar sphere surface area were determined by MetaMorph imaging software. Data are reported as means ± SD (n = 6). **P < 0.01. D: MMP mRNAs from AT II epithelial cells of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were analyzed by real-time PCR (n = 4 to 6). The enzymatic activities of MMP9 and MMP12 in BALF from doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were analyzed by an MMP assay kit (n = 4 to 6). Treatment duration: 4 months. Data are reported as means ± SD. *P < 0.05. **P < 0.01; −DOX, doxycycline-untreated bitransgenic mice. +DOX, doxycycline-treated bitransgenic mice.
MMPs are major tissue remodeling enzymes. Their up-regulation is associated with emphysema formation and progression.20 In 4-month doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, mRNA expression levels of MMP2, -7, -8, -9, and -12 were highly induced in AT II epithelial cells (Figure 2D). No change was observed in CCSP-rtTA single transgenic mice (see Supplemental Figure S1B at http://ajp.amjpathol.org). In our previous study, at least MMP12 was demonstrated to be a direct PPARγ negatively regulated downstream target gene in lung epithelial cells.2 To determine whether MMP concentrations were increased in BALF, enzymatic activities of MMP9 and MMP12 were analyzed. BALF from 4-month doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice was collected. The enzymatic activities were analyzed by cleavage of a fluorescence quenched substrate. The MMP9 specific activity was significantly higher in doxycycline-treated bitransgenic mice (1.93 ± 0.1 ng/μL) than in untreated control mice (0.30 ± 0.08 ng/μL). The MMP12 specific activity was also significantly higher in doxycycline-treated bitransgenic mice (1.52 ± 0.14 ng/μL) than in untreated control mice (0.22 ± 0.03 ng/μL) (Figure 2D). No statistically significant change was observed in CCSP-rtTA single transgenic mice (see Supplemental Figure S1C at http://ajp.amjpathol.org). Thus, MMP synthesis and secretion were induced by inactivation of PPARγ in AT II epithelial cells that contributed to emphysema formation.
ChIP of Proinflammatory Genes in AT II Epithelial Cells of Bitransgenic Mice
Because PPARγ is an anti-inflammatory molecule, it was important to evaluate whether the proinflammatory molecules were up-regulated in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. These molecules are potentially important for triggering and recruiting immune cells into the lung to initiate inflammation and emphysema. PPARγ binds to specific DNA sites (PPAR E) on the promoters after ligand binding and prevents transcriptional events of proinflammatory molecules. Because the Api6, IL-1β, IL-6, MMP12, and TNF-α genes are downstream of PPARG,6–8 it is possible that overexpression of dnPPARγ in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice interferes with endogenous PPARγ function at the transcriptional level by competition, with DNA binding on the same PPAR E sites of these genes' promoters. To test this assumption, ChIP assay was performed to investigate the association between dnPPARγ and PPAR E sites within the Api6, IL-1β, IL-6, MMP12, and TNF-α promoters in 2-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic lung. Pathologically, overexpression of MMP12 and Api6 in mice caused chronic inflammation, emphysema, and bronchioalveolar adenocarcinoma.10,11 The putative PPAR E sites within the promoters of Api6, IL-1β, IL-6, MMP12, and TNF-α were identified (Figure 3A). Anti-Flag polyclonal antibody was used to immunoprecipitate the dnPPARγ-Flag/DNA complex. In doxycycline-treated mice, the signals of precipitated promoter DNA were stronger than those of untreated controls, according to quantitative real-time PCR analysis (Figure 3B). The results showed that dnPPARγ-Flag fusion protein was able to bind to the promoters of these proinflammatory genes.
Figure 3.
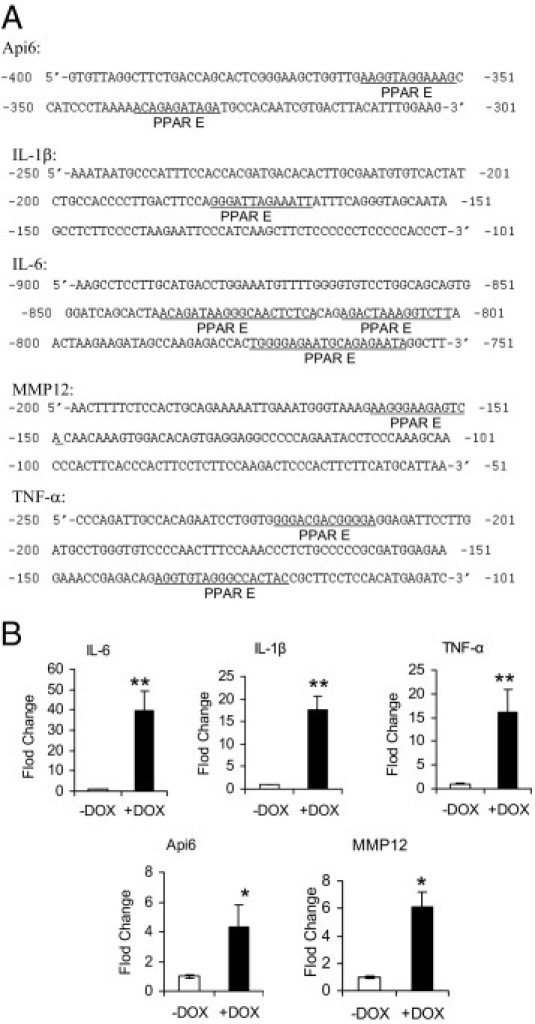
ChIP assay of dnPPARγ-Flag fusion protein on the promoters of Api6, IL-1β, IL-6, MMP12 and TNF-α inflammatory molecule genes in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: The amplified regions that contain putative PPARγ DNA binding sites (PPAR E) within the promoters of genes encoding Api6, IL-1β, IL-6, MMP12, and TNF-α. B: Statistical real-time PCR analyses of ChIP assay from dnPPARγ-Flag fusion protein and promoters of Api6, IL-1β, IL-6, MMP12, and TNF-α (mice, n = 4 to 6). Relative binding levels were determined by 2(−ΔΔCt), in which ΔΔCt = ΔCt(+DOX) − ΔCt(−DOX). The testing sample Ct value was normalized by each input DNA fraction Ct value, as ΔCt = Ct(testing) − (Ct(input) − log2(input dilution factor)). Data are reported as means ± SD. *P < 0.05; **P < 0.01. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
Overexpression of Proinflammatory Molecules in the Lung of Bitransgenic Mice
To determine whether dnPPARγ overexpression indeed up-regulates proinflammatory cytokines in vivo, total RNAs were purified from AT II epithelial cells of 2-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. The expression levels of multiple cytokines were quantitatively determined by real-time PCR. Among these, mRNA expression levels of IFN-α, IFN-γ, IL-1β, IL-6, IL-13, and TNF-α in doxycycline-treated mice were increased, compared with doxycycline-untreated mice (Figure 4A). No change was observed in CCSP-rtTA single transgenic mice (see Supplemental Figure S1D at http://ajp.amjpathol.org). In BALF, protein concentrations of secreted IL-1β, IL-6, and TNF-α were steadily increased as measured by ELISA, compared with untreated mice (Figure 4B).
Figure 4.
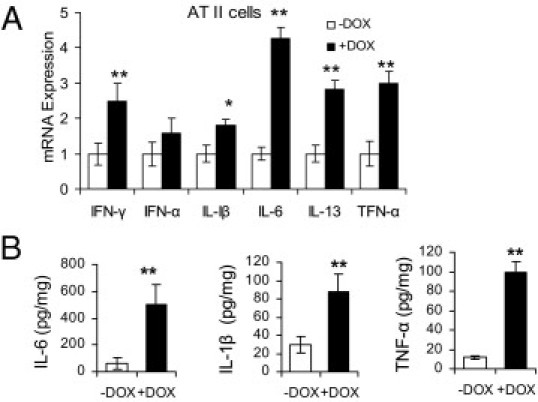
Overexpression of inflammatory cytokines in AT II epithelial cells and BALF of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: Real-time PCR analysis of mRNA expression levels of inflammatory cytokines in AT II epithelial cells of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice that were treated or untreated with doxycycline for 2 months (n = 5). The mRNA expression level of GAPDH was used for normalization. Therefore, ΔCt = Ct(testing) − Ct(GAPDH). Relative expression levels were determined by the 2−ΔΔCt method, in which ΔΔCt = ΔCt(+DOX) − ΔCt(−DOX). Data are reported as means ± SD. *P < 0.05; **P < 0.01. B: The protein expression levels of IL-1β, IL-6, and TNF-α in BALF from the lungs of 4-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice by ELISA (n = 5 to 6). Data are reported as means ± SD. **P < 0.01. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
Overexpression of dnPPARγ Increases Myeloid Cell Infiltration in the Lung of Bitransgenic Mice
Up-regulation of proinflammatory cytokines suggests that overexpression of dnPPARγ may induce inflammation that contributes to emphysema. To determine whether overexpression of dnPPARγ induces inflammatory cell infiltration in the lung of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice as a result of proinflammatory cytokine up-regulation, peripheral blood mononuclear cells and lung mononuclear cells were isolated from doxycycline-treated or untreated mice (treatment duration of 1, 4, or 8 months) and stained with fluorochrome-conjugated anti-mouse CD11b (for monocytes) and Gr-1 (for neutrophils) antibodies for FACS analysis. Compared with doxycycline-untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, the absolute numbers and (excepting 1 month treatment) percentage numbers of immature CD11b+GR-1+ MDSCs were steadily increased in the blood and lung of 1-month, 4-month, and 8-month doxycycline-treated mice (Figure 5, A and B). No change was observed in CCSP-rtTA single transgenic mice (see Supplemental Figure S1E at http://ajp.amjpathol.org). Pathologically, clustered inflammatory cell accumulation was observed in some lung areas of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Figure 5C). IHC showed positive staining of myeloid-specific Mac2 antibody on these cells. Kwik-Diff staining showed significant increase of inflammatory cells in the BALF of doxycycline-treated mice (Figure 5D), which resembled the morphological characteristics of MDSCs.4,21,22 This suggests that myeloid cells were blocked at the immature stage from differentiating into mature macrophages and neutrophils after dnPPARγ overexpression in AT II epithelial cells. This is similar to observations in lal−/− mice.4
Figure 5.
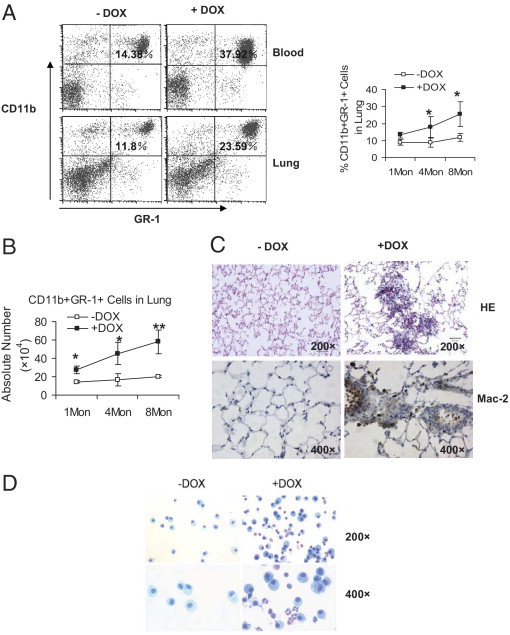
Myeloid cell populations in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Single-cell suspensions were prepared from the blood and lung of 1-month, 4-month, and 8-month doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: The percentage number of Gr-1+CD11b+ cell population was analyzed by FACS. A representative dot-plot analysis of 4-month doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice is presented. Significant differences between doxycycline-treated and untreated groups at various time points were identified by two-way analysis of variance. Data are reported as means ± SD (n = 4 to 6). *P < 0.05. B: Absolute numbers of total Gr-1+CD11b+ cells from the lungs of doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Treatment duration: 1 month, 4 months, or 8 months (1Mon, 4Mon, 8Mon). Significant differences between doxycycline-treated and untreated groups at various time points were identified by two-way analysis of variance. Data are reported as means ± SD (n = 4 to 6). *P < 0.05; **P < 0.01. C: H&E staining (HE) and immunohistochemical staining with Mac 2 antibody (Mac-2) of lung tissue sections from representative 4-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice with inflammatory cell infiltration. Original magnification as indicated on each image. D: Kwik-Diff staining of BALF cells from 11-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice. Original magnification is indicated for each row.
Overexpression of dnPPARγ Suppresses T-Cell Proliferation in Bitransgenic Mice
To assess T-cell proliferation, blood and lung mononuclear cells were isolated from 1-month, 4-month, and 8-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice and stained with fluorochrome-conjugate anti-mouse CD3, CD4, and CD8 antibodies for FACS analysis. In contrast to myeloid cells, the percentage numbers of CD3+ cells in the blood and lung were significantly reduced in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, compared with doxycycline-untreated controls (Figure 6, A and C). Similarly, the percentage numbers of CD4+ and CD8+ cells in the blood and lung were reduced in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Figure 6, B and C). No change was observed in CCSP-rtTA single transgenic mice (see Supplemental Figure S1, F and G, at http://ajp.amjpathol.org). In these assays, the absolute numbers of CD3+, CD4+, and CD8+ T cells were also reduced (Figure 6D). In some of the 1-month doxycycline-treated samples, the differences of percentage numbers and absolute numbers between treated and untreated CD3+, CD4+, and CD8+ T cells showed no statistical significance. This is again similar to observations in lal−/− mice.4
Figure 6.
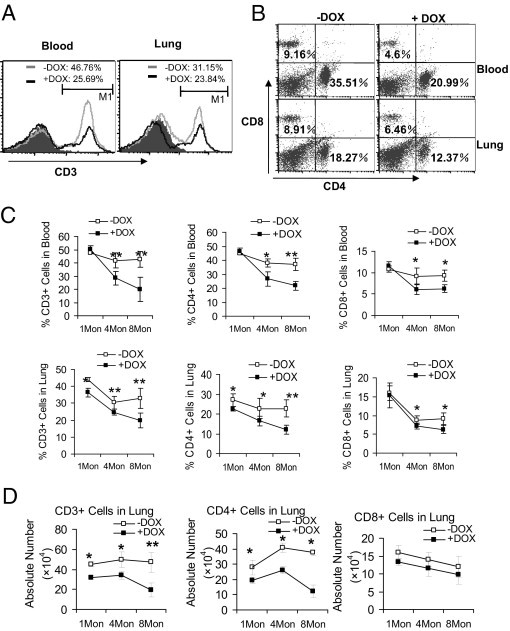
T-cell populations in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Single-cell suspensions were prepared from the blood and lung of 1-month, 4-month, and 8-month (1Mon, 4Mon, 8Mon) doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: The percentage number of CD3+ cell population was analyzed by FACS. A representative histogram analysis of 4-month doxycycline-treated and untreated mice is presented. B: The percentage numbers of CD4+, and CD8+ cell populations were analyzed by FACS. A representative dot-plot analysis of 4-month doxycycline-treated and untreated mice is presented. C: Percentage numbers of total CD3+, CD4+, and CD8+ cells from the blood and lungs of doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Treatment duration: 1 month, 4 months, or 8 months (1Mon, 4Mon, 8Mon). Significant differences between doxycycline-treated and untreated groups at various time points were identified by two-way analysis of variance. Data are reported as means ± SD (n = 4 to 6). *P < 0.05; **P < 0.01. D: Absolute numbers of total CD3+, CD4+, and CD8+ cells from the lungs of 1-month, 4-month, and 8-month (1Mon, 4Mon, 8Mon) doxycycline-treated and untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. Significant differences between doxycycline-treated and untreated groups at various time points were identified by two-way analysis of variance. Data are reported as means ± SD (n = 4 to 6). *P < 0.05; **P < 0.01. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice
MDSCs Inhibit T-Cell Proliferation and Function
In lal−/− mice, T-cell population reduction and functional impairment was due in part to MDSC suppression.4 To measure the effect of CD11b+Gr-1+ cells on T-cell proliferation in CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, CFSE-labeled wild-type splenic CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAb in the presence of CD11b+Gr-1+ cells that were isolated from the bone marrow, spleen, and lung of doxycycline-treated or untreated mice. After 4 days coculturing, T-cell proliferation was measured by CFSE labeling dilution and flow cytometry. T cell receptor-stimulated T-cell proliferation (represented by multiple peaks) was suppressed by CD11b+Gr-1+ cells from the lung of doxycycline-treated bitransgenic mice, compared with CD11b+Gr-1+ cells of untreated bitransgenic mice (Figure 7A). In the PBS negative control group, no proliferation was observed. There was no change in CD4+ T-cell proliferation in the CFSE labeling study under coculturing with MDSCs from the bone marrow or spleen, regardless of doxycycline treatment (data not shown). Thus, the defect caused by dnPPARγ overexpression in AT II epithelial cells was restricted to the lung.
Figure 7.
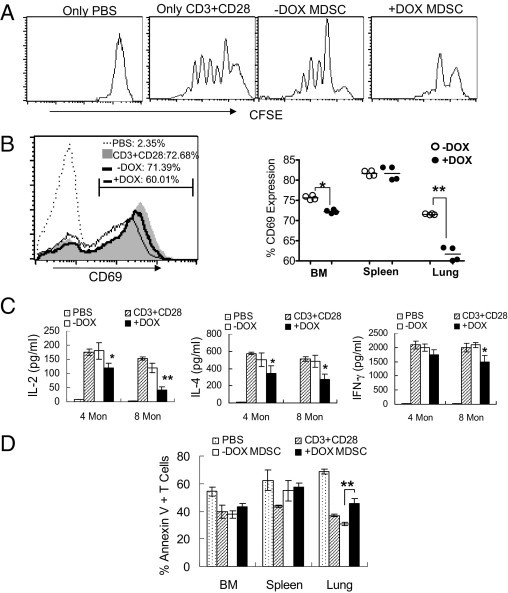
The suppressive function of MDSCs on CD4+ T cells. A: CFSE-labeled wild-type splenic CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAb for 4 days in the presence or absence of CD11b+Gr-1+ MDSCs from the lung of doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. The ratio between MDSCs and splenic CD4+ T cells was 1:5. Proliferation of labeled CD4+ T cells was analyzed by flow cytometry. Peaks represent cell division cycles. PBS was negative control. B: Wild-type CD4+ T cells were stimulated with anti-CD3 and anti-CD28 mAb for 2 days in the presence or absence of CD11b+Gr-1+ MDSCs from doxycycline-treated or untreated bitransgenic mice. Cells were stained with anti-CD4 and anti-CD69 antibodies for flow cytometry analysis. B, left: A representative study of lung MDSCs on CD69 expression. Dotted bar: PBS stimulation negative control. Shaded bar: anti-CD3 plus anti-CD28 mAb stimulation positive control. White bar: MDSCs from doxycycline-untreated bitransgenic mice. Black bar: MDSCs from doxycycline-treated bitransgenic mice. B, right: Statistical analyses of CD69 expression by MDSCs from the bone marrow, spleen, and lung of doxycycline-treated or untreated bitransgenic mice. The percentage number of CD69 in CD4+ T cells is presented as mean ± SD (n = 4 to 6). *P < 0.05; **P < 0.01. C: The concentrations of IL-2, IL-4, and IFN-γ in the above cultured medium were measured by ELISA analysis. Significant differences between doxycycline-treated and untreated groups at various time points were identified by two-way analysis of variance. Data are reported as means ± SD (n = 4 to 6). *P < 0.05; **P < 0.01. D: Prepared as described for panel A, CD4+ T cells were labeled with annexin V and CD4 antibodies for flow cytometry analysis. The percentage of annexin V-labeled CD4+ T cells is reported as means ± SD (mice, n = 5). *P < 0.05; **P < 0.01. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
A similar observation was made in the expression assay for the T-cell activation marker CD69. After 48 hours coculturing, CD11b+Gr-1+ cells from the lung of doxycycline-treated bitransgenic mice suppressed anti-CD3 Ab plus anti-CD28 Ab-stimulated CD69 expression, compared with CD11b+Gr-1+ cells of untreated bitransgenic mice (Figure 7B). In this assay, CD11b+Gr-1+ cells from the bone marrow of doxycycline-treated bitransgenic mice showed slight suppression on CD69 expression, whereas CD11b+Gr-1+ cells from the spleen of doxycycline-treated bitransgenic mice showed no suppression. In functional analysis, secretion of lymphokines was measured in MDSCs/CD4 T-cell coculturing medium by ELISA. CD11b+Gr-1+ cells from the lungs of doxycycline-treated mice showed reduced secretion of IL-2, IL-4, and IFN-γ after T-cell receptor activation, compared with CD11b+Gr-1+ cells of untreated bitransgenic mice (Figure 7C).
To determine whether CD4+ T-cell reduction is associated with apoptosis, CD4+ T cells were analyzed by annexin V staining. Only CD4+ T cells that were cocultured with CD11b+Gr-1+ cells from the lungs of doxycycline-treated bitransgenic mice showed increased apoptotic activity, compared with CD11b+Gr-1+ cells of untreated bitransgenic mice (Figure 7D). Thus, dnPPARγ-induced myeloid defect had a profound influence on T-cell proliferation and function.
Overexpression of dnPPARγ Activates Intracellular Signaling Molecules in the Lung of Bitransgenic Mice
In addition to immune response, nonimmune responses were investigated, such as intracellular signaling pathways that are activated by dnPPARγ overexpression in AT II epithelial cells. Single cells from the lung of 4-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice were costained with fluorochrome-conjugated SP-C antibody (AT II epithelial marker) and anti-phospho-Stat1, anti-phospho-Stat2, anti-phospho-Stat3, anti-phospho-Stat4, anti-phospho-Stat5, anti-phospho-Stat6, anti-phospho-Erk1/2, or anti-phospho-p38 antibody. Costained cells were analyzed by flow cytometry. Antibodies against these proteins were also included for total protein measurement by flow cytometry, for normalization. In the present study, AT II epithelial cells with activation of Stat1, Stat2, Stat3, Stat4, Erk1/2, and p38 and deactivation of Stat5 and Stat6 were observed in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Table 2). No change was observed in CCSP-rtTA single transgenic mice (see Supplemental Table S1 at http://ajp.amjpathol.org). These results indicate that inactivation of the PPARγ pathway not only up-regulated proinflammatory cytokines but also activated a unique set of intracellular signaling molecules in AT II epithelial cells. When CD11b+Gr-1+ MDSCs were analyzed, activation of Stat3, Erk1/2, and p38 was observed in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, compared with untreated bitransgenic mice (Table 3).
Table 2.
Phosphorylated Activation of Intracellular Signaling Molecules in Lung Alveolar Type II Epithelial Cells from CCSP-rtTA/(tetO)7-CMV-dnPPARγ Bitransgenic Mice
| Percentage ratio (%) |
MFI ratio (%) |
|||
|---|---|---|---|---|
| −DOX | +DOX | −DOX | +DOX | |
| pStat1 | 10.37 ± 3.18 | 47.32 ± 5.92⁎⁎ | 22.42 ± 9.18 | 66.65 ± 16.11⁎⁎ |
| pStat2 | 9.54 ± 4.23 | 33.47 ± 6.12⁎⁎ | 12.62 ± 2.79 | 27.78 ± 5.45⁎⁎ |
| pStat3 | 6.47 ± 2.1 | 39.01 ± 8.31⁎⁎ | 11.22 ± 3.13 | 41.22 ± 7.22⁎⁎ |
| pStat4 | 11.18 ± 2.88 | 42.11 ± 4.35⁎⁎ | 11.90 ± 2.74 | 14.72 ± 3.85 |
| pStat5 | 23.79 ± 7.15 | 9.50 ± 4.69⁎ | 30.09 ± 10.65 | 13.48 ± 3.01⁎ |
| pStat6 | 25.81 ± 6.8 | 10.07 ± 4.77⁎ | 20.20 ± 5.22 | 18.48 ± 4.52 |
| pErk | 10.87 ± 1.55 | 33.78 ± 3.33⁎⁎ | 18.86 ± 3.96 | 51.30 ± 7.76⁎⁎ |
| pP38 | 14.51 ± 3.56 | 41.11 ± 10.49⁎⁎ | 24.79 ± 5.56 | 59.05 ± 9.55⁎⁎ |
−DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
Cells isolated from the lung were stained with anti-SP-C antibody, followed by intranuclear staining with anti-phospho-Stat1-6, anti-phospho-Erk, or anti-phospho-P38 antibody, and Stat1-6, Erk, or P38 antibody as described under Materials and Methods. Numbers of percentage ratio and mean fluorescent intensity (MFI) ratio between phospho-proteins and total proteins in gated SP-C positive cells from four independent experiments (n = 4) were calculated and statistically analyzed.
P < 0.05;
P < 0.01.
Table 3.
Phosphorylated Activation of Intracellular Signaling Molecules in Myeloid-Derived Suppressor Cells from CCSP-rtTA/(tetO)7-CMV-dnPPARγ Bitransgenic Mice
| Blood |
Lung |
|||
|---|---|---|---|---|
| −DOX | +DOX | −DOX | +DOX | |
| pStat3 | 11.24 ± 3.12 | 39.78 ± 6.31⁎ | 16.87 ± 4.15 | 20.1 ± 3.12 |
| pErk | 13.58 ± 4.88 | 40.85 ± 5.87⁎ | 12.32 ± 3.23 | 33.55 ± 4.45⁎ |
| pP38 | 15.46 ± 4.37 | 27.46 ± 4.04⁎ | 16.11 ± 4.1 | 27.05 ± 2.13⁎ |
−DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
Cells isolated from the blood and lung were stained with anti-CD11b and GR-1 antibodies, followed by intranuclear staining with anti-phospho-Stat3, anti-phospho-Erk, or anti-phospho-P38 antibody, and Stat3, Erk, or P38 antibody as described under Materials and Methods. Numbers of percentage ratio between phospho-proteins and total proteins in gated CD11b and Gr-1 double positive cells from four independent experiments (n = 4) were calculated and statistically analyzed.
P < 0.05.
It appears that inflammatory molecules induced by dnPPARγ overexpression in the milieu of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic lung activated oncogenic intracellular signaling molecules in AT II epithelial cells through the autocrine mechanism and those in CD11b+Gr-1+ MDSCs through the paracrine mechanism.
BMSC Conversion into AT II Epithelial Cells in the Lung of Bitransgenic Mice
Bone marrow stem cells (BMSCs) are known to function in repair of injured AT II cells in emphysema. In our previous study, LAL deficiency caused inflammation-induced BMSC conversion into AT II epithelial cells.17 Because PPARγ serves as a downstream effector of LAL, it is conceivable that BMSC-to-AT II cell conversion is regulated by PPARγ. To test this assumption, the purified adherent BMSCs from hSP-B 1.5-kb lacZ transgenic mice were injected into 2-month doxycycline-treated or untreated CCSP-rtTA/(tetO)7-CMV-dnPPARγ recipient bitransgenic mice. AT II epithelial cells were isolated from recipient mice 2 months later and were double-stained with rabbit anti-proSP-C antibody (AT II cell marker) and mouse β-galactosidase antibody (Figure 8A). Compared with doxycycline-untreated bitransgenic mice, treated bitransgenic mice showed approximately 10% double-positive AT II epithelial cells. To further quantify the conversion rate of hSP-B 1.5-kb lacZ adherent BMSCs, the purified AT II epithelial cells from the lung of recipient mice (double-stained with proSP-C antibody and β-galactosidase antibody) were analyzed by flow cytometry (Figure 8B). In this method, approximately 12% AT II epithelial cells showed β-galactosidase and proSP-C double-positive in doxycycline-treated mice, compared with untreated mice.
Figure 8.
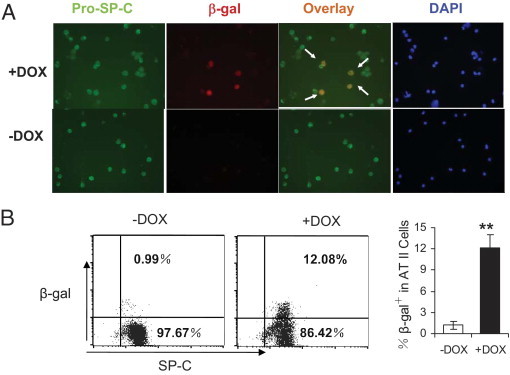
BMSCs conversion into AT II epithelial cells in the lung of CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice. A: Double-immunofluorescence staining of proSP-C and β -galactosidase in purified AT II epithelial cells from CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice that were injected with hSP-B 1.5-kb lacZ BMSCs. Arrows point to colocalized proSP-C and β-galactosidase positive AT II epithelial cells. DAPI represents nuclear staining. B: FACS analysis of AT II epithelial cells from the lungs of recipient CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice after hSP-B 1.5-kb lacZ BMSC injection. The cells were double-labeled with anti-SP-C and β-galactosidase antibodies. Data are reported as means ± SD (n = 6). **P < 0.01. −DOX, doxycycline-untreated bitransgenic mice; +DOX, doxycycline-treated bitransgenic mice.
Discussion
We have previously reported that blockage of PPARγ ligand synthesis during LAL deficiency caused exuberant inflammation and emphysema in the lung,1 implicating PPARγ inactivation as responsible for these phenotypes. To identify the pathophysiological roles of PPARγ in the lung, a doxycycline-controlled CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mouse model was generated to overexpress dnPPARγ in lung epithelial cells (Figure 1A) and so block the function of endogenous PPARγ. In this bitransgenic model, doxycycline treatment induced dnPPARγ-Flag fusion protein expression in AT II epithelial cells at both mRNA and protein levels (Figure 1, B–D). Flow cytometry analysis showed dnPPARγ-Flag fusion protein expression associated with AT II epithelial cells (Figure 1D). This is consistent with our previous observation, in which the same CCSP-rtTA system dictated expression of Stat3C-Flag fusion protein and MMP12-Flag fusion protein in AT II epithelial cells.11,14 As a consequence, emphysema was developed in the bitransgenic lung. This is in agreement with a previous finding.23 To elucidate the relationship between inflammation and dnPPARγ-induced emphysema, various proinflammatory cytokines were investigated. The rationale for this is based on two facts: emphysema is a chronic inflammatory disease and PPARγ is an anti-inflammation transcription factor after ligand binding.
In doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice, overexpression of dnPPARγ significantly increased concentrations of IL-1β, IL-6, and TNF-α in BALF at an early time point (<2 months of doxycycline treatment) (Figure 4B). Real-time PCR analysis revealed that at least AT II epithelial cells were the source cells for synthesis of these and other proinflammatory molecules, after suppression of the endogenous PPARγ anti-inflammatory function (Figure 4A). ChIP assay indicated that augment of these molecules was due to directly DNA binding of dnPPARγ on the promoters of these genes (Figure 3). Up-regulation of IL-6 and IL-13 has been reported to cause emphysema in the lung.24,25 Severity of elastase-induced emphysema was decreased in IL-1β and TNF-α receptor-deficient mice.26
dnPPARγ can induce proinflammatory gene expression by two mechanisms at the transcriptional level. First, PPARγ suppresses gene expression by antagonizing the activities of AP-1, Stat1, Stat3, and NFκB through physical interaction.9,27,28 It is conceivable that overexpression of dnPPARγ interferes with the PPARγ antagonizing function on these factors to promote proinflammatory gene expression. Alternatively, dnPPARγ may suppress the endogenous PPARγ function by defectiveness of ligand-dependent coactivator recruitment and ligand-dependent release of corepressor.19 For example, the nuclear receptor-interacting MED/TRAP220 subunit of the coactivator complex is required for PPARγ function.29,30
Up-regulation of these proinflammatory cytokines subsequently triggered accumulation of MDSCs in the lung and circulation system 4 months after doxycycline treatment (Figure 5). IL-1β is capable of inducing CD11b+Gr-1+ MDSC accumulation in mice.31,32 IL-6 can partially restore the accumulation of CD11b+Gr-1+ MDSC in IL-1 receptor (IL-1R)–deficient mice.33 In CD11b+Gr-1+ MDSCs from dnPPARγ overexpressed bitransgenic mice, multiple intracellular signaling molecules were overactivated (Table 3). It is known that MDSCs suppress T-cell proliferation and function to subvert immune surveillance and prevent the immune system from eliminating unwanted pathogenic events.21,22,34 Indeed, decreased numbers of CD3+, CD4+, and CD8+ T cells were observed in doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic lungs in correlation with CD11b+Gr-1+ MDSCs expansion 4 months after doxycycline treatment (Figure 6). This observation was further supported by the in vitro coculturing study, in which CD11b+Gr-1+ MDSCs isolated from dnPPARγ overexpressing bitransgenic mice significantly reduced proliferation, lymphokine production, and CD69 expression of wild-type CD4+ T cells that were stimulated by anti-CD3 mAb plus anti-CD28 mAb (Figure 7).
This observation is consistent with clinical studies in which CD4+ and CD8+ T cells were significantly reduced in the peripheral blood and BALF of patients with chronic obstructive pulmonary disease (COPD).35,36 The severity of COPD is directly correlated with decreased numbers of CD4+ and CD8+ T cells in peripheral blood mononuclear cells.37,38 Although MDSC-induced T-cell immunosuppression was initially reported in cancer, the present study demonstrates a link between MDSC and emphysema, a major phenotype in COPD. COPD and lung cancer are major diseases associated with smoking in humans. Because MDSCs are heterogenous populations, it remains to be determined which MDSC subpopulations induce tumor and which subpopulations induce emphysema.
In AT II epithelial cells, a set of intracellular signaling molecules that include Stat1, Stat2, Stat3, Stat4, Erk1/2, and p38 showed increased phosphorylation (activation) by dnPPARγ overexpression (Table 2). This can be a result of up-regulation of proinflammatory cytokine stimuli through autocrine pathways. Activation of these pathways can further exacerbate inflammation and pathogenic phenotypes by stimulating proinflammatory downstream genes. This is supported by clinical observations. For example, up-regulation of Stat3 and its downstream genes served as biomarkers for COPD diagnosis in humans.39 Activation of STAT-4 and Erk in the lung tissue was observed in emphysema patients, compared with lungs of control subjects.40,41
One important group of PPARγ negatively regulated genes in the lung is the MMPs.1,2 MMP up-regulation facilitates degradation of extracellular matrix to weaken the interstitial alveolar structure, resulting in pulmonary emphysema.20 For example, gene ablation of MMP12 conferred resistance to emphysema.42 Likewise, overexpression of MMP12 caused emphysema and lung cancer.11 MMP12 gene regulation can be directly inhibited by PPARγ in lung epithelial cells and is down-regulated in PPARγ ligand-treated lal−/− mice.2 After dnPPARγ overexpression, the mRNA levels of MMP12, along with MMP2, MMP7, MMP8, and MMP9, were all up-regulated in AT II epithelial cells from doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice (Figure 2D). As a result, the enzymatic activities of MMP9 and MMP12 were increased in BALF of doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic mice that contributed to emphysema formation (Figure 2D).
Emphysema can trigger infiltration of BMSCs into the lung to repair injured AT II cells.43 It would therefore be intriguing to see whether dnPPARγ-induced emphysema causes BMSC infiltration into the lung to convert into AT II cells. We have previously reported that inactivation of the endogenous PPARγ function by blocking ligand synthesis promoted BMSCs to convert into AT II epithelial cells in lal−/− mice as part of pathogenesis.17 Based on the observations of double-immunofluorescence staining and FACS analysis in the present study, BMSCs from hSP-B 1.5-kb lacZ donor mice showed the ability to convert into AT II epithelial cells during pulmonary inflammation and injury in the doxycycline-treated CCSP-rtTA/(tetO)7-CMV-dnPPARγ bitransgenic lung (Figure 8A). Conversion did not occur in doxycycline-untreated bitransgenic mice. Although it is generally thought that BMSCs can regenerate injured AT II epithelial cells during lung injury, for regenerative medicine one concern is that BMSCs may contribute to emphysema and cancer development, because BMSCs possess T-cell immune suppressive function similar to that exhibited by MDSCs.44,45
In summary, overexpression of dnPPARγ relieved endogenous PPARγ suppression of the transcriptional activities of proinflammatory genes, including cytokines and MMPs. This action promoted inflammatory cell infiltration (especially MDSCs) into the lung and changed the regional microenvironment. In the nonimmune response, AT II epithelial cells were changed, as evident from activation of multiple intracellular signaling molecules. As a consequence, emphysema was formed. The action also promoted conversion of BMSCs into AT II epithelial cells during injury. Infiltration of MDSCs and BMSCs reduced T-cell proliferation and function in the lung. These observations support the concept that the LAL/ligands/PPARγ axis plays a critical role in maintaining alveolar homeostasis by suppressing pulmonary inflammation.
Acknowledgments
We thank Dr. Xuemei Lian, Ph.D. (Cincinnati Children's Hospital Medical Center, Cincinnati, OH), for assistance with Kwik-Diff staining and Miss Beilin Li (Indiana University and Perdue University) for animal genotyping.
Footnotes
Supported by NIH grants HL087001 (H.D.), CA138759 (C.Y.), and HL061803 and HL067862 (C.Y and H.D.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.046.
Contributor Information
Cong Yan, Email: coyan@iupui.edu.
Hong Du, Email: hong.du@cchmc.org.
Supplemental data
Characterization of doxycycline-untreated and treated CCSP-rtTA single transgenic mice. A: Expression of the dnPPARγ-Flag fusion protein in the AT II epithelial cells from the lung of doxycycline-untreated (−DOX) or treated (+DOX)CCSP-rtTA single transgenic mice by FACS. The cells were double-labeled with anti-SP-C (AT II marker) and anti-Flag antibodies. CD11b-labeled macrophages served as the negative control. n = 3. There was no statistical significance between treated and untreated groups. B: Real-time PCR analysis of mRNA expression levels of MMPs in AT II epithelial cells of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice (n = 4). The mRNA expression level of GAPDH was used for normalization. ΔCt = Ct(testing) − Ct(GAPDH). The average fold changes were determined by 2(−ΔΔCt), in which ΔΔCt = ΔCt (+DOX) − ΔCt (−DOX). There was no statistical significance between treated and untreated groups. C: The enzymatic activities of MMP9 and MMP12 in BALF from doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by an MMP assay kit (n = 3). Data are reported as means ± SD. There was no statistical significance between treated and untreated groups. D: Real-time PCR analysis of mRNA expression levels of inflammatory cytokines in AT II epithelial cells of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice (n = 5). There was no statistical significance between treated and untreated groups. E: The percentage number of CD11b+ Gr-1+ cell populations in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by FACS. n = 3. There was no statistical significance between treated and untreated groups. A representative dot plot and statistical analysis are presented. F: The percentage number of CD3+ cell population in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice was analyzed by FACS. There was no statistical significance between treated and untreated groups. A representative profile and statistical analysis are presented. G: The percentage numbers of CD4+ and CD8+ cell populations in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by FACS. There was no statistical significance between treated and untreated groups. A representative dot plot and statistical analysis are presented.
References
- 1.Lian X., Yan C., Yang L., Xu Y., Du H. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L801–L807. doi: 10.1152/ajplung.00335.2003. [DOI] [PubMed] [Google Scholar]
- 2.Lian X., Yan C., Qin Y., Knox L., Li T., Du H. Neutral lipids and peroxisome proliferator-activated receptor-{gamma} control pulmonary gene expression and inflammation-triggered pathogenesis in lysosomal acid lipase knockout mice. Am J Pathol. 2005;167:813–821. doi: 10.1016/s0002-9440(10)62053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan C., Lian X., Li Y., Dai Y., White A., Qin Y., Li H., Hume D.A., Du H. Macrophage-specific expression of human lysosomal acid lipase corrects inflammation and pathogenic phenotypes in lal−/− mice. Am J Pathol. 2006;169:916–926. doi: 10.2353/ajpath.2006.051327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qu P., Shelley W.C., Yoder M.C., Wu L., Du H., Yan C. Critical roles of lysosomal acid lipase in myelopoiesis. Am J Pathol. 2010;176:2394–2404. doi: 10.2353/ajpath.2010.091063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qu P., Du H., Wilkes D.S., Yan C. Critical roles of lysosomal acid lipase in T cell development and function. Am J Pathol. 2009;174:944–956. doi: 10.2353/ajpath.2009.080562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li A.C., Brown K.K., Silvestre M.J., Willson T.M., Palinski W., Glass C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marx N., Kehrle B., Kohlhammer K., Grüb M., Koenig W., Hombach V., Libby P., Plutzky J. PPAR activators as antiinflammatory mediators in human T lymphocytes: Implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–710. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang C., Ting A.T., Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 9.Wang L.H., Yang X.Y., Zhang X., Huang J., Hou J., Li J., Xiong H., Mihalic K., Zhu H., Xiao W., Farrar W.L. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity. 2004;20:205–218. doi: 10.1016/s1074-7613(04)00030-5. [DOI] [PubMed] [Google Scholar]
- 10.Qu P., Du H., Li Y., Yan C. Myeloid-specific expression of Api6/AIM/Sp alpha induces systemic inflammation and adenocarcinoma in the lung. J Immunol. 2009;182:1648–1659. doi: 10.4049/jimmunol.182.3.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu P., Du H., Wang X., Yan C. Matrix metalloproteinase 12 overexpression in lung epithelial cells plays a key role in emphysema to lung bronchioalveolar adenocarcinoma transition. Cancer Res. 2009;69:7252–7261. doi: 10.1158/0008-5472.CAN-09-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L., Yan D., Yan C., Du H. Peroxisome proliferator-activated receptor gamma and ligands inhibit surfactant protein B gene expression in the lung. J Biol Chem. 2003;278:36841–36847. doi: 10.1074/jbc.M304156200. [DOI] [PubMed] [Google Scholar]
- 13.Tichelaar J.W., Lu W., Whitsett J.A. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–11864. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Du H., Qin Y., Roberts J., Cummings O.W., Yan C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007;67:8494–8503. doi: 10.1158/0008-5472.CAN-07-0647. [DOI] [PubMed] [Google Scholar]
- 15.Rice W.R., Conkright J.J., Na C.L., Ikegami M., Shannon J.M., Weaver T.E. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol. 2002;283:L256–L264. doi: 10.1152/ajplung.00302.2001. [DOI] [PubMed] [Google Scholar]
- 16.Yan C., Naltner A., Conkright J., Ghaffari M. Protein-protein interaction of retinoic acid receptor alpha and thyroid transcription factor-1 in respiratory epithelial cells. J Biol Chem. 2001;276:21686–21691. doi: 10.1074/jbc.M011378200. [DOI] [PubMed] [Google Scholar]
- 17.Yan C., Lian X., Dai Y., Wang X., Qu P., White A., Qin Y., Du H. Gene delivery by the hSP-B promoter to lung alveolar type II epithelial cells in LAL-knockout mice through bone marrow mesenchymal stem cells. Gene Ther. 2007;14:1461–1470. doi: 10.1038/sj.gt.3303006. [DOI] [PubMed] [Google Scholar]
- 18.Yang L., Naltner A., Kreiner A., Yan D., Cowen A., Du H., Yan C. An enhancer region determines hSP-B gene expression in bronchiolar and ATII epithelial cells in transgenic mice. Am J Physiol Lung Cell Mol Physiol. 2003;284:L481–L488. doi: 10.1152/ajplung.00280.2002. [DOI] [PubMed] [Google Scholar]
- 19.Gurnell M., Wentworth J.M., Agostini M., Adams M., Collingwood T.N., Provenzano C., Browne P.O., Rajanayagam O., Burris T.P., Schwabe J.W., Lazar M.A., Chatterjee V.K. A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J Biol Chem. 2000;275:5754–5759. doi: 10.1074/jbc.275.8.5754. [DOI] [PubMed] [Google Scholar]
- 20.Shapiro S.D., Senior R.M. Matrix metalloproteinases: Matrix degradation and more. Am J Respir Cell Mol Biol. 1999;20:1100–1102. doi: 10.1165/ajrcmb.20.6.f151. [DOI] [PubMed] [Google Scholar]
- 21.Ostrand-Rosenberg S., Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon D.M., Arikan M.C., Srisuma S., Bhattacharya S., Tsai L.W., Ingenito E.P., Gonzalez F., Shapiro S.D., Mariani T.J. Epithelial cell PPARγ contributes to normal lung maturation. FASEB J. 2006;20:1507–1509. doi: 10.1096/fj.05-5410fje. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn C., 3rd, Homer R.J., Zhu Z., Ward N., Flavell R.A., Geba G.P., Elias J.A. Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (IL)-11 and IL-6 in the lung. Am J Respir Cell Mol Biol. 2000;22:289–295. doi: 10.1165/ajrcmb.22.3.3690. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino T., Kato S., Oka N., Imaoka H., Kinoshita T., Takei S., Kitasato Y., Kawayama T., Imaizumi T., Yamada K., Young H.A., Aizawa H. Pulmonary inflammation and emphysema: role of the cytokines IL-18 and IL-13. Am J Respir Crit Care Med. 2007;176:49–62. doi: 10.1164/rccm.200603-316OC. [DOI] [PubMed] [Google Scholar]
- 26.Lucey E.C., Keane J., Kuang P.P., Snider G.L., Goldstein R.H. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1beta receptor-deficient mice. Lab Invest. 2002;82:79–85. doi: 10.1038/labinvest.3780397. [DOI] [PubMed] [Google Scholar]
- 27.Ricote M., Li A.C., Willson T.M., Kelly C.J., Glass C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 28.Castrillo A., Mojena M., Hortelano S., Bosca L. Peroxisome proliferator-activated receptor-gamma-independent inhibition of macrophage activation by the non-thiazolidinedione agonist L-796,449: Comparison with the effects of 15-deoxy-delta(12,14)-prostaglandin J(2) J Biol Chem. 2001;276:34082–34088. doi: 10.1074/jbc.M102472200. [DOI] [PubMed] [Google Scholar]
- 29.Ge K., Cho Y.W., Guo H., Hong T.B., Guermah M., Ito M., Yu H., Kalkum M., Roeder R.G. Alternative mechanisms by which mediator subunit MED1/TRAP220 regulates peroxisome proliferator-activated receptor gamma-stimulated adipogenesis and target gene expression. Mol Cell Biol. 2008;28:1081–1091. doi: 10.1128/MCB.00967-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge K., Guermah M., Yuan C.X., Ito M., Wallberg A.E., Spiegelman B.M., Roeder R.G. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature. 2002;417:563–567. doi: 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- 31.Bunt S.K., Sinha P., Clements V.K., Leips J., Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 32.Tu S., Bhagat G., Cui G., Takaishi S., Kurt-Jones E.A., Rickman B., Betz K.S., Penz-Oesterreicher M., Bjorkdahl O., Fox J.G., Wang T.C. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunt S.K., Yang L., Sinha P., Clements V.K., Leips J., Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sica A., Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barcelo B., Pons J., Fuster A., Sauleda J., Noguera A., Ferrer J.M., Agusti A.G. Intracellular cytokine profile of T lymphocytes in patients with chronic obstructive pulmonary disease. Clin Exp Immunol. 2006;145:474–479. doi: 10.1111/j.1365-2249.2006.03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta J., Chattopadhaya D., Bhadoria D.P., Qadar Pasha M.A., Gupta V.K., Kumar M., Dabur R., Yadav V., Sharma G.L. T lymphocyte subset profile and serum alpha-1-antitrypsin in pathogenesis of chronic obstructive pulmonary disease. Clin Exp Immunol. 2007;149:463–469. doi: 10.1111/j.1365-2249.2007.03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Stefano A., Capelli A., Lusuardi M., Caramori G., Balbo P., Ioli F., Sacco S., Gnemmi I., Brun P., Adcock I.M., Balbi B., Barnes P.J., Chung K.F., Donner C.F. Decreased T lymphocyte infiltration in bronchial biopsies of subjects with severe chronic obstructive pulmonary disease. Clin Exp Allergy. 2001;31:893–902. doi: 10.1046/j.1365-2222.2001.01098.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhu X., Gadgil A.S., Givelber R., George M.P., Stoner M.W., Sciurba F.C., Duncan S.R. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol. 2009;182:3270–3277. doi: 10.4049/jimmunol.0802622. [DOI] [PubMed] [Google Scholar]
- 39.Qu P., Roberts J., Li Y., Albrecht M., Cummings O.W., Eble J.N., Du H., Yan C. Stat3 downstream genes serve as biomarkers in human lung carcinomas and chronic obstructive pulmonary disease. Lung Cancer. 2009;63:341–347. doi: 10.1016/j.lungcan.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Stefano A., Caramori G., Capelli A., Gnemmi I., Ricciardolo F.L., Oates T., Donner C.F., Chung K.F., Barnes P.J., Adcock I.M. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 2004;24:78–85. doi: 10.1183/09031936.04.00080303. [DOI] [PubMed] [Google Scholar]
- 41.Mercer B.A., Kolesnikova N., Sonett J., D'Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 2004;279:17690–17696. doi: 10.1074/jbc.M313842200. [DOI] [PubMed] [Google Scholar]
- 42.Hautamaki R.D., Kobayashi D.K., Senior R.M., Shapiro S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 43.Weiss D.J., Kolls J.K., Ortiz L.A., Panoskaltsis-Mortari A., Prockop D.J. Stem cells and cell therapies in lung biology and lung diseases. Proc Am Thorac Soc. 2008;5:637–667. doi: 10.1513/pats.200804-037DW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alison M.R., Lim S., Houghton J.M. Bone marrow-derived cells and epithelial tumours: more than just an inflammatory relationship. Curr Opin Oncol. 2009;21:77–82. doi: 10.1097/CCO.0b013e32831de4cf. [DOI] [PubMed] [Google Scholar]
- 45.Djouad F., Plence P., Bony C., Tropel P., Apparailly F., Sany J., Noël D., Jorgensen C. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–3844. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of doxycycline-untreated and treated CCSP-rtTA single transgenic mice. A: Expression of the dnPPARγ-Flag fusion protein in the AT II epithelial cells from the lung of doxycycline-untreated (−DOX) or treated (+DOX)CCSP-rtTA single transgenic mice by FACS. The cells were double-labeled with anti-SP-C (AT II marker) and anti-Flag antibodies. CD11b-labeled macrophages served as the negative control. n = 3. There was no statistical significance between treated and untreated groups. B: Real-time PCR analysis of mRNA expression levels of MMPs in AT II epithelial cells of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice (n = 4). The mRNA expression level of GAPDH was used for normalization. ΔCt = Ct(testing) − Ct(GAPDH). The average fold changes were determined by 2(−ΔΔCt), in which ΔΔCt = ΔCt (+DOX) − ΔCt (−DOX). There was no statistical significance between treated and untreated groups. C: The enzymatic activities of MMP9 and MMP12 in BALF from doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by an MMP assay kit (n = 3). Data are reported as means ± SD. There was no statistical significance between treated and untreated groups. D: Real-time PCR analysis of mRNA expression levels of inflammatory cytokines in AT II epithelial cells of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice (n = 5). There was no statistical significance between treated and untreated groups. E: The percentage number of CD11b+ Gr-1+ cell populations in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by FACS. n = 3. There was no statistical significance between treated and untreated groups. A representative dot plot and statistical analysis are presented. F: The percentage number of CD3+ cell population in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice was analyzed by FACS. There was no statistical significance between treated and untreated groups. A representative profile and statistical analysis are presented. G: The percentage numbers of CD4+ and CD8+ cell populations in the blood and lung of doxycycline-untreated (−DOX) and treated (+DOX) CCSP-rtTA single transgenic mice were analyzed by FACS. There was no statistical significance between treated and untreated groups. A representative dot plot and statistical analysis are presented.