

Abstract
An approach that facilitates rapid isolation and characterization of tumor cells with enhanced metastatic potential is highly desirable. Here, we demonstrate that plating GI-101A human breast cancer cells on hard (0.9%) agar selects for the subpopulation of metastasis-initiating cells. The agar-selected cells, designated GI-AGR, were homogeneous for CD44+ and CD133+ and five times more invasive than the parental GI-101A cells. Moreover, mice injected with GI-AGR cells had significantly more experimental brain metastases and shorter overall survival than did mice injected with GI-101A cells. Comparative gene expression analysis revealed that GI-AGR cells were markedly distinct from the parental cells but shared an overlapping pattern of gene expression with the GI-101A subline GI-BRN, which was generated by repeated in vivo recycling of GI-101A cells in an experimental brain metastasis model. Data mining on 216 genes shared between GI-AGR and GI-BRN breast cancer cells suggested that the molecular phenotype of these cells is consistent with that of cancer stem cells and the aggressive basal subtype of breast cancer. Collectively, these results demonstrate that analysis of cell growth in a hard agar assay is a powerful tool for selecting metastasis-initiating cells in a heterogeneous population of breast cancer cells, and that such selected cells have properties similar to those of tumor cells that are selected based on their potential to form metastases in mice.
The vast majority of cancer deaths result from progressive growth of metastases that are resistant to conventional therapies.1 Metastases originate from a selected subpopulation of cells that reside in a biologically heterogeneous primary tumor.2,3 Results from experimental4 and clinical5 examinations indicate that most metastases are clonal in origin and that the metastatic process is highly selective.6 Studies have also shown profound differences between local and disseminated cancers,7 suggesting that information on primary tumors alone may not be sufficient to determine optimal therapeutic interventions. For this reason, researchers have directed considerable effort toward improving understanding of the molecular phenotypes of metastasis-initiating tumor cells.
One widely used experimental approach to isolate populations of tumor cells with enhanced metastatic potential is an in vivo selection process in which tumor cells are implanted into syngeneic or immunodeficient mice and metastasis is allowed to occur. Tumor cells from the resultant metastatic lesions can be isolated and expanded to establish cell sublines, some of which may have higher metastatic capacity than the parental tumor-cell population.8 Comparative gene expression profiling of parental tumor cells and their metastatic subpopulations has yielded invaluable information regarding the genetic determinants critical for organ-specific metastasis.9 For example, Kang et al10 compared the transcriptional profiles of parental MDA-231 human breast cancer cells with those of a bone-colonizing variant of this cell line and reported the underlying gene expression signature required for organ tropism to bone. Investigators used a similar approach to identify genes whose expression is critical for metastasis of breast cancer cells to the brain11 and lungs.12
Nevertheless, although this in vivo selection technique has provided new insight into the cellular and molecular mechanisms that control site-specific metastasis, there are some disadvantages. Perhaps the principal drawback of in vivo selection is that it can be time-consuming, often requiring several rounds of tumor-cell implantation and in vitro expansion to enrich for populations of cells with enhanced metastatic capacity. For example, a previous study showed that 10 cycles of selection were needed to generate the murine B16-F10 melanoma cell line with enhanced ability to generate lung metastases.13 Another highly invasive melanoma cell variant required six selection cycles before it could undergo full characterization.14 In addition, repeated cycling of tumor cells in mice is not a practical approach to examining tumor samples collected from patients. Clearly, an in vitro method that facilitates the rapid isolation, identification, and characterization of metastasis-initiating cells is highly desirable.
Researchers have widely used cloning techniques with semisolid medium, such as evaluating cell growth in soft agar, to study the biology of murine and human progenitor cells.15 Agar assays can also distinguish tumor cells from nontransformed cells, because cells lacking the ability to undergo anchorage-independent growth are unable to thrive on an agar substrate.16,17 Previous work in our laboratory demonstrated that the growth of nonmetastatic tumor cells on agar could be restricted when the concentration of the agar medium was increased from 0.3% (soft agar) to 0.6% (hard agar).17 Moreover, the growth capacity of human tumor cells on hard agar correlated with their metastatic potential in vivo.18 In those studies, however, we were unable to perform a systematic molecular comparison of metastatic tumor cells selected on hard agar with those selected using an in vivo procedure.
In the present study, we isolated a subpopulation of GI-101A human breast cancer cells (GI-AGR cells) capable of growing on 0.9% hard agar and compared them with a subpopulation of GI-101A cells obtained in vivo using several rounds of selection in the brains of nude mice (GI-BRN cells). Our results suggest that these two methods yielded cells with common molecular characteristics and that selection of cancer cells based on their ability to grow on hard agar is a rapid approach to isolating cells that may ultimately form metastases. We look forward to extension of this method to other types of tumor cells and, more importantly, clinical biopsy specimens for identification of new molecular targets for therapy.
Materials and Methods
Breast Cancer Cell Line and Sublines
The human breast cancer cell line GI-101A19 was provided by Dr. Janet E. Price (The University of Texas M.D. Anderson Cancer Center). The cell line was tested at the M.D. Anderson Characterized Cell Line Core Facility using short tandem repeats DNA profiling. The results of that analysis indicated that there was no untoward match between this newly accessioned cell line and an existing DNA profile. GI-101A cells were maintained in Dulbecco's minimum essential medium (DMEM; Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), L-glutamine, and a 5% (v/v) insulin selenium-transferrin supplement (Sigma-Aldrich, St. Louis, MO). The GI-101A sublines GI-AGR and GI-BRN were maintained in identical media and were cultured as monolayers in a 37°C humidified incubator containing a mixture of 5% CO2 and 95% air.
Mice
Female athymic nude mice (NCI-nu) were purchased from the Animal Production Area of the NIH National Cancer Institute (Frederick, MD). The mice were housed and maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and were used in accordance with institutional guidelines.
Tumor-Cell Growth in Agar and Establishment of the GI-101A Hard Agar-Selected Subline GI-AGR
Agar assays were performed as described previously.18 In brief, 1 mL of DMEM containing 10% FBS and 0.6% agar was plated into individual wells of six-well plates (BD Biosciences, San Jose, CA). GI-101A cells were harvested via brief exposure to a solution containing 0.25% trypsin/0.02% EDTA (v/v) and then resuspended in DMEM containing 10% FBS. The resulting cell mixture was then passaged through a 40-μm cell strainer (BD Biosciences) to generate a single-cell suspension. Cells were seeded at a density of 5 × 103 cells/well in 1 mL of DMEM containing 10% FBS and 0.3%, 0.6%, or 0.9% agar. This cell-containing mixture was then pipetted gently over the bottom layer of agar in the wells. One milliliter of DMEM containing 10% FBS was periodically added to the wells to keep the agar surface hydrated. The numbers and diameters of tumor colonies were calculated when the tumor colonies became visible. Colonies growing on 0.9% agar that exceeded 200 μm in diameter were harvested using pipet tips and were transferred to a 3.5-cm plastic culture dish (Corning Life Sciences, Lowell, MA). This cell subline was designated as GI-AGR, to distinguish it from the parental GI-101A cell line.
Experimental Brain Metastasis Model and Establishment of the Subline GI-BRN
To identify parental GI-101A cells capable of producing experimental brain metastases in mice, eight female nude mice were injected with GI-101A cells via the common carotid artery as described previously.20 The mice were euthanized by injecting sodium pentobarbital (Abbott Laboratories, Des Plaines, IL) when they became moribund or on day 200 after tumor-cell injection. Visible metastases were placed in a medium containing DMEM supplemented with 0.2% type IV collagenase (Sigma-Aldrich), incubated in a 37°C water bath for 1 hour, and then centrifuged, resuspended in culture medium, and passaged through a 40-μm mesh. The cells were expanded and injected into female mice, and the cycle was repeated two additional times. The resulting GI-101A tumor cells that were recycled to the brain were termed GI-BRN cells.
Tumor-Cell Invasion
To determine the invasive capacity of GI-101A, GI-AGR, and GI-BRN cells, we seeded them at a density of 1 × 105 cells/well into 8-μm inserts precoated with Matrigel (BD Biosciences). The cells were added to the inserts in 100 μL of serum-free DMEM and then placed in 24-well plates containing 600 μL of DMEM plus 10% FBS. The assay was terminated after 36 hours, and the inserts were fixed and processed for cell counting. The cells were examined under a microscope, and the numbers of migrating cells in four high-power fields (×200 magnification) were recorded. The assay was performed in triplicate, and the experiment was repeated three times.
Comparative Gene Expression Analysis
Parental GI-101A, GI-AGR, and GI-BRN cells were used to generate total RNA samples for analysis. Samples were labeled and hybridized to Sentrix Human-6 v2 Expression BeadChips according to the manufacturer's protocol (Illumina, San Diego, CA). In brief, cDNA was generated from total RNA using an Illumina TotalPrep RNA amplification kit (Applied Biosystems, Foster City, CA). Next, in vitro transcription was performed to incorporate biotin-labeled nucleotides into cRNA for 4 hours at 37°C. A total of 1500 ng of biotin-labeled cRNA was hybridized to Sentrix Human-6 v2 Expression BeadChips at 58°C for 16 hours. Hybridized biotinylated cRNA was detected using 1 μg/mL cyanide 3-streptavidin (GE Healthcare, Piscataway, NJ), and the BeadChips were scanned using a BeadArray Reader (Illumina) without any normalization or background subtraction. Gene expression data were normalized using the quantile normalization method with the LIMMA package in the R programming language (http://www.r-project.org). The level of expression of each gene was transformed into a log2 function for further analysis. To identify differentially expressed genes in the different groups of samples, a random-variance t-test was performed and gene expression differences were considered statistically significant if the P value was <0.001. Gene expression assays were performed in quadruplicate.
Semiquantitative and Quantitative RT-PCR Analysis
Total RNA was isolated from GI-101A, GI-AGR, and GI-BRN cells using standard molecular biological techniques. First-strand cDNA was synthesized using the SuperScript first-strand synthesis system according to the manufacturer's instructions (Invitrogen). PCR analysis was performed for a total of 25 to 28 cycles. For quantitative PCR reactions, 1:10 dilutions of cDNA products were amplified using SYBR Green PCR master mix (Applied Biosystems) and analyzed using an ABI Prism 7500 Fast System (Applied Biosystems). The following four sets of primers were used to amplify the specific human genes: Forkhead box A1 (FOXA1) 5′-GTGGGTCCAGGATGTTAGGA-3′ (forward) and 5′-CCGCAGTCATGCTGTTCAT-3′ (reverse); γ-aminobutyric acid A receptor, pi (GABRP) 5′-CTCTCCAAATCCAGCCAGAG-3′ (forward) and 5′-ATGATTGGCTCATACAACCACA-3′ (reverse); msh homeobox 1 (MSX1) 5′-AAGTTCCGCCAGAAGCAGTA-3′ (forward) and 5′-TCAGGTGGTACATGCTGTAG-3′ (reverse); and basonuclin (BNC1) 5′-AGCTCAGATGAAGACATGCC-3′ (forward) and 5′-CTTTGAAGATGACAGATGTCTGGG-3′ (reverse).
Western Blot Analysis
Western blot analysis was performed as previously described.20 Western analyses of proteins were performed by using dickkopf-related protein-3 (3 Dkk-3; R&D Systems, Minneapolis, MN), anti-secreted frizzled-related protein 1 (sFRP1) (Cell Signaling Technology, Danvers, MA), anti-β-actin (Sigma-Aldrich), and the corresponding horseradish peroxidase-conjugated antibodies.
Expression of CD44 and CD133 in GI-101A, GI-AGR, and GI-BRN Cells
Cultured parental GI-101A, GI-AGR, and GI-BRN cells in log-phase growth were harvested via brief exposure to a 0.25% trypsin/0.02% EDTA solution (v/v). Harvested cells were centrifuged for 5 minutes at 200 × g and then prepared for sorting. The cells were labeled by resuspending the pellet in 2% DMEM containing 2 μg/mL phycoerythrin-conjugated CD133 antibody (Miltenyi Biotec, Auburn, CA) and 2 μg/mL fluorescein isothiocyanate-conjugated CD44 antibody (BD Biosciences). Cells were incubated in this solution for 45 minutes at 4°C, washed twice, and resuspended in DMEM. Additional GI-101A cells labeled with phycoerythrin- and fluorescein isothiocyanate-conjugated isotype standards (BD Biosciences) were used to assess the level of background intensity. Cell staining was evaluated using a Beckman Epics Elite flow cytometer (Beckman Coulter, Fullerton, CA). Gating parameters were adjusted based on the fluorescence histograms of the positive and negative controls.
Expression of CD133 in GI-101A Primary Breast Tumors and Brain Metastases
To produce primary breast tumors, female nude mice were injected in the mammary fat pad with 1 × 106 parental GI-101A cells. Four weeks later, the tumors were harvested as described previously.18 Paraffin-embedded GI-101A tumors harvested from the mammary fat pads and brains of nude mice were processed and immunolabeled with an antibody directed against CD133 or an isotype matched control. A goat anti-rabbit Alexa 594 antibody (A-11037, 1:1500) was used for visualization, and images were captured with a Zeiss Axioplan fluorescent microscope (Carl Zeiss, Göttingen, Germany). Cells that were positive for CD133 were identified by red fluorescence.
Expression of CD44 and CD133 in Clinical Samples of Breast Cancer Brain Metastasis
Five clinical cases of grade 3 invasive ductal carcinomas metastatic to the brain were provided by Dr. Dario Marchetti (Baylor College of Medicine) with the approval of the Institutional Review Board. Paraffin-embedded tissues were sectioned (6-μm thick) and used to detect expression of CD44 and CD133. Tissue sections were mounted on positively charged Superfrost slides (Fisher Scientific, Pittsburgh, PA) and dried overnight. The sections were deparaffinized in xylene, dehydrated in a graded series of alcohol [100%, 95%, and 80% ethanol/water (v/v)], and rehydrated in PBS (pH 7.5). Antigen retrieval was performed using heat retrieval with Diva Decloaker solution (Biocare Medical, Concord, CA). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol. Samples were incubated in a protein-blocking solution (5% normal horse serum and 1% normal goat serum in PBS) and then overnight at 4°C with the individual primary antibody in blocking solution. Control samples were not incubated with primary antibodies. The following primary antibodies were used for these studies: CD133 (19898; Abcam, Cambridge, MA) and CD44 (51037; Abcam). Both antibodies were used at a concentration of 1:100. Slides were rinsed three times (five minutes each) in PBS and then incubated with MACH4 HRP polymer (Biocare Medical) for 30 minutes at ambient temperature. A positive reaction was detected by exposure to stable 3,3′-diaminobenzidine for 5 to 10 minutes. Slides were counterstained with Gill's number 3 hematoxylin and viewed with a Nikon Microphot-FXA photomicroscope (Nikon Instruments, Tokyo, Japan) equipped with a Leica DFC320 digital camera (Leica Microsystems, Wetzlar, Germany) and representative images from each tumor sample were captured using Adobe Photoshop CS3 software.
Experimental Lung and Liver Metastasis Models
To determine whether the hard agar assay selected for metastatic cells per se, or for brain metastatic cells specifically, we assessed the potential of GI-101A parental cells and GI-AGR cells to form metastases in the liver and lungs of nude mice. To evaluate liver metastasis, we created a small incision in the left abdominal flank of anesthetized (methoxyflurane) mice and injected either GI-101A or GI-AGR tumor cells (5 × 105 cells/40 μL HBSS) into the spleen of mice (10 mice in each group). The spleen was returned to the abdomen and the wound was closed in one layer with wound clips. Intravenous injection was used to evaluate the formation of lung metastases in mice. GI-101A or GI-AGR tumor cells (1 × 106 cells/200 μL HBSS) were injected into the lateral vein of nude mice (10 mice in each group) with a 27-gauge needle. The mice were monitored daily, and all mice were euthanized 115 days following tumor cell injection. The liver and lungs of mice were removed and placed in Bouin's fixative for 24 hours. The number of visible liver and lung metastases was determined with the aid of a dissecting microscope. Tissue sections were stained with H&E for histological analysis.
Statistical Analysis
The significance of differences in invasion and colony and metastasis formation were analyzed using Student's two-tailed t-test. Survival rate estimates and median survival durations were determined using the Kaplan-Meier method. The survival data were tested for significance using the Mantel-Cox log-rank test.
Results
Generation of the Hard Agar GI-AGR Subline
We tested the anchorage-independent growth capacity of parental GI-101A cells by plating them on agar at increasing concentrations (0.3%, 0.6%, and 0.9%) supplemented with 10% FBS. On 0.3% agar, tumor cells formed many colonies that varied in size (Figure 1, A and B); however, the number of colonies formed on 0.6% agar was dramatically reduced, and the cells generated very few colonies on 0.9% agar. The colonies that formed on 0.9% hard agar were isolated and expanded in cell culture and thereafter referred to as GI-AGR.
Figure 1.
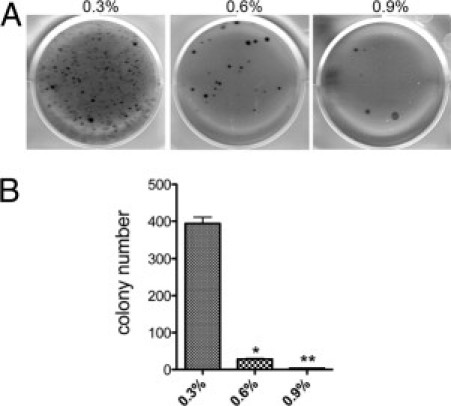
Plating on hard agar selects for a small population of GI-101A breast cancer cells. A: GI-101A cells were plated at 5000 cells/well on 0.3%, 0.6%, or 0.9% agar. Cell growth on the agar was evaluated after 60 days. B: The number of colonies on each agar surface was recorded in triplicate; data are reported as means ± SD. The numbers of colonies that formed on 0.6% agar (*P < 0.0005) and 0.9% agar (**P = 0.0001) were significantly lower than those that formed on 0.3% agar.
Invasive and Metastatic Capacity of GI-101A Parental and GI-AGR Breast Cancer Cells
We found that the GI-AGR cells were almost five times more invasive than the parental GI-101A cells (Figure 2A). We next examined the ability of GI-101A and GI-AGR cells to form experimental brain metastases following injection into the common carotid artery in nude mice. The GI-101A cells produced metastases in four of eight mice (50%), whereas the GI-AGR cells formed brain metastases in seven of eight mice (88%) (Figure 2B). Moreover, the median overall survival duration in mice injected with GI-AGR cells was significantly shorter than that in mice injected with GI-101A cells, most likely because of the larger sizes and greater numbers of brain metastases formed by the GI-AGR cells (Figure 2C).
Figure 2.
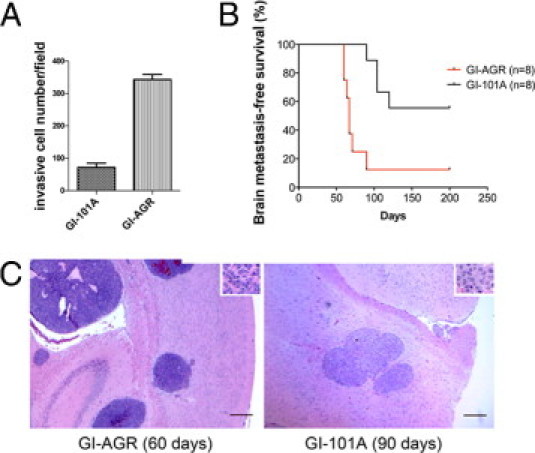
Plating on 0.9% agar selects for GI-101A cells that are invasive and metastatic. A: GI-101A and GI-AGR cells were plated onto the upper chambers of 8-μm Matrigel-coated inserts and the number of invading cells was recorded. The assay was repeated three times; results of a representative experiment are shown. B: Kaplan-Meier plot comparing survival durations in mice injected with parental GI-101A cells and GI-AGR cells. C: H&E staining of brain metastases in mice formed by GI-AGR cells 60 days after injection (left) and GI-101A cells 90 days after injection (right) of the cells into the common carotid artery. Insets: Tumor cells at higher magnification. Original magnification, ×20. Scale bars = 50 μm.
Generation of the GI-BRN Subline through in Vivo Selection
To identify the subpopulation of parental GI-101A cells capable of forming experimental brain metastases in nude mice, we initially injected eight mice with GI-101A cells via the common carotid artery. We euthanized mice when they began to exhibit gait and balance difficulties and harvested the brains. We isolated and expanded the metastatic cells as described above and injected them into the common carotid arteries of nude mice. We repeated this in vivo cycling two additional times and designated the final cell line established using this process as GI-BRN.
We next compared the ability of GI-BRN cells to form colonies on agar with that of the parental GI-101A and GI-AGR cells. Similar to our initial examination, GI-101A cells formed numerous colonies on 0.3% agar, but the number of colonies was dramatically lower when we plated the cells on 0.9% agar (Figure 3). The colonies formed by GI-BRN and GI-AGR cells on 0.3% agar were much larger than those formed by GI-101A cells. When we plated the cells on 0.6% and 0.9% agar, both GI-BRN cells (selected in vivo) and GI-AGR cells (selected in vitro) produced more and larger colonies than did the parental cells.
Figure 3.

GI-AGR and GI-BRN cells have higher capacities for growth on hard agar than do GI-101A cells. GI-101A, GI-AGR, and GI-BRN cells were cultured on 0.3%, 0.6%, or 0.9% agar in six-well plates (5 × 103 cells/well) for 30 days. Scale bar = 100 μm.
Comparative Transcriptional Profiling of GI-101A, GI-AGR, and GI-BRN Cells
To examine the molecular basis for the correlation between breast cancer cell growth on hard agar and the ability of these cells to produce brain metastases in mice, we conducted gene expression profiling of the parental GI-101A, GI-AGR, and GI-BRN cells using an Illumina bead-array gene expression platform with 48,000 gene features. To prevent any potential false-positive genes resulting from technical variance, all of these experiments were performed in quadruplicate. The results revealed fundamental differences in gene expression between GI-101A and GI-AGR cells. Using P < 0.001 and fold-change >2 as selection criteria, we found that a total of 402 genes were differentially expressed in GI-101A and GI-AGR tumor cells (Figure 4A). We obtained strikingly similar results when we compared the patterns of gene expression in GI-101A and GI-BRN cells: a total of 373 genes were differentially expressed in these two cell lines. We then analyzed both of the differential expression data sets to identify those genes that were common to both GI-AGR and GI-BRN cells. This examination revealed that 216 genes (>50% of the differentially expressed genes) were expressed at similar levels in GI-AGR and GI-BRN cells (Figure 4A; see also Supplemental Table S1 at http://ajp.amjpathol.org), suggesting that these genes play a role in the formation of brain metastases.
Figure 4.
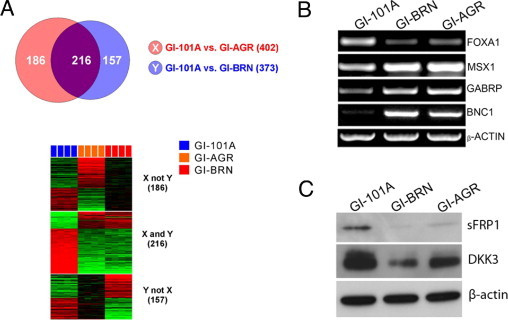
Comparative transcriptional profiling of GI-101A, GI-AGR, and GI-BRN cells. A: Using selection criteria of P < 0.001 and fold-change in gene expression >2, a total of 402 genes were differentially expressed in parental GI-101A and GI-AGR cells; 373 genes were differentially expressed in GI-101A cells and GI-BRN cells, and 216 of the differentially expressed genes were expressed in both GI-AGR and GI-BRN cells. B: RT-PCR analysis of the expression of FOXA1, MSX1, GABRP, and BNC1 in GI-101A, GI-AGR, and GI-BRN cells. β-actin was used as a loading control. C: Western blot analysis showed that expression of sFRP1 and DKK3 was significantly lower in GI-BRN and GI-AGR cells than in parental GI-101A cells.
Data mining was performed on the 216 genes similarly expressed in GI-AGR and GI-BRN cells. The results revealed significant increases in the expression of genes that are characteristic of the basal subtype of breast carcinoma, including MSX1,21 GABRP,22 SERPINB5,23 ANXA8,24 and KRT1425 (Table 1). The basal subtype of breast cancer is usually associated with a poor prognosis,26 whereas the luminal subtype generally has a more favorable prognosis.27 Genes associated with the luminal subtype of breast cancer, such as FOXA1,28 SCGB2A1,29 and SCGB1D2,29 were found to be down-regulated in the GI-AGR and GI-BRN cells. We validated the expression of several of these genes, including FOXA1, MSX1, GABRP, and BNC1, using RT-PCR analysis (Figure 4B). We readily detected FOXA1 expression in parental GI-101A cells, but it was not expressed in either GI-AGR or GI-BRN cells. MSX1, GABRP, and BNC1 were minimally expressed in GI-101A cells, but were highly expressed in both GI-AGR and GI-BRN cells. This examination also suggested that the Wnt signaling pathway was dysregulated in GI-AGR and GI-BRN cells. We evaluated the expression of two proteins that are considered negative regulators of Wnt signaling (ie, sFRP1 and DKK3) and found that both had significantly lower expression in GI-AGR and GI-BRN cells than in parental GI-101A cells (Figure 4C).
Table 1.
Expression of Differentiation-Related Genes in GI-AGR and GI-BRN Human Breast Cancer Cells
| Unique identifier | Gene symbol | Gene name | log2 ratio changes |
|
|---|---|---|---|---|
| GI-AGR vs GI-101A | GI-BRN vs GI-101A | |||
| ILMN_1777397 | MSX1 | msh homeobox 1 | 2.41 | 1.34 |
| ILMN_1689146 | GABRP | gamma-aminobutyric acid (GABA) A receptor, pi | 2.33 | 2.00 |
| ILMN_1786720 | PROM1 | prominin 1 (PROM1) | 1.59 | 1.25 |
| ILMN_1793888 | SERPINB5 | serpin peptidase inhibitor, clade B (ovalbumin), member 5 | 1.27 | 2.31 |
| ILMN_1778087 | ANXA8 | annexin A8 | 1.08 | 1.14 |
| ILMN_1665035 | KRT14 | keratin 14 | 1.01 | 1.30 |
| ILMN_1766650 | FOXA1 | forkhead box A1 | −1.84 | −1.53 |
| ILMN_1732398 | SCGB2A1 | secretoglobin, family 2A, member 1 | −2.31 | −1.88 |
| ILMN_1714536 | SCGB1D2 | secretoglobin, family 1D, member 2 | −2.69 | −2.43 |
In Vitro and in Vivo Expression of CD44 and CD133 in GI-AGR and GI-BRN Cells
The results of the expression analysis showed that the GI-AGR and GI-BRN cells had high levels of expression of CD44, and CD133 antigens. To verify these gene expression data at the protein level, we performed fluorescence-activated cell sorting analysis of parental GI-101A, GI-AGR, and GI-BRN cells that had been incubated with antibodies against CD44 and CD133. The GI-101A cells had high levels of expression of CD44, but were heterogeneous in their expression of CD133 (Figure 5A). In contrast, 90% of the GI-AGR and GI-BRN cells had high levels of expression of both CD44 and CD133, suggesting that both the in vitro and in vivo selection processes enriched for cell populations that coexpress CD44 and CD133.
Figure 5.
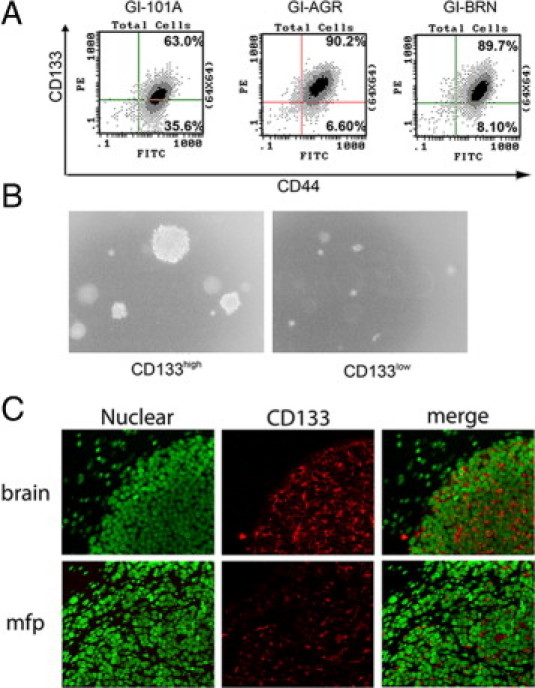
Elevated expression of CD133 in GI-101A cells correlates with growth on hard agar and brain metastasis of these cells. A: GI-101A, GI-AGR, and GI-BRN cells were labeled with fluorescein isothiocyanate-conjugated CD44 and phycoerythrin-conjugated CD133 antibodies and then analyzed using flow cytometry. B: CD133high GI-101A cells formed larger and more numerous colonies than CD133low cells when plated on 0.9% agar. C: Immunofluorescent staining for CD133 in brain metastases and mammary fat pad tumors (mfp) formed by GI-101A cells in mice.
We next asked whether cells with high levels of CD133 expression were the authentic progenitor cells that grow on hard agar and for generation of brain metastases. First, we performed fluorescence activation-based cell sorting to separate parental GI-101A cells into two distinct populations, based on their levels of CD133 expression (CD133high and CD133low). We then placed identical numbers of cells in both of the populations on 0.9% agar and evaluated their growth. The CD133high cells exhibited much greater growth capacity than did the CD133low cells (Figure 5B). Next, we examined the expression of CD133 in brain metastases that were generated by injection of parental GI-101A cells into the common carotid artery in nude mice. Most of the tumor cells isolated from the brain metastases expressed CD133 (Figure 5C). To determine whether parental GI-101A cells expressed CD133 merely because they grew in nude mice, we implanted these cells into the mammary fat pad in female nude mice. We then immunostained the resultant tumors with an anti-CD133 antibody. The CD133 expression in the GI-101A cells that grew in these tumors was highly heterogeneous, compared with that in the cells growing in brain metastases (Figure 5C). These results suggest that primary GI-101A-induced tumors contained subpopulations of cells that expressed CD133 and that these cells were the brain metastasis-initiating cells.
CD44 and CD133 Expression in Clinical Samples of Breast Cancer Brain Metastasis
To determine whether CD44 and CD133 expression is a molecular feature of clinical breast cancer brain metastases, we performed immunohistochemical staining on samples collected from five women with the disease. In each of the samples, we observed expression of both CD44 and CD133 antigens (Figure 6, A and B). Expression of CD44 and CD133 was heterogeneous, in that the intensity of staining was variable, with some samples exhibiting more robust labeling than others. We also noted that not all of the breast cancer cells in the brain specimens expressed CD44 and CD133. In two different cases, CD133 also localized to tumor-associated vascular endothelial cells (data not shown).
Figure 6.
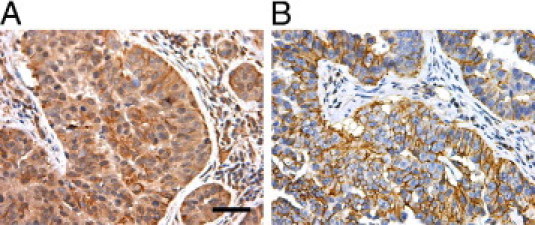
Representative immunohistochemical staining for CD133 (A) and CD44 (B) in clinical samples of human breast cancer brain metastases. Original magnification, ×200. Scale bar = 100 μm.
Experimental Metastasis of Parental GI-101A and GI-AGR Cells to Liver and Lung
We next asked whether the hard agar assay selected for cells with enhanced ability to metastasize to other organs that are relevant to breast cancer metastasis, or if the assay was selective only for cells with potential for forming brain metastases. To address this question, we used experimental models of lung and liver metastasis as described under Materials and Methods, and evaluated metastasis formation 115 days after injection of GI-101A and GI-AGR tumor cells. We selected this time point to make meaningful comparisons with the data from the experimental brain metastasis model (Figure 2B). Both the GI-101A and GI-AGR cancer cells were capable of forming experimental brain metastases within this time frame. We did not detect any evidence of liver metastases (microscopic or macroscopic) in mice (0 of 10 mice) that were injected with GI-101A cells, but 4 of 10 mice injected with GI-AGR cells had liver metastases (P < 0.05; data not shown). Similarly, no evidence of lung metastases was observed in mice injected with GI-101A cells (0 of 10 mice), but 3 of 10 mice injected with GI-AGR cells had lung metastases (P = 0.06; data not shown). Although these experiments showed that the hard agar-selected GI-AGR cells have the potential to experimentally metastasize to other target organs, they also suggest that the brain may offer a more conducive microenvironment for growth of GI-101A and GI-AGR cells.
Discussion
The development of improved therapies for metastasis is one of the primary goals of cancer research. All primary neoplasms are biologically heterogeneous, and the process of metastasis is highly selective1–3,6 Therefore, an in vitro method that can rapidly isolate the precursor cells of metastasis is highly desirable. Here, we have shown that culturing a heterogeneous population of breast cancer cells on 0.9% agar selects for the metastasis-initiating population of the cells and that much of the gene expression analyses of these cells are consistent with those of breast cancer cells selected after successive in vivo cycling in an experimental model of brain metastasis. Similarly, examination of the in vivo selected breast cancer subline GI-BRN revealed that the ability of these cells to grow on hard agar was enhanced. These findings demonstrate that this hard agar assay is a powerful tool for rapidly identifying breast cancer cells capable of generating metastases.
Recent studies suggest that tumors are organized hierarchically into clonally derived populations of cells with different capacities for cell division, and that only small fractions of tumor cells are capable of extensive renewal.30 Tumor- and metastasis-initiating cells have been the focus of much recent investigation, because they are thought to express genes that promote resistance to chemotherapy and programmed cell death.31 Metastatic tumor cells in both mice and humans have also been shown to express genes that are characteristic of stem cells.32 The present results suggest that both the in vitro and in vivo selection processes enrich for subpopulations of breast cancer cells that express CD44 and CD133, two of the markers that have been used to identify stem cells.33 We also found that both CD44 and CD133 were expressed in clinical samples of human breast cancer brain metastases, which lends further credibility to the hard agar assay and the GI-101A cell line used in our study.
CD133 is a cell surface glycoprotein that is thought to participate in maintenance of the topology of the cell membrane.34 CD133 may also be used as a marker to identify tumor precursor cells from several different tumors, including those originating in the brain,35 colon,36 pancreas,37 and lung.38 CD44 is a transmembrane glycoprotein that plays a key regulatory role in a number of diverse processes, such as organ development, neuronal axon guidance, immune regulation, and hematopoiesis.39 Investigators have also used CD44 to isolate tumor-initiating cells from breast40 and prostate41 cancers.
The fact that the hard agar selection process could select for tumor breast cancer cells that coexpress putative stem cell markers was, in itself, not surprising. Indeed, studies by Hamburger and Salmon42 more than three decades ago demonstrated that culture of tumor cells on soft agar could be used to isolate tumor stem cells from different cancers. However, whether the genetic makeup of metastatic cells is identical to that of tumor-initiating cells remains unclear. Recent evidence suggests that a subset of metastasis genes may be superimposed on the tumor-initiating phenotype. For example, a recent study demonstrated that, whereas CD133+ pancreatic tumor cells deficient in expression of the chemokine receptor CXCR4 were capable of forming tumors, only CD133+/CXCR4+ pancreatic tumor cells produced hepatic metastases in mice.43 Reports also indicate that heterogeneity may exist in tumor-initiating cell populations, and that the phenotypes among cancer subtypes or even tumors of the same subtype may not be uniform.30,44 Indeed, an examination of stem cell markers in several breast cancers concluded that expression of the markers differed greatly among breast cancer cell lines as well as primary breast tumors.45 The hard agar assay described here is an unbiased approach, in that it does not rely on previously defined markers for selection of breast cancer cells and thus effectively negates the issue of tumor-cell heterogeneity. Moreover, this assay appears to select only cells capable of initiating metastases.
The gene expression analysis of GI-AGR and GI-BRN cells suggests that expression of genes associated with poorly differentiated basal-subtype breast cancers is up-regulated in metastatic cells, whereas expression of genes characteristic of well-differentiated breast cancers is down-regulated. For example, MSX1 is a member of the homeobox gene family that normally regulates the growth and development of the mammary gland.21 Dysregulated expression of MSX1 has been implicated in breast cancer progression, because of its ability to up-regulate expression of the cell cycle-regulatory protein cyclin D1,46 and we found that MSX1 was highly expressed in GI-AGR and GI-BRN cells, whereas it was minimally expressed by GI-101A parental tumor cells. Alternatively, expression of FOXA1, a member of the forkhead family of transcription factors that is thought to prevent metastatic progression47 was significantly lower in GI-AGR and GI-BRN cells than in GI-101A cells. We also found that the Wnt signaling pathway is deregulated in GI-AGR and GI-BRN cells. Signaling mediated by Wnt proteins regulates a number of mammalian developmental processes, including cell differentiation, stem cell self-renewal, and epithelial-mesenchymal interactions. Inappropriate activation of the Wnt signaling pathway is a major feature of human neoplasia, and oncogenic activation of this pathway can occur at many levels.48 In the present study, expression of the two negative regulators of Wnt signaling, sFRP1 and DKK3, was significantly lower in the GI-AGR and GI-BRN cells than in the GI-101A cells.
Notably, the patterns of gene expression in metastatic GI-AGR and GI-BRN cells were not completely identical, in that 47% of the genes were differentially expressed in these two cell lines. One possible explanation is related to the microenvironment in which the tumor cells grew. The tissue microenvironment can have a profound influence on the pattern of gene expression in a tumor.49 In a study in which human renal carcinoma cells were implanted into different organs in mice, expression of the angiogenic protein basic fibroblast growth factor was 10 to 20 times higher in growing kidney tumors than in growing skin tumors.50 A systematic examination of the genes differentially expressed in GI-AGR and GI-BRN cells could prove informative.
Although the present results demonstrate that the hard agar assay can be used to isolate the breast cancer tumor cell variants that possess enhanced ability to metastasize, the underlying mechanism of this cell-selection process remains unclear. Culturing tumor cells on a hard agar medium selects for variants with increased ability to undergo anchorage-independent growth, which for many years has been linked with the process of metastasis.17 High mechanical pressure in the hard agar network may play an important role in the selection of tumor-cell subpopulations. Prolonged exposure to elevated mechanical pressure has been shown to diminish apoptosis51 and stimulate cell division52 of some tumor cells and, moreover, can decrease survival duration in some tumor models.53
It is widely appreciated that the outcome of metastasis is determined by the interactions that take place between specific populations of metastatic cells and their organ microenvironment.1 The results of the present studies with GI-101A breast cancer cells indicate that the hard agar assay selects for cells that are capable of growth in multiple target organs of breast metastasis (brain, liver, lung). The data also suggest that GI-101A cells may possess some degree of intrinsic tropism for brain tissue. Indeed, parental GI-101A cells formed experimental brain metastases in 50% of injected mice, but were not successful in creating discernible metastases in either the liver or lungs of nude mice within a similar time frame. The hard agar assay selected cells with enhanced metastatic potential for the brain and increased ability for generating metastases in other target organs, such as liver and lung. Our findings warrant extension of the hard agar assay to other tumor cell lines, and to clinical specimens for rapid identification and expression profiling of metastasis-initiating cells.
Acknowledgments
We thank Don Norwood for critical editorial review and Lola López for expert assistance with the preparation of this manuscript and Donna Reynolds for technical expertise (all from the M.D. Anderson Cancer Center, Houston, TX.).
Footnotes
Supported in part by M.D. Anderson's Cancer Center support core grant CA16672 and by grant 1U54CA143837 from the National Cancer Institute, NIH.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.047.
Supplemental data
References
- 1.Langley R.R., Fidler I.J. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- 2.Fidler I.J., Kripke M.L. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 3.Chiang A.C., Massagué J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talmadge J.E., Wolman S.R., Fidler I.J. Evidence for the clonal origin of spontaneous metastases. Science. 1982;217:361–363. doi: 10.1126/science.6953592. [DOI] [PubMed] [Google Scholar]
- 5.Wang X., Wang M., MacLennan G.T., Abdul-Karim F.W., Eble J.N., Jones T.D., Olobatuyi F., Eisenberg R., Cummings O.W., Zhang S., Lopez-Beltran A., Montironi R., Zheng S., Lin H., Davidson D.D., Cheng L. Evidence for common clonal origin of multifocal lung cancers. J Natl Cancer Inst. 2009;101:560–570. doi: 10.1093/jnci/djp054. [DOI] [PubMed] [Google Scholar]
- 6.Fidler I.J. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 7.Stoecklein N.H., Hosch S.B., Bezler M., Stern F., Hartmann C.H., Vay C., Siegmund A., Scheunemann P., Schurr P., Knoefel W.T., Verde P.E., Reichelt U., Erbersdobler A., Grau R., Ullrich A., Izbicki J.R., Klein C.A. Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell. 2008;13:441–453. doi: 10.1016/j.ccr.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Palmieri D., Chambers A.F., Felding-Habermann B., Huang S., Steeg P.S. The biology of metastasis to a sanctuary site. Clin Cancer Res. 2007;12:1656–1662. doi: 10.1158/1078-0432.CCR-06-2659. [DOI] [PubMed] [Google Scholar]
- 9.Gupta G.P., Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Kang Y., Siegel P.M., Shu W., Drobnjak M., Kakonen S.M., Cordón-Cardo C., Guise T.A., Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 11.Bos P.D., Zhang X.H., Nadal C., Shu W., Gomis R.R., Nguyen D.X., Minn A.J., van de Vijver M.J., Gerald W.L., Foekens J.A., Massagué J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minn A.J., Gupta G.P., Siegel P.M., Bos P.D., Shu W., Giri D.D., Viale A., Olshen A.B., Gerald W.L., Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fidler I.J., Nicolson G.L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst. 1976;57:1199–1202. doi: 10.1093/jnci/57.5.1199. [DOI] [PubMed] [Google Scholar]
- 14.Poste G., Doll J., Hart I.R., Fidler I.J. In vitro selection of murine B16 melanoma variants with enhanced tissue-invasive properties. Cancer Res. 1980;40:1636–1644. [PubMed] [Google Scholar]
- 15.Buick R.N., Fry S.E., Salmon S.E. Application of in vitro soft agar techniques for growth of tumor cells to the study of colon cancer. Cancer. 1980;45:1238–1242. doi: 10.1002/1097-0142(19800315)45:5+<1238::aid-cncr2820451333>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 16.Maruyama K., East J.L., Wagner S.H., Dmochowski L. In vitro transformation of cells from human neoplasms. Bibl Haematol. 1975;40:85–92. doi: 10.1159/000397521. [DOI] [PubMed] [Google Scholar]
- 17.Cifone M.A., Fidler I.J. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci USA. 1980;77:1039–1043. doi: 10.1073/pnas.77.2.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L., Price J.E., Fan D., Zhang R.D., Bucana C.D., Fidler I.J. Correlation of growth capacity of human tumor cells in hard agarose with their in vivo proliferative capacity at specific metastatic sites. J Natl Cancer Inst. 1989;81:1406–1412. doi: 10.1093/jnci/81.18.1406. [DOI] [PubMed] [Google Scholar]
- 19.Lev D.C., Kiriakova G., Price J.E. Selection of more aggressive variants of the gI101A human breast cancer cell line: a model for analyzing the metastatic phenotype of breast cancer. Clin Exp Metastasis. 2003;20:515–523. doi: 10.1023/a:1025837631179. [DOI] [PubMed] [Google Scholar]
- 20.Langley R.R., Fan D., Guo L., Zhang C., Lin Q., Brantley E.C., McCarty J.H., Fidler I.J. Generation of an immortalized astrocyte cell line from H-2Kb-tsA58 mice to study the role of astrocytes in brain metastasis. Int J Oncol. 2009;35:665–672. doi: 10.3892/ijo_00000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh K., Ginsburg E., Vonderhaar B.K. Msx-1 and Msx-2 in mammary gland development. J Mammary Gland Biol Neoplasia. 2004;9:195–205. doi: 10.1023/B:JOMG.0000037162.84758.b5. [DOI] [PubMed] [Google Scholar]
- 22.Symmans W.F., Fiterman D.J., Anderson S.K., Ayers M., Rouzier R., Dunmire V., Stec J., Valero V., Sneige N., Albarracin C., Wu Y., Ross J.S., Wagner P., Theriault R.L., Arun B., Kuerer H., Hess K.R., Zhang W., Hortobagyi G.N., Pusztai L. A single-gene biomarker identifies breast cancers associated with immature cell type and short duration of prior breastfeeding. Endocr Relat Cancer. 2005;12:1059–1069. doi: 10.1677/erc.1.01051. [DOI] [PubMed] [Google Scholar]
- 23.Jones C., Mackay A., Grigoriadis A., Cossu A., Reis-Filho J.S., Fulford L., Dexter T., Davies S., Bulmer K., Ford E., Parry S., Budroni M., Palmieri G., Neville A.M., O'Hare M.J., Lakhani S.R. Expression profiling of purified normal human luminal and myoepithelial breast cells: identification of novel prognostic markers for breast cancer. Cancer Res. 2004;64:3037–3045. doi: 10.1158/0008-5472.can-03-2028. [DOI] [PubMed] [Google Scholar]
- 24.Sorlie T., Tibshirani R., Parker J., Hastie T., Marron J.S., Nobel A., Deng S., Johnsen H., Pesich R., Geisler S., Demeter J., Perou C.M., Lønning P.E., Brown P.O., Børresen-Dale A.L., Botstein D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abd El-Rehim D.M., Pinder S.E., Paish C.E., Bell J., Blamey R.W., Robertson J.F., Nicholson R.I., Ellis I.O. Expression of luminal and basal cytokeratins in human breast carcinoma. J Pathol. 2004;203:661–671. doi: 10.1002/path.1559. [DOI] [PubMed] [Google Scholar]
- 26.Rakha E.A., Reis-Filho J.S., Ellis I.O. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 27.Brenton J.D., Carey L.A., Ahmed A.A., Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin Oncol. 2005;23:7350–7360. doi: 10.1200/JCO.2005.03.3845. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi N., Ito E., Azuma S., Honma R., Yanagisawa Y., Nishikawa A., Kawamura M., Imai J., Tatsuta K., Inoue J., Semba K., Watanabe S. FoxA1 as a lineage-specific oncogene in luminal type breast cancer. Biochem Biophys Res Commun. 2008;365:711–717. doi: 10.1016/j.bbrc.2007.11.064. [DOI] [PubMed] [Google Scholar]
- 29.Lacroix M. Significance, detection and markers of disseminated breast cancer cells. Endocr Relat Cancer. 2006;13:1033–1067. doi: 10.1677/ERC-06-0001. [DOI] [PubMed] [Google Scholar]
- 30.Visvader J.E., Lindeman G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 31.Hirschmann-Jax C., Foster A.E., Wulf G.G., Nuchtern J.G., Jax T.W., Gobel U., Goodel M.A., Brenner M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glinsky G.V., Berezovska O., Glinskii A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke M.F., Dick J.E., Dirks P.B., Eaves C.J., Jamieson C.H., Jones D.L., Visvader J., Weissman I.L., Wahl G.M. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 34.Mizrak D., Brittan M., Alison M.R. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 35.Singh S.K., Clarke I.D., Terasaki M., Bonn V.E., Hawkins C., Squire J., Dirks P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 36.Ricci-Vitiani L., Lombardi D.G., Pilozzi E., Biffoni M., Todaro M., Peschle C., De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 37.Hermann P.C., Huber S.L., Herrler T., Aicher A., Ellwart J.W., Guba M., Bruns C.J., Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S., Roz E., Caserini R., Lo Vullo S., Camerini T., Mariani L., Delia D., Calabrò E., Pastorino U., Sozzi G. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponta H., Sherman L., Herrlich P.A. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 40.Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [Erratum appeared in Proc Natl Acad Sci U S A 2003, 27;100:6890] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins A.T., Berry P.A., Hyde C., Stower M.J., Maitland N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 42.Hamburger A.W., Salmon S.E. Primary bioassay of human tumor stem cells. Science. 1977;197:461–463. doi: 10.1126/science.560061. [DOI] [PubMed] [Google Scholar]
- 43.Hermann P.C., Huber S.L., Herrler T., Aicher A., Ellwart J.W., Guba M., Bruns C.J., Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Stingl J., Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7:791–799. doi: 10.1038/nrc2212. [DOI] [PubMed] [Google Scholar]
- 45.Hwang-Verslues W., Kuo W.-H., Chang P.H., Pan C.C., Wang H.H., Tsai S.T., Jeng Y.M., Shew J.Y., Kung J.T., Chen C.H., Lee E.Y., Chang K.J., Lee W.H. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS One. 2009;4:e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu G., Lee H., Price S.M., Shen M.M., Abate-Shen C. Msx homeobox genes inhibit differentiation through upregulation of cyclin D1. Development. 2001;128:2373–2384. doi: 10.1242/dev.128.12.2373. [DOI] [PubMed] [Google Scholar]
- 47.Nakshatri H., Badve S. FOXA1 as a therapeutic target for breast cancer. Expert Opin Ther Targets. 2007;11:507–514. doi: 10.1517/14728222.11.4.507. [DOI] [PubMed] [Google Scholar]
- 48.Smalley M.J., Dale T.C. Wnt signalling in mammalian development and cancer. Cancer Metastasis Rev. 1999;18:215–230. doi: 10.1023/a:1006369223282. [DOI] [PubMed] [Google Scholar]
- 49.Nakamura T., Fidler I.J., Coombes K.R. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res. 2007;67:139–148. doi: 10.1158/0008-5472.CAN-06-2563. [DOI] [PubMed] [Google Scholar]
- 50.Singh R.K., Bucana C.D., Gutman M., Fan D., Wilson M.R., Fidler I.J. Organ site-dependent expression of basic fibroblast growth factor in human renal cell carcinoma cells. Am J Pathol. 1994;145:365–374. [PMC free article] [PubMed] [Google Scholar]
- 51.Helmlinger G., Netti P.A., Lichtenbeld H.C., Melder R.J., Jain R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nature Biotechnol. 1997;15:778–783. doi: 10.1038/nbt0897-778. [DOI] [PubMed] [Google Scholar]
- 52.Nathan S.S., DiResta G.R., Casas-Ganem J.E., Hoang B.H., Sowers R., Yang R., Huvos A.G., Gorlick R., Healey J.H. Elevated physiologic tumor pressure promotes proliferation and chemosensitivity in human osteosarcoma. Clin Cancer Res. 2005;11:2389–2397. doi: 10.1158/1078-0432.CCR-04-2048. [DOI] [PubMed] [Google Scholar]
- 53.Craig D.H., Owen C.R., Conway W.C., Walsh M.F., Downey C., Basson M.D. Colchicine inhibits pressure-induced tumor cell implantation within surgical wounds and enhances tumor-free survival in mice. J Clin Invest. 2008;118:3170–3180. doi: 10.1172/JCI34279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.