

Abstract
Although the early growth response-2 (Egr-2, alias Krox20) protein shows structural and functional similarities to Egr-1, these two related early-immediate transcription factors are nonredundant. Egr-2 plays essential roles in peripheral nerve myelination, adipogenesis, and immune tolerance; however, its regulation and role in tissue repair and fibrosis remain poorly understood. We show herein that transforming growth factor (TGF)-β induced a Smad3-dependent sustained stimulation of Egr2 gene expression in normal fibroblasts. Overexpression of Egr-2 was sufficient to stimulate collagen gene expression and myofibroblast differentiation, whereas these profibrotic TGF-β responses were attenuated in Egr-2–depleted fibroblasts. Genomewide transcriptional profiling revealed that multiple genes associated with tissue remodeling and wound healing were up-regulated by Egr-2, but the Egr-2–regulated gene expression profile overlapped only partially with the Egr-1–regulated gene profile. Levels of Egr-2 were elevated in lesional tissue from mice with bleomycin-induced scleroderma. Moreover, elevated Egr-2 was noted in biopsy specimens of skin and lung from patients with systemic sclerosis. These results provide the first evidence that Egr-2 is a functionally distinct transcription factor that is both necessary and sufficient for TGF-β–induced profibrotic responses and is aberrantly expressed in lesional tissue in systemic sclerosis and in a murine model of scleroderma. Together, these findings suggest that Egr-2 plays an important nonredundant role in the pathogenesis of fibrosis. Targeting Egr-2 might represent a novel therapeutic strategy to control fibrosis.
Scleroderma or systemic sclerosis (SSc) is an acquired connective tissue disease of unknown etiology associated with progressive fibrosis in the skin and internal organs.1–3 There is no effective therapy to prevent or control the progression of fibrosis in SSc. Transforming growth factor (TGF)-β is a potent inducer of collagen synthesis, myofibroblast differentiation, and epithelial-mesenchymal transition; and is implicated in both physiological and pathological tissue repair.4,5 The intracellular signaling pathways that mediate fibrotic TGF-β responses remain incompletely understood.
The early growth response (Egr) transcription factors belong to a family comprising Egr-1 (alias Krox-20), Egr-2 (alias Krox-24), Egr-3, and Egr-4, along with their endogenous inhibitors, NGFI-A binding protein (NAB1 and NAB2).6,7 These pleiotropic early-immediate gene products are induced in response to multiple extracellular signals, such as growth factors, cytokines, hypoxia, and mechanical forces associated with injury and cellular stress. Egr-1, Egr-2, and Egr-3 share conserved zinc finger DNA-binding domains that recognize a 9-bp GC-rich DNA sequence called Egr-binding element (EBS) found in multiple target gene promoters.8 The induction of Egr-1 is characteristically rapid and transient.9 In contrast to Egr-1, which has been extensively investigated, the regulation and function of Egr-2 and Egr-3 remain less well characterized. These transcription factors show a high degree of homology to each other and to Egr-1 but are regulated by distinct intracellular signaling pathways in a cell type–specific manner. Moreover, it is becoming clear that individual members of the Egr gene family serve nonredundant and occasionally even opposing biological functions. The functional divergence is highlighted by genetic studies that showed that, although Egr-1–null mice have no spontaneous phenotype,10 targeting Egr-2 resulted in early mouse lethality due to defective myelination.11 Functional divergence is also evident in immune regulation: Egr-2 appears to be required for immune tolerance, whereas Egr-1 enhances T-cell activity in response to T-cell receptor engagement.12,13 These and similar observations underline the significant differences that exist between these two structurally related members of the Egr transcription factor family.
In previous studies14 investigating the expression and function of Egr-1 in the context of fibrogenesis, we showed that Egr-1 was required for TGF-β–induced fibroblast activation and played an important role in both physiological and pathological fibrogenesis. The expression of Egr-1 was rapidly induced by TGF-β in skin and lung fibroblasts, and Egr-1 was sufficient to stimulate the transcription of COL1A2 and other TGF-β–inducible genes.14,15 Moreover, mice with targeted deletion of Egr-1 were protected from bleomycin-induced fibrosis.16 In contrast to Egr-1, the expression, regulation, and physiological roles of Egr-2 in connective tissue metabolism remain incompletely understood. Therefore, the present studies focused on Egr-2 in the context of TGF-β–mediated fibrogenesis in vivo and in vitro. The results demonstrate that Egr-2 is markedly elevated in skin and lung biopsy specimens from patients with SSc and in a mouse model of scleroderma. In explanted normal fibroblasts, TGF-β stimulated the expression of Egr-2; however, the kinetics and signal transduction pathways were distinct from those associated with Egr-1. Overexpression of Egr-2 in these cells was associated with induction of profibrotic gene responses. Taken together, these findings identify Egr-2 as a novel TGF-β–inducible mediator of profibrotic responses that is aberrantly expressed in SSc and murine fibrosis and suggest an important nonredundant functional role for Egr-2 in pathogenesis.
Materials and Methods
Cell Culture and Reagents
Primary cultures of skin fibroblasts were established by explantation from biopsy specimens of healthy adults (n = 3) or patients with diffuse cutaneous SSc (n = 3) or from neonatal foreskin specimens.17 The protocols for skin biopsies were approved by the Institutional Review Board at Northwestern University, Chicago, IL. Fibroblasts were maintained in Eagle's minimum essential medium or Dulbecco's modified Eagle medium, supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland), 50 μg/mL penicillin, and 50 μg/mL streptomycin, in a humidified atmosphere of 5% CO2 at 37°C; and studied between passages 2 and 8.17 At confluence, serum-free media supplemented with 0.1% bovine serum albumin (BSA) were added to the cultures for 16 hours before the addition of TGF-β2 or TGF-β1 (both from Peprotech, Rocky Hill, NJ). Mouse embryonic fibroblasts explanted from Smad3−/− or wild-type embryos were maintained in Dulbecco's modified Eagle medium. In selected experiments, LY294002 (Cell Signaling, Danvers, MA), SB431542 (Glaxo Smith Kline, King of Prussia, PA), or U0126 (Cell Signaling) was added to the cultures.
RNA Isolation and Real-Time Quantitative PCR
At the end of each experiment, cultures were harvested and RNA was isolated using minikits (RNeasy Plus; Qiagen, Valencia, CA) and examined by real-time quantitative PCR (qPCR).18 RNA, 1 μg, was used for cDNA synthesis in a 20-μL reaction volume using supermix (cDNA Synthesis Supermix; Quanta Biosciences, Gaithersburg, MD): 8 μL cDNA, 2 μL primers (2 μmol/L each), and 10 μL ×2 mix (Power SYBR Mastermix; Applied Biosystems, Foster City, CA). qPCR was performed in triplicate on a thermocycler (ABI 7300; Applied Biosystems). Data were normalized to 18S RNA, and fold change was represented as follows: 2−ΔΔCt (2−[(Ct target-Ct 18S)treatment-(Ct target-Ct 18S)nontreatment]). The primers used for qPCR are listed in Table 1.18
Table 1.
Primers Used for qPCR
| Gene name | Primer sequences | RNA accession no. |
|---|---|---|
| 18S ribosomal RNA | Olg1 5′-CCCCATGAACGAGGGAATT-3′ | NR_003286 |
| Olg2 5′-GGGACTTAATCAACGCAAGCTT-3′ | ||
| GAPDH | Olg3 5′-CATGAGAAGTATGACAACAGCCT-3′ | NM_002046 |
| Olg4 5′-AGTCCTTCCACGATACCAAAGT-3′ | ||
| COL1A2 | Olg10 5′-CGGACGACCTGGTGAGAGA-3′ | NM_000089 |
| Olg11 5′-CATTGTGTCCCCTAATGCCTT-3′ | ||
| ACTA2 (α-SMA) | Olg20 5′-CAGGGCTGTTTTCCCATCCAT-3′ | NM_001613 |
| Olg21 5′-GCCATGTTCTATCGGGTACTTC-3′ | ||
| CTGF (CCN2) | Olg18 5′-AGCTGACCTGGAAGAGAACATTAAG-3′ | NM_001901 |
| Olg19 5′-GATAGGCTTGGAGATTTTGGGAGTA-3′ | ||
| Egr-1 | Olga 5′-TGCGGCAGAAGGACAAGAAAGC-3′ | NM_001964 |
| Olgb 5′-TGAGGAAGGGAAGCTGCTGACC-3′ | ||
| Egr-2 | Olg26 5′-AACGGAGTGGCCGGAGAT-3′ | NM_000399 |
| Olg27 5′-ATGGGAGATCCAACGACCTCTT-3′ | ||
| Egr-3 | Olg31 5′-GCGACCTCTACTCAGAGCC-3′ | NM_004430 |
| Olg32 5′-ATGGGGAAGAGATTGCTGTCC-3′ | ||
| COL1A1 | Olg149 5′-GCTGGTGTGATGGGATTC-3′ | NM_000088 |
| Olg150 5′-GGGAACACCTCGCTCT-3′ | ||
| Pai1 | Olg151 5′-CGCCTCTTCCACAAATCA-3′ | NM_000602 |
| Olg162 5′-GCAGTTCCAGGATGTCGTA-3′ | ||
| Plod2 | Olg179 5′-CTTTAGTGTGGATGCAGATGTTG-3′ | NM_182943 |
| Olg180 5′-GACTCAATGCTCCCCAGAAA-3′ | ||
| Col5A1 | Olg230 5′-TCGCTTACAGAGTCACCAAAG-3′ | NM_000093 |
| Olg231 5′-GTTGTAGATGGAGACCAGGAAG-3′ | ||
| Itgb1 | Olg222 5′-CAGCGCATATCTGGAAATTTGG-3′ | NM_002211 |
| Olg223 5′-TCTCCAGCAAAGTGAAACCC-3′ |
Primer sequences were obtained using computer software (Primer Express 3.0; Primerbank Boston, MA) or were from published sequences.14,18 Most primer pairs span two exons, except the intronless 18S ribosomal RNA. Primers were tested by dissociation curve analysis to ensure only a single amplicon. The base pairs that the forward primer starts and reverse primer ends are based on RNA sequence. All primers are located inside the coding sequence.
Plasmids and Transient Transfection Assays
The Egr–2–luciferase (luc) construct harbors a truncated human Egr2 gene promoter with 5′ ends at –1.3 kb fused to firefly luc.19 pCMV Egr-1 or pCMV Egr-2 contains the full-length Egr-1 or Egr-2 coding sequence, respectively.20 772COL1A2–CAT harbors the truncated human COL1A2 promoter with 5′ ends at −772 bp fused to chloramphenicol acetyltransferase (CAT).21 p[EBS]4-luc harbors four copies of the minimal Egr-1–responsive element linked to luc.22 ASMA–luc harbors a 1.1-kb promoter fragment from the mouse alpha smooth muscle actin (ASMA; gene symbol, ACTA) gene upstream of luc.23 Fibroblasts at early confluence were transfected with the indicated constructs using a kit (SuperFect Transfection kit; Qiagen). Cultures were incubated in serum-free media containing 0.1% BSA for 16 hours, followed by TGF-β2 for a further 24 hours. Cultures were harvested and whole cell lysates were assayed for their chloramphenicol acetyltransferase activities by liquid scintillation counting or luc activities using the dual-luc reporter assay system (Promega, Madison, WI). In each experiment, Renilla luc pRL-TK (Promega) was cotransfected as a control for transfection efficiency.24 Transient transfection experiments were performed in triplicate and repeated at least twice, with consistent results.
Short-Interfering RNA-Mediated Knockdown and Adenovirus Infection
Duplex oligo short-interfering RNAs (siRNAs) specific to human Smad3 (Pierce, Rockford, IL) or Egr-2 (Dharmacon, Lafayette, CO) and scrambled control siRNA were used to deplete endogenous Smad3 or Egr-2. Skin fibroblasts in serum-free media containing 0.1% BSA were transfected with indicated siRNA at a final concentration of 10 nmol/L. Sixteen hours later, fresh media containing TGF-β were added and the incubations were continued for a further 24 hours. Knockdown efficiency was determined by measuring endogenous protein or mRNA levels by Western analysis or real-time qPCR. For adenovirus (Ad) infection, confluent fibroblasts in serum-free media were infected with Ad–Egr-2 (Vector Biolabs, Philadelphia, PA) or Ad GFP (green fluorescent protein) and incubated for up to 48 hours before harvesting.
Confocal Immunofluorescence Microscopy
Fibroblasts (10,000 cells per well) were seeded onto eight-well chamber-glass slides (Lab-Tek II; Nalge Nunc International, Naperville, IL) and incubated in serum-free Eagle's minimum essential medium with 0.1% BSA for 16 hours. Fresh media with TGF-β2 (10 ng/mL) were added, and the incubations were continued for a further 1 to 24 hours. At the end of the experiments, cells were fixed, permeabilized, and incubated with primary antibodies to Egr-2 at 1:200 dilution (Covance, Princeton, NJ) or to type I collagen at 1:100 dilution (Southern Biotech, Birmingham, AL).14 Cells were then washed with PBS and incubated with secondary antibodies at 1:200 dilution (Alexa Fluor 488 and 594; Invitrogen, Carlsbad, CA) and viewed under a confocal microscope (Nikon C1Si).25
Western Analysis
At the end of each experiment, fibroblasts were harvested, lysed in radioimmunoprecipitation assay buffer supplemented with 1× protease inhibitor cocktail and whole cell lysates (100 μg), and subjected to Western analysis using antibodies to Egr-2 (Covance), type I collagen (Southern Biotech), and glyceraldehyde-3-phosphate dehydrogenase (Zymed, San Francisco, CA). Images were detected by electrochemiluminescence reagents (Pierce) and developed on film.
Microarray Procedures and Data Analysis
To examine genomewide changes in gene expression induced by Egr-2, serum-starved confluent fibroblasts were infected with Ad–Egr-1, Ad–Egr-2, or Ad-GFP using multiplicity of infection 50, previously shown to be optimal for high-level expression. Close to 100% of infected fibroblasts showed strong GFP expression by immunofluorescence microscopy. After 48 hours of incubation, total RNA was isolated from three independent cultures using kits (RNeasy Mini Plus Kits; Qiagen). The integrity of RNA was determined using a bioanalyzer (Agilent, Santa Clara, CA). Fluorescently labeled cDNA was prepared using labeling kits (Ambion, Austin, TX), followed by hybridization to microarray chips (Illumina Human HT-12 Version 3) containing 44,000 probes (Illumina, San Diego, CA).
Raw signal intensities for each probe were obtained using data analysis software (Beadstudio; Illumina) and imported to the Bioconductor lumi package for transformation and normalization.26–28 The data were preprocessed using a variance stabilization transformation method,28 followed by quantile normalization. Data from probes that produced signals near or lower than background levels (estimated based on Illumina negative control probes) with all samples were discarded. Genes that showed greater than twofold up- or down-regulation in Egr-2–expressing fibroblasts compared with controls at 48 hours were subjected to further analysis.
IHC in a Scleroderma Model
To examine the tissue expression of Egr-2 in vivo, scleroderma was induced in mice by bleomycin injections (Jackson Laboratory, Bar Harbor, ME).29 The animal protocols were institutionally approved by the Northwestern University Animal Care and Use Committee. Filter-sterilized bleomycin (20 μg per mouse dissolved in PBS) (Mayne Pharma, Paramus, NJ) or PBS was administered by daily s.c. injections into the shaved backs of 6- to 8-week-old female BALB/c mice.30 Each experimental group consisted of at least five mice. At the end of the experiments, mice were sacrificed and lesional skin was harvested.25 Sections (4 μm) were deparaffinized, rehydrated, and immersed in TBS–T buffer (Tris-buffered saline and 0.1% Tween-20); and treated with target retrieval solution (DAKO, Carpinteria, CA) at 95°C for 10 minutes, followed by incubation with primary antibodies against Egr-2 (Covance) at 1:200 dilution (anti–Egr-2 antibody was used for both mouse and human sections). Secondary antibodies (Histomouse-Max kit; Zymed) at 1:200 dilution were used to detect bound antibodies. Substitution of the primary antibodies with isotype-matched irrelevant IgG served as negative controls. After counterstaining with hematoxylin, sections were mounted (Permount; Fisher Scientific, Pittsburgh, PA) and viewed under a microscope (Olympus BH-2).25 Unambiguously immunopositive fibroblastic cells were counted by a blinded observed. Results are shown as the mean ± SEM from a representative section from each of five mice per experimental group.
Immunodetection of Egr-2 in Skin and Lung Biopsy Specimens
Skin biopsy specimens from the affected forearm from patients with early diffuse cutaneous SSc or healthy adult control subjects were obtained under protocols approved by the Institutional Review Boards for Human Studies at Thomas Jefferson University, Philadelphia, PA, or Northwestern University. Paraffin-embedded or frozen sections (4-μm thick) were immunostained with the same primary antibody to Egr-2 (Covance) as used in the mouse studies (previously described), followed by incubation with rabbit IgG secondary antibodies (Alexa Fluor 488; Invitrogen). The entire skin section was scanned at ×20 magnification at four different microscopic fields spanning the dermis under a confocal laser scanning microscope (Zeiss LSM 510 META). The computer then generated 2.5-dimensional images plotting the fluorescence intensity of each microscopic field. Sections were analyzed with software (Image J) that calculated the sum of the intensity of each pixel in a given microscopic field as the integrated density of fluorescence. Each biopsy specimen is represented as the mean of four integrated density of fluorescence values per specimen.31
Egr-2 expression was also examined in lungs from patients with SSc-associated pulmonary fibrosis undergoing lung transplantation and in normal donor lungs. The biopsy protocols were approved by the Institutional Review Board for Human Studies at the University of Pittsburgh, Pittsburgh. Paraffin-embedded sections of lung tissue (6 μm) were immunostained with antibodies to Egr-2 (Covance) at 1:200 dilution, followed by incubation with biotinylated anti-rabbit IgG secondary antibodies (sc-2089; Santa Cruz Biotechnology, Santa Cruz, CA) at 1:200 dilution. Tissue sections from a lung tumor were used as positive control, and substitution of nonspecific rabbit IgG for Egr-2 antibody was used as a negative control.32 Images were taken using a microscope (Zeiss Axioskop) with a CRi Nuance spectral camera.
Statistical Analysis
Each experiment was repeated at least three times. Statistical analysis was performed (GraphPad Prism 4; GraphPad Software, La Jolla, CA) using the Student's t-test. The results are shown as the mean ± SEM. P < 0.05 was considered statistically significant.
Results
TGF-β2 Stimulates Egr2 Gene Expression and Egr-2 Promoter Activity
Previously, TGF-β induced rapid and transient Egr-1 expression in skin and lung fibroblasts.14 In the present studies, we focused our attention on the regulation of Egr-2. The results of qPCR showed that TGF-β2 stimulated Egr-2 mRNA expression in foreskin fibroblasts in a time- and dose-dependent manner (Figure 1, A and B). An identical stimulation of mRNA expression was seen in normal adult skin fibroblasts and fibroblasts from a patient with diffuse cutaneous SSc (Figure 1C). The levels of Egr-2 showed a similar time-dependent increase in TGF-β2–treated fibroblasts (Figure 1D). TGF-β1 elicited a comparable stimulation of Egr-2 (data not shown). To further characterize the regulation of Egr-2, transcriptional stimulation by TGF-β was examined using an Egr-2 reporter construct transiently transfected into mouse 3T3 cells. The results showed that TGF-β induced a dose-dependent increase in Egr-2 promoter activity in 3T3 fibroblasts (Figure 1E).
Figure 1.
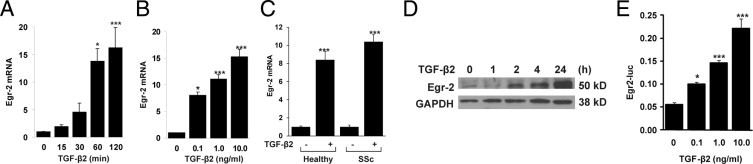
TGF-β stimulates Egr-2 expression. Confluent foreskin fibroblasts (A, B, and D) or healthy adult skin and lesional SSc skin fibroblasts (C) were incubated in serum-free media with TGF-β2 (10 ng/mL, unless otherwise indicated) for the indicated periods (A and D) or for 120 minutes (B and C). A through C: Total RNA was subjected to qPCR. The results represent the mean ± SEM of triplicate determinations from three independent experiments. *P < 0.05; ***P < 0.001. D: Whole cell lysates were subjected to Western analysis. Representative autoradiograms are shown. E: NIH-3T3 fibroblasts were transiently transfected with Egr-2–luc and incubated with TGF-β2 for 24 hours. Cell lysates were assayed for their luc activities.
Time-course experiments were performed to compare the Egr-2 response with that of the Egr-1 response. The results showed that the stimulation of Egr-2 mRNA and protein by TGF-β was somewhat delayed compared with the rapid stimulation of Egr-1, but the response was substantially more durable (Figure 2, A and B). To further examine the regulation of Egr-2, fibroblasts incubated with TGF-β2 for various periods were examined by confocal immunofluorescence microscopy. The results showed that, although only low levels of Egr-2 could be seen localized in both the cytoplasm and nucleus of untreated fibroblasts, incubation with TGF-β2 for 24 hours resulted in a long-lived increase and nuclear accumulation of Egr-2 (Figure 2C).
Figure 2.
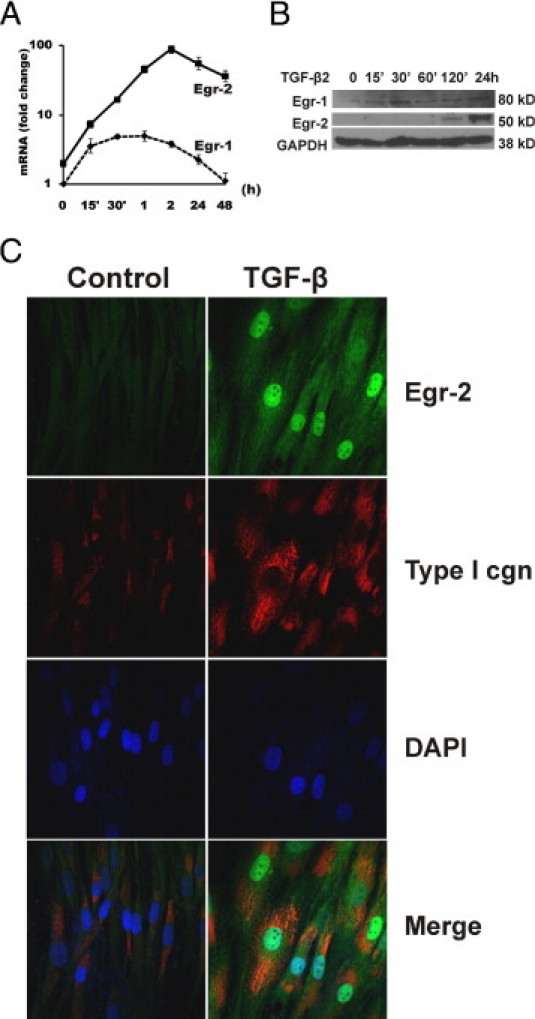
Sustained Egr-2 stimulation by TGF-β. Foreskin fibroblasts at confluence were incubated in serum-free media with TGF-β (10 ng/mL or as indicated) for the indicated periods (A and B) or for 24 hours (C). A: qPCR. The results represent the mean ± SEM of triplicate determinations from three independent experiments. B: Western analysis. C: Confocal microscopy. Cells were fixed and immunostained with antibodies to Egr-2 (green), type I collagen as a positive control (red), or DAPI (blue). Representative immunofluorescence photomicrographs are shown. Original magnification, ×100.
Intracellular Pathways Mediating Egr-2 Stimulation
Next, a series of studies was undertaken to characterize the intracellular signaling mechanisms underlying the stimulation of Egr-2 expression by TGF-β. Pretreatment of the fibroblast cultures with either U0126 (a MAP/ERK inhibitor) or SB431542 (a TGF-β type 1 receptor kinase inhibitor) dramatically attenuated the stimulation of Egr-2 mRNA (Figure 3A). In contrast, LY294002 (a phosphoinositol-3-kinase inhibitor) had only a partial effect. Because these results implicated canonical Smad signaling in TGF-β2–induced Egr-2 regulation, we proceeded to examine the role of Smad3 in their response using two complementary approaches. First, we used RNA interference to suppress endogenous Smad3. Transfection of fibroblasts with Smad3-specific siRNA consistently achieved a >50% reduction in Smad3 mRNA and protein expression, whereas a scrambled siRNA had no effect (Figure 3B and data not shown). TGF-β–induced stimulation of Egr-2 mRNA was approximately 50% lower in fibroblasts with Smad3-specific knockdown, whereas little change in Egr-2 stimulation was noted in fibroblasts transfected with scrambled siRNA, suggesting an important role for Smad3 in mediating the Egr-2 response. Second, we examined the effect of TGF-β in mouse fibroblasts lacking Smad3. These experiments showed that the magnitude of Egr-2 stimulation by TGF-β was dramatically reduced in Smad3−/− fibroblasts compared with Smad3+/+ control fibroblasts (Figure 3, C and D). Together, these results indicate that the canonical Smad pathway plays a dominant role in the regulation of Egr-2 by TGF-β.
Figure 3.
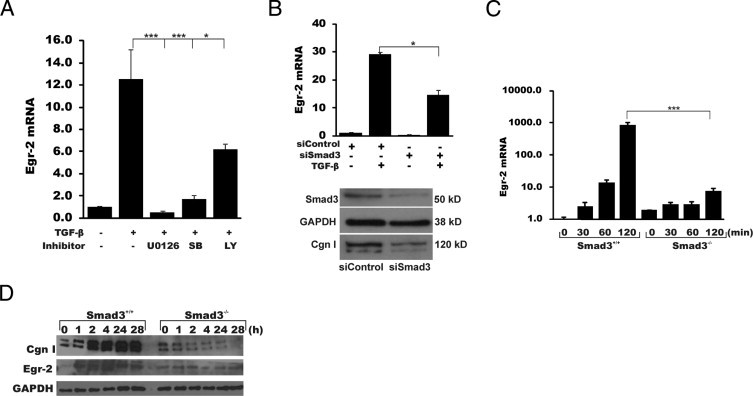
Stimulation of Egr-2 expression via Smad3 and extracellular signal–regulated kinase 1/2. A: Confluent fibroblasts were preincubated for 30 minutes with SB431542 (SB), U0126, or LY294002 (LY), followed by TGF-β (10 ng/mL) for a further 120 minutes before RNA isolation. qPCR was performed. The results represent the mean ± SEM of triplicate determinations from three independent experiments. B: Fibroblasts were transiently transfected with Smad3-specific siRNA or scrambled control siRNA in parallel and incubated with TGF-β (10 ng/mL) for a further 120 minutes before RNA isolation and qPCR analysis. *P < 0.05. The lower panel shows the results of Western analysis. C and D: Fibroblasts from Smad3+/+ or Smad3-/- mouse embryos were incubated in parallel with TGF-β2 (10 ng/mL) for up to 120 minutes before RNA isolation and qPCR analysis (C) or up to 28 hours before isolation of whole cell lysates for Western analysis (D). ***P < 0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; cgn, collagen.
In previous studies using genomewide expression microarray to examine changes in gene expression induced by Ad–Egr-1 infection for 48 hours, we identified Egr-2 as a direct target gene for Egr-1 in fibroblasts (S.B., S.L., and J.V.; data not shown). The levels of Egr-2 mRNA were 10-fold greater in Egr-1–overexpressing fibroblasts compared with fibroblasts infected with Ad-GFP. Analysis of the data showed that Egr-2 mRNA levels increased for up to 24 hours after Egr-1 overexpression in normal fibroblasts and then declined after 48 hours (Figure 4A). In contrast, when induced by TGF-β, Egr-2 expression showed sustained elevation (Figure 2). qPCR analysis confirmed that wild-type Egr-1 and a mutant form of Egr-1 lacking the Nab binding domain (discussed later) stimulated Egr-2 mRNA expression (Figure 4B).
Figure 4.
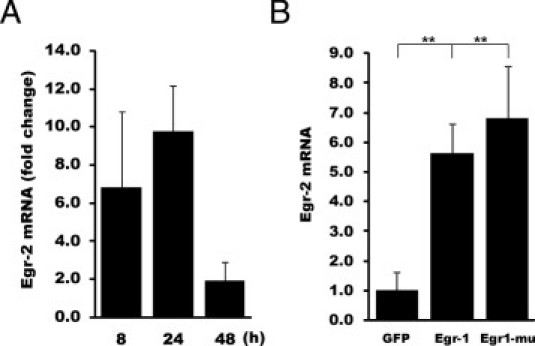
Egr-1 stimulates Egr-2 expression. A and B: Foreskin fibroblasts at confluence were infected with Ad–Egr-1 or Ad-GFP, followed by incubation in serum-free media for the indicated periods. RNA was isolated and subjected to qPCR analysis. A: Analysis using microarrays (Illumina). Egr-2 mRNA signal intensity for each point was determined. Results, indicating fold change in Egr-1–infected fibroblasts compared with Ad-GFP–infected fibroblasts, are expressed as mean ± SEM from triplicate determinations. B: qPCR was performed. The results represent the mean ± SEM of triplicate determinations from three independent experiments. **P < 0.01.
The transient transfection assays showed that overexpressed Egr-1 stimulated the activity of p[EBS]4-luc but had no effect on Egr-2 promoter-driven luc activity. As a positive control, we show that p[EBS]4-luc activity in the same cells was induced by TGF-β. In contrast, TGF-β in the same experiments caused a greater than fourfold stimulation of Egr-2–luc activity. Because the proximal 1.3 kb of the Egr-2 promoter lacks a perfect consensus Egr-1 binding site, we speculate that stimulation of Egr-2 expression by Egr-1 might be mediated through more upstream enhancer elements in the Egr-2 promoter. The related supportive data are in Supplemental Figure S1 (available at http://ajp.amjpathol.org). Results also showed that, although overexpressed Egr-2 stimulated the activity of p[EBS]4-luc, Nab2 attenuated the response, indicating that, similar to Egr-1, Egr-2 is also a subject of negative regulation by Nab2 (see Supplemental Figure S2 at http://ajp.amjpathol.org).
Egr-2 Induces Fibrotic Gene Expression
Next, we turned our attention to the biological responses modulated by Egr-2 in the context of TGF-β–mediated fibrogenesis. Fibroblasts were infected with Ad–Egr-2, and whole cell lysates were prepared after 48 hours of incubation. The results of Western analysis showed that overexpressed Egr-2 caused a dose-dependent augmentation of type I collagen and ASMA protein levels, with a greater than threefold increase at 48 hours (Figure 5A). In transient transfection assays, overexpressed Egr-2 significantly enhanced the activity of reporter genes driven by COL1A2 and ASMA (gene symbol, ACTA) promoters (Figure 5, B and C).
Figure 5.
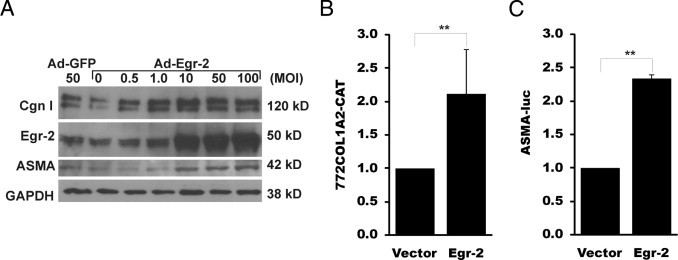
Overexpressed Egr-2 stimulates profibrotic gene expression. A: Confluent fibroblasts were infected with increasing concentrations of Ad–Egr-2 or control virus. After 48 hours' incubation, fibroblasts were harvested and whole cell lysates were subjected to Western analysis. Representative immunoblots are shown. B and C: Fibroblasts were transiently cotransfected with pCMV–Egr-2, along with 772COL1A2–chloramphenicol acetyltransferase (CAT) (B) or ASMA-luc (C). After 48 hours' incubation, whole cell lysates were assayed for their CAT or luc activities. The results represent the means of triplicate determinations from two independent experiments. **P < 0.01. cgn indicates; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; cgn, collagen; MOI, multiplicity of infection.
Egr-2 Is Required for TGF-β Stimulation of Collagen and ASMA (gene symbol, ACTA) Gene Expression
An RNA interference strategy was used to investigate the requirement for Egr-2 in TGF-β–induced fibrotic responses. Fibroblasts were transiently transfected with Egr-2–specific siRNA or scrambled control siRNA and incubated with TGF-β for 24 hours. qPCR analysis showed that basal Egr-2 mRNA was reduced by approximately 80%, and Egr-2 stimulation by TGF-β was significantly attenuated in fibroblasts transfected with si-Egr-2 (Figure 6). These cells showed significantly reduced stimulation of collagen, ASMA, and connective tissue growth factor mRNA expression by TGF-β, whereas basal levels of Egr-1 mRNA were moderately elevated. The attenuated TGF-β response in fibroblasts with reduced Egr-2, despite elevated levels of Egr-1, suggests that Egr-2 plays a more important role in mediating the response.
Figure 6.
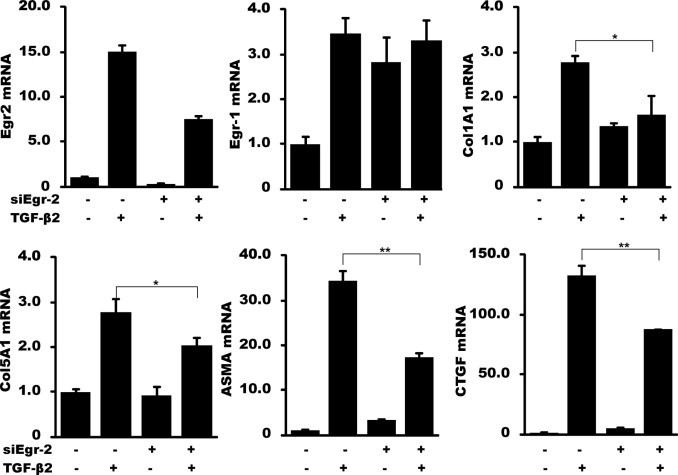
Egr-2 is required for maximal TGF-β responses. Fibroblasts were transiently transfected with Egr-2–specific siRNA or scrambled control siRNA. After incubation with 10 ng/mL TGF-β for 24 hours, RNA was isolated and analyzed by qPCR. The results are shown as fold change compared with control siRNA and represent the mean ± SEM of triplicate determinations from three independent experiments. *P <0.05; **P <0.01. CTGF, connective tissue growth factor.
Egr-2 Regulates a Tissue Remodeling Gene Program
To further characterize the importance of Egr-2 on the regulation of fibrotic responses, genomewide changes induced by Egr-2 overexpression were examined by transcriptional profiling. Thus, foreskin fibroblasts were infected with Ad–Egr-2 or control virus; total RNA was harvested; and, after amplification and labeling, total RNA was hybridized to whole-genome cDNA microarrays (Illumina) containing 44,000 probes. An analysis of the results showed that at 48 hours, Egr-2 had a broad effect on fibroblast gene expression. Close to 700 transcripts showed a greater than twofold change (314 increased and 363 decreased) (Figure 7A). Table 2 and Table 3 showed the 40 genes with the greatest change. The expression of Egr-1 was reduced, whereas Egr-2 mRNA levels were increased in Egr-2–overexpressing fibroblasts. The complete list of Egr-2–regulated genes is shown in Supplemental Table S1 (available at http://ajp.amjpathol.org). Gene ontology analysis revealed multiple biological functions that were significantly enriched with Egr-2–regulated genes (Figure 7B). qPCR was used to validate Egr-2–induced changes in the expression of selected genes (Figure 7C). All three genes (ie, Plod2, Col5A1, and integrin β1) were up-regulated in fibroblasts infected with Ad–Egr-2, whereas Egr-1 mRNA was reduced (Figure 7C). Nab1, Nab2, and Egr-3 have little change in Egr-2–overexpressing fibroblasts. Other fibrotic genes, such as connective tissue growth factor, and Pai1 mRNA also increase in Egr-2–overexpressing fibroblasts (see Supplemental Table S1 at http://ajp.amjpathol.org).
Figure 7.
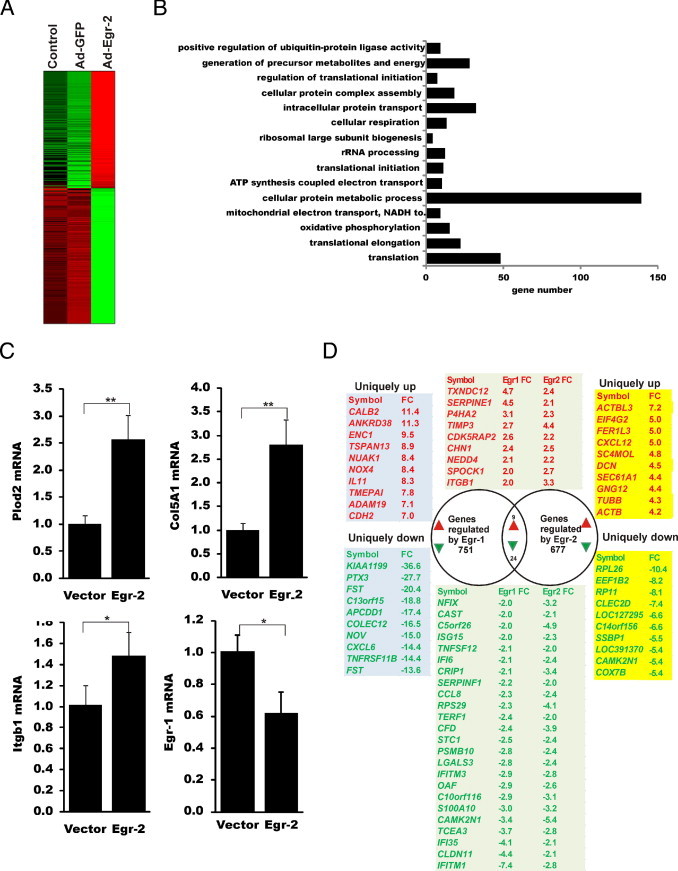
Genomewide expression changes induced by Egr-2. Fibroblasts were transfected with Ad-GFP or Ad–Egr-2 or left untransfected (control) for 48 hours before RNA isolation and processing for microarray analysis. A: Heat map. The genes that showed greater than twofold up- or down-regulation by Ad–Egr-2 compared with Ad-GFP were further analyzed. B: The top gene ontology categories were regulated by Egr-2. C: qPCR validation for selected Egr-2–regulated genes. D: Overlap between genes regulated by Egr-1 (751) and Egr-2 (677). *P < 0.05; **P < 0.01. rRNA, ribosomal RNA; FC, fold change; Plod, procollagen-lysine 1, 2-oxoglutarate 5-dioxygenase; Itgb, integrin β.
Table 2.
Top 40 Genes Up-Regulated by Egr-2
| Gene | EntrezID | Description | FC |
|---|---|---|---|
| ACTBL3 | 440915 | Actin, β-like 3 | 7.2 |
| EIF4G2 | 1982 | Eukaryotic translation initiation factor 4 γ, 2 | 5.0 |
| FER1L3 | 26509 | Fer-1–like 3, myoferlin (Caenorhabditis elegans) | 5.0 |
| CXCL12 | 6387 | Chemokine ligand 12 (stromal cell–derived factor 1) | 5.0 |
| SC4MOL | 6307 | Sterol-C4-methyl oxidaselike | 4.8 |
| DCN | 1634 | Decorin | 4.5 |
| SEC61A1 | 29927 | Sec61 α 1 subunit (Saccharomyces cerevisiae) | 4.4 |
| GNG12 | 55970 | Guanine nucleotide–binding protein (G protein), γ 12 | 4.4 |
| TIMP3 | 7078 | TIMP metallopeptidase inhibitor 3 | 4.4 |
| TUBB | 203068 | Tubulin, β | 4.3 |
| ACTB | 60 | Actin, β | 4.2 |
| CSNK1A1 | 1452 | Casein kinase 1, α 1 | 4.1 |
| TUBA1A | 7846 | Tubulin, α 1a | 4.0 |
| SNX6 | 58533 | Sorting nexin 6 | 4.0 |
| FAM65A | 79567 | Family with sequence similarity 65, member A | 3.9 |
| C1R | 715 | Complement component 1, r subcomponent | 3.9 |
| C17orf70 | 80233 | Chromosome 17 open reading frame 70 | 3.8 |
| FSTL1 | 11167 | Follistatinlike 1 | 3.7 |
| C1S | 716 | Complement component 1, s subcomponent | 3.7 |
| CTNNB1 | 1499 | Catenin (cadherin-associated protein), β 1, 88 kDa | 3.7 |
| SEC23A | 10484 | Sec23 homolog A (S. cerevisiae) | 3.7 |
| SLC7A5 | 8140 | Solute carrier family 7 | 3.6 |
| EHD1 | 10938 | EH-domain containing 1 | 3.6 |
| TCEA1 | 6917 | Transcription elongation factor A (SII), 1 | 3.6 |
| ATP2B4 | 493 | ATPase, Ca2+ transporting, plasma membrane 4 | 3.6 |
| VCP | 7415 | Valosin-containing protein | 3.6 |
| C1R | 715 | Complement component 1, r subcomponent | 3.5 |
| CBX6 | 23466 | Chromobox homolog 6 | 3.5 |
| RNF150 | 57484 | Ring finger protein 150 | 3.5 |
| TNC | 3371 | Tenascin C | 3.4 |
| MAP1B | 4131 | Microtubule-associated protein 1B | 3.4 |
| FBLN1 | 2192 | Fibulin 1 | 3.4 |
| SAFB | 6294 | Scaffold attachment factor B | 3.4 |
| NEK7 | 140609 | NIMA-related kinase 7 | 3.4 |
| ACTG1 | 71 | Actin, γ 1 | 3.4 |
| TUBB6 | 84617 | Tubulin, β 6 | 3.3 |
| ADAM10 | 102 | ADAM metallopeptidase domain 10 | 3.3 |
| CTNNB1 | 1499 | Catenin (cadherin-associated protein), β 1, 88 kDa | 3.3 |
| ITGB1 | 3688 | Integrin, β 1 | 3.3 |
| MSRB3 | 253827 | Methionine sulfoxide reductase B3 | 3.2 |
FC, fold change.
Table 3.
Top 40 Genes Down-Regulated by Egr-2
| Gene | EntrezID | Description | FC | Gene | EntrezID | Description | FC |
|---|---|---|---|---|---|---|---|
| RPL26 | 6154 | Ribosomal protein L26 | −10 | UQCRB | 7381 | Ubiquinol–cytochrome | −4.7 |
| EEF1B2 | 1933 | Eukaryotic translation elongation factor 1 β 2 | −8.2 | c reductase binding protein | |||
| RP11 | 442454 | Ubiquinol–cytochrome c reductase binding | −8.1 | ATP5H | 10476 | ATP synthase, H+ transporting | −4.7 |
| protein pseudogene | MT1A | 4489 | Metallothionein 1A | −4.7 | |||
| CLEC2D | 29121 | C-type lectin domain family 2, member D | −7.4 | HINT1 | 3094 | Histidine triad nucleotide | −4.7 |
| LOC127295 | 127295 | Similar to 60S ribosomal | −6.6 | binding protein 1 | |||
| protein L36 | GSTO1 | 9446 | Glutathione | −4.6 | |||
| C14orf156 | 81892 | Chromosome 14 open reading frame 156 | −6.6 | S–transferase ω 1 | |||
| SSBP1 | 6742 | Single-stranded DNA binding protein 1 | −5.5 | EIF3K | 27335 | Eukaryotic translation | −4.5 |
| LOC391370 | 391370 | Similar to hCG1818387 | −5.4 | initiation factor 3, | |||
| CAMK2N1 | 55450 | Calcium/calmodulin- | −5.4 | subunit K | |||
| dependent protein kinase II inhibitor 1 | ATP5H | 10476 | ATP synthase, H+ transporting | −4.5 | |||
| COX7B | 1349 | Cytochrome c oxidase subunit VIIb | −5.4 | RPS26L1 | 441502 | Ribosomal protein S26-like 1 | −4.5 |
| RPL36 | 25873 | Ribosomal protein L36 | −5.4 | DRAP1 | 10589 | DR1-associated | −4.5 |
| NDUFB3 | 4709 | NADH dehydrogenase (ubiquinone) 1 β subcomplex, 3, 12 kDa | −5.2 | protein 1 (negative cofactor 2 α) | |||
| HCFC1R1 | 54985 | Host cell factor C1 regulator 1 (XPO1 dependent) | −5.2 | C20orf52 | 140823 | Chromosome 20 open reading frame 52 | −4.5 |
| COX5A | 9377 | Cytochrome c oxidase subunit Va | −5.2 | HCG2P7 | 80867 | HLA complex group 2 | −4.4 |
| COX7C | 1350 | Cytochrome c oxidase | −5.1 | pseudogene 7 | |||
| subunit VIIc | CISD1 | 55847 | CDGSH iron sulfur | −4.4 | |||
| NDUFB3 | 4709 | NADH dehydrogenase | −5.1 | domain 1 | |||
| (ubiquinone) 1 β subcomplex, 3, 12 kDa | NDUFA3 | 4696 | NADH dehydrogenase | −4.4 | |||
| SELM | 140606 | Selenoprotein M | −5.0 | (ubiquinone) 1 α | |||
| FXYD5 | 53827 | FXYD domain–containing ion transport regulator 5 | −4.9 | subcomplex, 3, 9 kDa | |||
| PFDN5 | 5204 | Prefoldin subunit 5 | −4.9 | UXT | 8409 | Ubiquitously | −4.4 |
| C5orf26 | 114915 | Chromosome 5 open reading frame 26 | −4.8 | expressed transcript | |||
| RPS17 | 6218 | Ribosomal protein S17 | −4.8 | LOC440737 | 440737 | Similar to | −4.3 |
| LY96 | 23643 | Lymphocyte antigen 96 | −4.8 | ribosomal | |||
| LOC441377 | 441377 | Similar to ribosomal | −4.8 | protein L35 | |||
| protein S26 | NDUFS4 | 4724 | NADH | −4.3 | |||
| LSM3 | 27258 | LSM3 homolog, U6 small nuclear | −4.7 | dehydrogenase (ubiquinone) | |||
| RNA associated (S. cerevisiae) |
FC, fold change.
Although Egr-2 shares substantial structural similarity with Egr-1, including conserved zinc fingers, these two related transcriptional factors show divergence of N- and C-terminal amino acid sequences. To compare gene expression regulated by Egr-1 and Egr-2, microarray analysis was performed. Changes in mRNA expression in fibroblasts infected with Ad–Egr-1 and Egr-2 were compared with virus-infected fibroblasts at 48 hours (Figure 7D). Egr-1 modulated the expression of 751 genes, and Egr-2 modulated the expression of 677 genes. However, only nine genes showed concordant up-regulation by both Egr-1 and Egr-2: PAI1, TIMP3, TXNDC12, P4HA2, CDK5RAP2, CHN1, NEDD4, SPOCK1, and ITGB1 (integrin β1). ASMA and connective tissue growth factor were both induced by Egr-1 and Egr-2, with less fold change (less than the cutoff). In contrast, the following 24 genes showed concordant down-regulation by both Egrs: NFIX, CAST, C5orf26, ISG15, TNFSF12, IFI6, CRIP1, SERPINF1, CCL8, RPS29, TERF1, CFD, STC1, PSMB10, LGALS3, IFITM3, OAF, C10orf116, S100A10, CAMK2N1, TCEA3, IFI35, CLDN11, and IFITM1.
To confirm the effect of Egr-2 overexpression in adult fibroblasts, normal adult and SSc fibroblasts were infected with Ad–Egr-2. Results showed that Col5A1 was induced by overexpression of Egr-2 in both normal adult or SSc fibroblasts (see Supplemental Figure S3 at http://ajp.amjpathol.org); the effect was similar compared with normal foreskin fibroblasts.
To investigate if Egr-2 sensitizes the TGF-β signaling pathway, fibroblasts were infected with Ad–Egr-2, followed by TGF-β treatment. Results showed that Col1A1 mRNA was induced by TGF-β by twofold and by 3.2-fold after Egr-2 and TGF-β treatment together (see Supplemental Figure S4 at http://ajp.amjpathol.org), suggesting an additive effect of Egr-2 and TGF-β. ASMA mRNA was induced by TGF-β or Egr-2 by sixfold or twofold, respectively; and by approximately 12-fold on Egr-2 overexpression and TGF-β treatment together (see Supplemental Figure S4 at http://ajp.amjpathol.org), suggesting a synergistic effect of Egr-2 and TGF-β. Taken together, these results suggest that Egr-2 sensitizes gene expression responsive to TGF-β.
Elevated Egr-2 Expression in Murine Scleroderma
In light of the profibrotic activities associated with Egr-2 and its involvements in mediating TGF-β responses in fibroblasts, we sought to characterize in vivo the tissue expression of Egr-2 associated with fibrosis. The s.c. injection of bleomycin in mice induces progressive sclerodermalike skin and lung pathological features because of TGF-β–mediated fibroblast activation.33 BALB/c mice were sacrificed after 21 daily injections of bleomycin or PBS, and lesional skin was examined. By IHC, Egr-2 was barely detectable in the dermis of PBS-injected mice or untreated mice (Figure 8 and data not shown). In contrast, a substantial increase in Egr-2 accumulation was noted in the lesional dermis from bleomycin-injected mice. Abundant Egr-2 immunostaining was observed in dermal fibroblastic cells [5.4 ± 2.6 immunopositive cells in PBS-injected mice versus 14.0 ± 6.1 cells in bleomycin-injected mice (mean ± SEM from five mice); P < 0.003]. Moreover, a significant increase in Egr-2 mRNA levels in the lesional dermis was demonstrated by qPCR (Figure 8B). Enhanced Egr-2 gene expression in these tissues probably reflects local TGF-β activity and in situ fibroblast stimulation.
Figure 8.
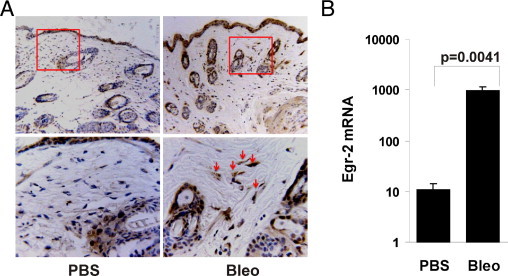
Elevated Egr-2 expression in mouse scleroderma. Dermal fibrosis was induced by daily s.c. injections of bleomycin (Bleo) for 28 days, and lesional skin was harvested. A: Sections were stained with antibodies against Egr-2 and counterstained with hematoxylin. Representative photomicrographs are shown. Original magnification: ×100 (upper panels); ×400 (lower panels). Lower panels in A represent the areas delineated by the squares in the upper panels. Arrows indicate immunopositive fibroblastic cells in the dermis. Omission of primary antibodies resulted in no staining (negative control). The number of immunopositive fibroblastic cells was determined in five separate microscopic fields by a blinded observer. B: Total RNA was isolated from the skin and subjected to qPCR. The results represent the mean ± SEM of three determinations from five mice per group.
Elevated Egr-2 Expression in SSc
To explore the involvement of Egr-2 in SSc, we proceeded to examine Egr-2 expression in lesional tissue from patients with diffuse cutaneous SSc. First, skin biopsy specimens from three patients with diffuse cutaneous SSc and three healthy controls, matched for age and sex, were studied in parallel. Immunofluorescence analysis showed that, in contrast to healthy dermis, in which little Egr-2 was detectable, in all three SSc skin biopsy specimens, many dermal fibroblastic cells were positive for Egr-2 (Figure 9A). Egr-2 could be detected in cells in both the papillary and reticular dermis and at the interface between the dermis and adipose layers. Interestingly, the epidermis showed rather strong Egr-2 expression in both healthy controls and patients with SSc. The proportion of Egr-2–positive dermal fibroblasts in SSc showed a 60% increase compared with healthy controls (Figure 9B).
Figure 9.
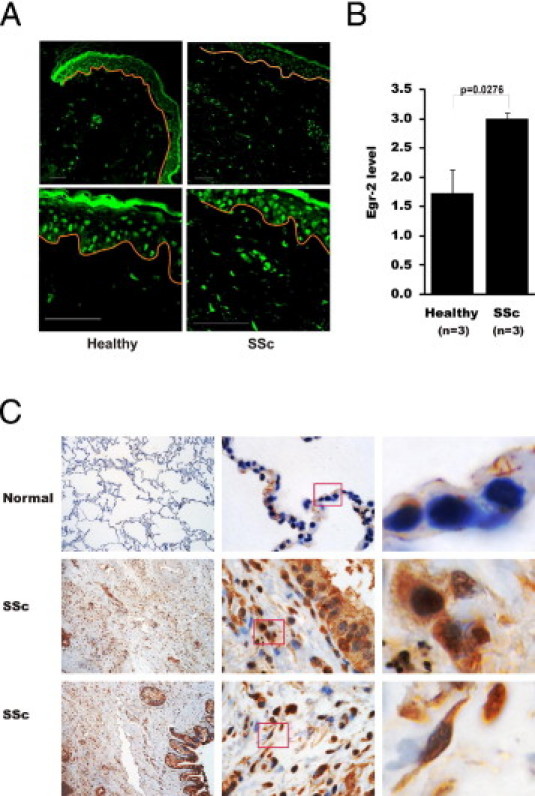
Elevated Egr-2 expression in SSc. A: Skin biopsy specimens from patients with SSc (n = 3) or healthy controls (n = 3) were stained with antibodies to Egr-2 and examined by immunofluorescence. Representative images are shown, with red lines delineating the epidermis. Original magnification: ×200 (upper panels); ×630 (lower panels). B: Quantification of Egr-2 immunofluorescence in the dermis (the Materials and Methods section provides details). Bars represent the mean ± SEM of three determinations. C: Lung tissues from nonfibrotic donors (n = 3) (normal) or patients with SSc who have pulmonary fibrosis (n = 3) (SSc) were examined. Representative images were captured by a microscope (Zeiss Axioskop) with a CRi Nuance spectral camera. Original magnification: ×100 (left); ×1000 (middle). Right: Panels showing the magnified images (from the boxes). Brown indicates Egr-2–positive cells. Nuclei are counterstained with hematoxylin (blue).
Fibrosis commonly affects the lungs in SSc and is associated with activation of parenchymal fibroblasts.34 To examine the expression of Egr-2 in SSc lungs, samples from three patients with SSc-associated pulmonary fibrosis and three normal donor controls were examined by IHC. Low levels of Egr-2 were detected in normal lung tissue (Figure 9C). In contrast, lung tissues from all three patients with SSc showed a marked increase in parenchymal Egr-2. Expression was most prominent at fibroblastic loci, in which Egr-2 could be detected in most blood vessels, macrophages, airway-lining cells, endothelial cells, interstitial fibroblasts, and alveolar epithelial cells (Figure 9C). Taken together, the results of the Egr-2 IHC from lesional skin and lungs provide correlative evidence, suggesting a potential correlative role of elevated Egr-2 in the development of fibrosis.
Discussion
Fibrosis, the hallmark of fibrosing disorders such as SSc, represents aberrant fibroblast activation with autonomous up-regulation of extracellular matrix gene expression and transition into myofibroblasts.35 TGF-β is capable of inducing the full repertoire of fibrotic responses and is implicated in the pathogenesis of SSc. Egr-1 was previously identified as an Smad-independent intracellular mediator of TGF-β signaling. Egr-1 expression was rapidly and transiently induced by TGF-β, and Egr-1 directly activated the transcription of COL1A2.15 The present studies reveal that Egr-2 is also stimulated by TGF-β but via distinct regulatory pathways and with distinct kinetics characterized by slower, but more durable, induction compared with Egr-1. Endogenous Egr-2 was required for fibrotic responses, and overexpressed Egr-2 expression in normal fibroblasts was sufficient to stimulate collagen gene expression and myofibroblast differentiation in the absence of TGF-β. Interestingly, although the expression of many genes was induced by Egr-2, the overlap between Egr-1– and Egr-2–regulated gene expression was only modest, indicating largely nonredundant function for these two structurally related TGF-β–inducible transcription factors. An abnormally elevated Egr-2 in lesional tissue biopsy specimens might suggest a novel role for Egr-2 in the pathogenesis of fibrosis in SSc and in a bleomycin-induced mouse model of scleroderma.
Egr-2 is a ubiquitous transcription factor whose expression is induced by synaptic activity in neurons,36 T-cell receptor ligation,37 and extracellular cues, such as the canonical Wnt3a in a variety of cell types.38 These responses are mediated through diverse signaling mechanisms involving Ca2+ flux, nuclear factor for activated T cell, and extracellular signal–regulated kinase cascades. Because T-cell receptor ligation acts as a potent stimulus for Egr-2 in lymphocytes, Egr-2 function has been studied most extensively in the context of immune tolerance and autoimmunity. In general, Egr-2 acts as a potent negative regulator of adaptive immune responses.39 The Egr2 gene has been a susceptibility locus for human autoimmune diseases.40 Mice with spontaneous lupuslike disease show deficient Egr-2 expression,41 and genetic targeting of Egr-2 is associated with expansion of autoreactive T cells, loss of self-tolerance, and development of autoimmunity.42
Although Egr-2 shows substantial structural similarity to Egr-1, including identical zinc finger DNA-binding domains, there is divergence at the C and N termini.43 Because Egr-1 and Egr-2 appear to bind to identical GC-rich DNA elements, the distinct or even opposing biological activities of Egr-1 and Egr-2 are thought to reflect the different repertoires of cofactors recruited into the transcriptional complex as the result of divergent non–DNA-binding domains.12,44 The functional distinction between Egr-1 and Egr-2 is highlighted by genetic targeting studies demonstrating that, although Egr-1–null mice show little spontaneous phenotype,10 loss of Egr-2 is associated with embryonic lethality.11 Moreover, biological responses can even be antagonistically regulated by Egr-1 and Egr-2. For instance, although Egr-1 enhances T-cell function,45 Egr-2 acts as a negative regulator.46 The functional divergence of Egr-1 and Egr-2 is also seen in adipogenesis, with Egr-2 stimulating preadipocyte differentiation and Egr-1 inhibiting the process.44 The Egr1 and Egr2 genes show divergent patterns of expression during embryogenesis.47 These observations highlight important differences in the regulation, expression, and function of Egr-1 and Egr-2.
In the present studies, we show that the expression of Egr-2 was markedly enhanced in normal fibroblasts stimulated by TGF-β. In contrast to Egr-1, whose expression was induced by TGF-β within 15 minutes but then declined to basal levels by 60 minutes, Egr-2 stimulation was delayed, with maximal effect at 120 minutes; and substantially elevated Egr-2 mRNA and protein accumulation persisted for as long as 48 hours. A comparison of promoter architecture revealed no significant similarity between Egr-1 and Egr-2. The divergent kinetics of induction suggest that Egr-1 and Egr-2 play distinct roles in injury and repair, with Egr-1 required for the early-immediate response and sustained Egr-2 expression driving persistent profibrotic gene expression. Consistent with this model, we show that Egr-1 itself stimulates the expression of Egr-2, which might, therefore, serve to amplify or sustain the response. The stimulation of Egr-2 expression by TGF-β occurs at the level of transcription; in contrast to Egr-1, Egr-2 appears to involve both canonical Smad pathways that are dominant and, potentially, non-Smad pathways, as shown by genetic targeting and RNA interference experiments.
In contrast to Egr-1, the biological functions of Egr-2 are not well understood. The present results indicate that Egr-2 is sufficient to induce collagen gene expression and ASMA (gene symbol, ACTA) promoter activity. Unbiased genomewide expression profiling indicated that Egr-2 stimulated the expression of multiple genes that play a role in fibrosis. These include structural extracellular matrix proteins, such as collagen V, tenascin, fibulin, and decorin; lysyl hydroxylase 2 (PLOD2), a key enzyme involved in the generation of hydroxylysine aldehyde–derived collagen cross-links48; TIMP1; soluble mediators, such as CXC chemokine ligand 12; c-Abl; and catenin. Each of these molecules is known to be abnormally expressed, and play a role, in the pathogenesis of fibrosis and SSc.49 In previous studies,14 Egr-1 similarly induced the expression of fibrotic genes.
An important consideration is the redundant versus unique expression and function of Egr-1 and Egr-2. Both show TGF-β inducibility, and the transcriptional function of both is abrogated by Nab2, a target gene for TGF-β. However, the pattern of gene expression regulated by these two transcription factors shows only a modest degree of overlap. Taken together with the different outcomes of genetic targeting of Egr-1 and Egr-2 in the mouse, these results indicate complementary, but largely nonredundant, biological functions for Egr-1 and Egr-2. The divergent kinetic profiles of induction of the Egr family members suggest a plausible mechanistic model (Figure 10). Early and short-lived Egr-1 induction by TGF-β results in direct stimulation of fibrotic responses, such as collagen gene transcription, but a more delayed and long-lasting stimulation of Egr-2 transcription. Thus, Egr-2 might serve as a critical nexus linking early short-term cellular responses to long-lived fibrotic responses characteristic of aberrant tissue repair and pathological fibrosis. Although Egr-2 expression is up-regulated transiently during normal wound healing,50–52 lesional skin and lung biopsy specimens from patients with SSc show sustained aberrant Egr-2 expression. An association between constitutive Egr-2 expression and pathological fibrosis was further illustrated in the bleomycin-induced model of scleroderma, in which marked elevation of Egr-2 protein and mRNA was evident in the lesional skin. Taken together, these results indicate that Egr-2 is a TGF-β–inducible transcription factor that plays an important nonredundant role in profibrotic responses and shows aberrant expression in fibrosis. Based on these findings, we propose that Egr-2 might serve as a critical switch in the cellular signaling machinery that transforms a short-term self-limited tissue response, leading to regeneration, into a deregulated persistent process that culminates in pathological fibrosis.
Figure 10.
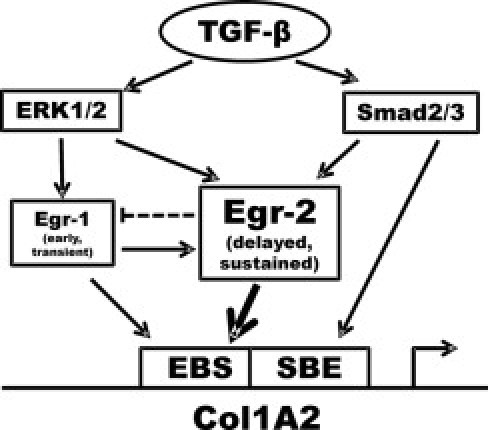
Proposed mechanistic model highlighting the role of Egr-2 in TGF-β–induced profibrotic responses. The expression of Egr-1 is rapidly and transiently induced by TGF-β via extracellular signal–regulated kinase (ERK) 1/2. In turn, Egr-1 induces Egr-2. The ERK1/2 and Smad2/3 pathways also stimulate Egr-2, leading to its sustained expression. Egr-1 and Egr-2 directly stimulate COL1A2 transcription. SBE, smad binding element.
Acknowledgments
We thank Drs. Vikas P. Sukhatme and Mary Goldring (Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA), Robert J. Schwartz (Baylor College of Medicine, Houston, TX), Gerald R. Crabtree (Stanford University, Palo Alto, CA), and Williams H. Schnaper (Northwestern University, Chicago, IL) for reagents; and Warren G. Tourtellotte, Anna P. Lam, and members of the Varga laboratory (Northwestern University, Chicago, IL) for helpful discussions.
Footnotes
Supported by grants from the Department of Defense (W81-XWH-06-01-0278) and the NIH (AR-04239) (J.V.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.01.035.
Supplementary data
The Egr-1 effect on Egr-2 proximal promoter. Fibroblasts were transiently cotransfected with Ad-Egr-1 along with Egr-2-luc (left panel) or p(EBS)4-luc (right panel), followed by incubation in media with TGF-β (10 ng/mL) for 24 hours. Whole cell lysates were then assayed for their luciferase activities. The results represent the means ± SEM of triplicate determinations from two independent experiments. *P < 0.05.
The Nab2 effect on Egr-2 acitivity. Confluent fibroblasts were transiently cotransfected with pCMV-Egr-2, and p[EBS]4-luc along with pCMV-Nab2 or empty vector. Following 48 hours of incubation, cell lysates were assayed for their luciferase activities. The results represent the means + SEM of triplicate determinations from two independent experiments.
The Egr-2 effect on gene expression in normal and SSc adult fibroblasts. Col5A1 was induced by overexpression of Egr-2 in both normal adult or SSc fibroblasts. Fibroblasts were infected with Ad-GFP or Ad-Egr-2 for 48 hours prior to RNA isolation for real-time PCR.
The effect of Egr-2 on TGF-β-responsive ASMA and Col1A1 gene expression. Foreskin fibroblasts at confluence in serum-free media were infected with Ad-GFP or Ad-Egr-2 (MOI=50) for 48 h followed by TGF-β2 (1 ng/mL) for a further 24 hours. Total RNA was subjected to real-time qPCR for Col1A1 (A) or ASMA (B). The results represent the means ± SEM of triplicate determinations.
References
- 1.Jimenez S.A., Derk C.T. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 2.Gabrielli A., Avvedimento E.V., Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 3.Abraham D.J., Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 2005;26:587–595. doi: 10.1016/j.it.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Varga J., Pasche B. Antitransforming growth factor-beta therapy in fibrosis: recent progress and implications for systemic sclerosis. Curr Opin Rheumatol. 2008;20:720–728. doi: 10.1097/BOR.0b013e32830e48e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heldin C.H., Landstrom M., Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Thiel G., Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
- 7.O'Donovan K.J., Tourtellotte W.G., Millbrandt J., Baraban J.M. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–173. doi: 10.1016/s0166-2236(98)01343-5. [DOI] [PubMed] [Google Scholar]
- 8.Christy B., Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proc Natl Acad Sci U S A. 1989;86:8737–8741. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sukhatme V.P., Kartha S., Toback F.G., Taub R., Hoover R.G., Tsai-Morris C.H. A novel early growth response gene rapidly induced by fibroblast, epithelial cell and lymphocyte mitogens. Oncogene Res. 1987;1:343–355. [PubMed] [Google Scholar]
- 10.Lee S.L., Sadovsky Y., Swirnoff A.H., Polish J.A., Goda P., Gavrilina G., Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 11.Le N., Nagarajan R., Wang J.Y., Araki T., Schmidt R.E., Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A. 2005;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins S., Lutz M.A., Zarek P.E., Anders R.A., Kersh G.J., Powell J.D. Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur J Immunol. 2008;38:528–536. doi: 10.1002/eji.200737157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basson M.A., Wilson T.J., Legname G.A., Sarner N., Tomlinson P.D., Tybulewicz V.L., Zamoyska R. Early growth response (Egr)-1 gene induction in the thymus in response to TCR ligation during early steps in positive selection is not required for CD8 lineage commitment. J Immunol. 2000;165:2444–2450. doi: 10.4049/jimmunol.165.5.2444. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya S., Chen S.J., Wu M., Warner-Blankenship M., Ning H., Lakos G., Mori Y., Chang E., Nihijima C., Takehara K., Feghali-Bostwick C., Varga J. Smad-independent transforming growth factor-beta regulation of early growth response-1 and sustained expression in fibrosis: implications for scleroderma. Am J Pathol. 2008;173:1085–1099. doi: 10.2353/ajpath.2008.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S.J., Ning H., Ishida W., Sodin-Semrl S., Takagawa S., Mori Y., Varga J. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. J Biol Chem. 2006;281:21183–21197. doi: 10.1074/jbc.M603270200. [DOI] [PubMed] [Google Scholar]
- 16.Wu M., Melichian D.S., de la Garza M., Gruner K., Bhattacharyya S., Barr L., Nair A., Shahrara S., Sporn P.H., Mustoe T.A., Tourtellotte W.G., Varga J. Essential roles for early growth response transcription factor Egr-1 in tissue fibrosis and wound healing. Am J Pathol. 2009;175:1041–1055. doi: 10.2353/ajpath.2009.090241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varga J., Brenner D., Phan S.H. Humana Press; Totowa, NJ: 2005. Fibrosis Research: Methods and Protocols. pp xvi, 392. [Google Scholar]
- 18.Fang F., Flegler A.J., Du P., Lin S., Clevenger C.V. Expression of cyclophilin B is associated with malignant progression and regulation of genes implicated in the pathogenesis of breast cancer. Am J Pathol. 2009;174:297–308. doi: 10.2353/ajpath.2009.080753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kao S.C., Wu H., Xie J., Chang C.P., Ranish J.A., Graef I.A., Crabtree G.R. Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science. 2009;323:651–654. doi: 10.1126/science.1166562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan L., Peng H., Osaki M., Choy B.K., Auron P.E., Sandell L.J., Goldring M.B. Egr-1 mediates transcriptional repression of COL2A1 promoter activity by interleukin-1beta. J Biol Chem. 2003;278:17688–17700. doi: 10.1074/jbc.M301676200. [DOI] [PubMed] [Google Scholar]
- 21.Ihn H., Ohnishi K., Tamaki T., LeRoy E.C., Trojanowska M. Transcriptional regulation of the human alpha2(I) collagen gene: combined action of upstream stimulatory and inhibitory cis-acting elements. J Biol Chem. 1996;271:26717–26723. doi: 10.1074/jbc.271.43.26717. [DOI] [PubMed] [Google Scholar]
- 22.Thiel G., Kaufmann K., Magin A., Lietz M., Bach K., Cramer M. The human transcriptional repressor protein NAB1: expression and biological activity. Biochim Biophys Acta. 2000;1493:289–301. doi: 10.1016/s0167-4781(00)00207-4. [DOI] [PubMed] [Google Scholar]
- 23.Min B.H., Foster D.N., Strauch A.R. The 5′-flanking region of the mouse vascular smooth muscle alpha-actin gene contains evolutionarily conserved sequence motifs within a functional promoter. J Biol Chem. 1990;265:16667–16675. [PubMed] [Google Scholar]
- 24.Fang F., Antico G., Zheng J., Clevenger C.V. Quantification of PRL/Stat5 signaling with a novel pGL4-CISH reporter. BMC Biotechnol. 2008;8:11. doi: 10.1186/1472-6750-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu M., Melichian D.S., Chang E., Warner-Blankenship M., Ghosh A.K., Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol. 2009;174:519–533. doi: 10.2353/ajpath.2009.080574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du P., Kibbe W.A., Lin S.M. nuID: a universal naming scheme of oligonucleotides for illumina, affymetrix, and other microarrays. Biol Direct. 2007;2:16. doi: 10.1186/1745-6150-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du P., Kibbe W.A., Lin S.M. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 28.Lin S.M., Du P., Huber W., Kibbe W.A. Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 2008;36:e11. doi: 10.1093/nar/gkm1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takagawa S., Lakos G., Mori Y., Yamamoto T., Nishioka K., Varga J. Sustained activation of fibroblast transforming growth factor-beta/Smad signaling in a murine model of scleroderma. J Invest Dermatol. 2003;121:41–50. doi: 10.1046/j.1523-1747.2003.12308.x. [DOI] [PubMed] [Google Scholar]
- 30.Lakos G., Takagawa S., Varga J. Animal models of scleroderma. Methods Mol Med. 2004;102:377–393. doi: 10.1385/1-59259-805-6:377. [DOI] [PubMed] [Google Scholar]
- 31.Galdo F.D., Sotgia F., de Almeida C.J., Jasmin J.F., Musick M., Lisanti M.P., Jimenez S.A. Decreased expression of caveolin 1 in patients with systemic sclerosis: crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008;58:2854–2865. doi: 10.1002/art.23791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu E., Feghali-Bostwick C.A. Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase- and phosphatidylinositol-3 kinase-dependent pathways. Am J Pathol. 2008;172:1580–1590. doi: 10.2353/ajpath.2008.071021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu M., Varga J. In perspective: murine models of scleroderma. Curr Rheumatol Rep. 2008;10:173–182. doi: 10.1007/s11926-008-0030-9. [DOI] [PubMed] [Google Scholar]
- 34.Shi-Wen X., Denton C.P., McWhirter A., Bou-Gharios G., Abraham D.J., du Bois R.M., Black C.M. Scleroderma lung fibroblasts exhibit elevated and dysregulated type I collagen biosynthesis. Arthritis Rheum. 1997;40:1237–1244. doi: 10.1002/1529-0131(199707)40:7<1237::AID-ART7>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 35.Varga J., Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy T.H., Worley P.F., Nakabeppu Y., Christy B., Gastel J., Baraban J.M. Synaptic regulation of immediate early gene expression in primary cultures of cortical neurons. J Neurochem. 1991;57:1862–1872. doi: 10.1111/j.1471-4159.1991.tb06396.x. [DOI] [PubMed] [Google Scholar]
- 37.Rengarajan J., Mittelstadt P.R., Mages H.W., Gerth A.J., Kroczek R.A., Ashwell J.D., Glimcher L.H. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity. 2000;12:293–300. doi: 10.1016/s1074-7613(00)80182-x. [DOI] [PubMed] [Google Scholar]
- 38.Leclerc N., Noh T., Cogan J., Samarawickrama D.B., Smith E., Frenkel B. Opposing effects of glucocorticoids and Wnt signaling on Krox20 and mineral deposition in osteoblast cultures. J Cell Biochem. 2008;103:1938–1951. doi: 10.1002/jcb.21587. [DOI] [PubMed] [Google Scholar]
- 39.Safford M., Collins S., Lutz M.A., Allen A., Huang C.T., Kowalski J., Blackford A., Horton M.R., Drake C., Schwartz R.H., Powell J.D. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 40.Myouzen K., Kochi Y., Shimane K., Fujio K., Okamura T., Okada Y., Suzuki A., Atsumi T., Ito S., Takada K., Mimori A., Ikegawa S., Yamada R., Nakamura Y., Yamamoto K. Regulatory polymorphisms in EGR2 are associated with susceptibility to systemic lupus erythematosus. Hum Mol Genet. 2010;19:2313–2320. doi: 10.1093/hmg/ddq092. [DOI] [PubMed] [Google Scholar]
- 41.Sela U., Dayan M., Hershkoviz R., Lider O., Mozes E. A peptide that ameliorates lupus up-regulates the diminished expression of early growth response factors 2 and 3. J Immunol. 2008;180:1584–1591. doi: 10.4049/jimmunol.180.3.1584. [DOI] [PubMed] [Google Scholar]
- 42.Zhu B., Symonds A.L., Martin J.E., Kioussis D., Wraith D.C., Li S., Wang P. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med. 2008;205:2295–2307. doi: 10.1084/jem.20080187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph L.J., Le Beau M.M., Jamieson G.A., Jr, Acharya S., Shows T.B., Rowley J.D., Sukhatme V.P. Molecular cloning, sequencing, and mapping of EGR2, a human early growth response gene encoding a protein with “zinc-binding finger” structure. Proc Natl Acad Sci U S A. 1988;85:7164–7168. doi: 10.1073/pnas.85.19.7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyle K.B., Hadaschik D., Virtue S., Cawthorn W.P., Ridley S.H., O'Rahilly S., Siddle K. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ. 2009;16:782–789. doi: 10.1038/cdd.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin J.X., Leonard W.J. The immediate-early gene product Egr-1 regulates the human interleukin-2 receptor beta-chain promoter through noncanonical Egr and Sp1 binding sites. Mol Cell Biol. 1997;17:3714–3722. doi: 10.1128/mcb.17.7.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lazarevic V., Zullo A.J., Schweitzer M.N., Staton T.L., Gallo E.M., Crabtree G.R., Glimcher L.H. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol. 2009;10:306–313. doi: 10.1038/ni.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Topilko P., Levi G., Merlo G., Mantero S., Desmarquet C., Mancardi G., Charnay P. Differential regulation of the zinc finger genes Krox-20 and Krox-24 (Egr-1) suggests antagonistic roles in Schwann cells. J Neurosci Res. 1997;50:702–712. doi: 10.1002/(SICI)1097-4547(19971201)50:5<702::AID-JNR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 48.van der Slot A.J., Zuurmond A.M., Bardoel A.F., Wijmenga C., Pruijs H.E., Sillence D.O., Brinckmann J., Abraham D.J., Black C.M., Verzijl N., DeGroot J., Hanemaaijer R., TeKoppele J.M., Huizinga T.W., Bank R.A. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003;278:40967–40972. doi: 10.1074/jbc.M307380200. [DOI] [PubMed] [Google Scholar]
- 49.Trojanowska M., Varga J. Molecular pathways as novel therapeutic targets in systemic sclerosis. Curr Opin Rheumatol. 2007;19:568–573. doi: 10.1097/BOR.0b013e3282e6f495. [DOI] [PubMed] [Google Scholar]
- 50.Grose R., Harris B.S., Cooper L., Topilko P., Martin P. Immediate early genes krox-24 and krox-20 are rapidly up-regulated after wounding in the embryonic and adult mouse. Dev Dyn. 2002;223:371–378. doi: 10.1002/dvdy.10064. [DOI] [PubMed] [Google Scholar]
- 51.Fang F., Zheng J., Galbaugh T.L., Fiorillo A.A., Hjort E.E., Zeng X., Clevenger C.V. Cyclophilin B as a co-regulator of prolactin-induced gene expression and function in breast cancer cells. J Mol Endocrinol. 2010;44:319–329. doi: 10.1677/JME-09-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang F., Rycyzyn M.A., Clevenger C.V. Role of c-Myb during prolactin-induced signal transducer and activator of transcription 5a signaling in breast cancer cells. Endocrinology. 2009;150:1597–1606. doi: 10.1210/en.2008-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Egr-1 effect on Egr-2 proximal promoter. Fibroblasts were transiently cotransfected with Ad-Egr-1 along with Egr-2-luc (left panel) or p(EBS)4-luc (right panel), followed by incubation in media with TGF-β (10 ng/mL) for 24 hours. Whole cell lysates were then assayed for their luciferase activities. The results represent the means ± SEM of triplicate determinations from two independent experiments. *P < 0.05.
The Nab2 effect on Egr-2 acitivity. Confluent fibroblasts were transiently cotransfected with pCMV-Egr-2, and p[EBS]4-luc along with pCMV-Nab2 or empty vector. Following 48 hours of incubation, cell lysates were assayed for their luciferase activities. The results represent the means + SEM of triplicate determinations from two independent experiments.
The Egr-2 effect on gene expression in normal and SSc adult fibroblasts. Col5A1 was induced by overexpression of Egr-2 in both normal adult or SSc fibroblasts. Fibroblasts were infected with Ad-GFP or Ad-Egr-2 for 48 hours prior to RNA isolation for real-time PCR.
The effect of Egr-2 on TGF-β-responsive ASMA and Col1A1 gene expression. Foreskin fibroblasts at confluence in serum-free media were infected with Ad-GFP or Ad-Egr-2 (MOI=50) for 48 h followed by TGF-β2 (1 ng/mL) for a further 24 hours. Total RNA was subjected to real-time qPCR for Col1A1 (A) or ASMA (B). The results represent the means ± SEM of triplicate determinations.