

Abstract
Oxidative stress plays a critical role in accelerating retinal pigment epithelial dysfunction and death in degenerative retinal diseases, including age-related macular degeneration. Given the key role of oxidative stress–induced retinal pigment epithelial cell death and secondary photoreceptor loss in the pathogenesis of age-related macular degeneration, we hypothesized that a novel thiol antioxidant, N-acetylcysteine amide (NACA), might ameliorate cellular damage and subsequent loss of vision. Treatment of human retinal pigment epithelial cells with NACA protected against oxidative stress–induced cellular injury and death. NACA acted mechanistically by scavenging existing reactive oxygen species while halting production of reactive oxygen species by reversing lipid peroxidation. Furthermore, NACA functioned by increasing the levels of reduced glutathione and the phase II detoxification enzyme glutathione peroxidase. Treatment of mice exposed to phototoxic doses of light with NACA maintained retinal pigment epithelial cell integrity and prevented outer nuclear layer cell death as examined by histopathologic methods and rescued photoreceptor function as measured by electroretinography. These observations indicate that NACA protects against oxidative stress–induced retinal pigment epithelial and photoreceptor cell death in vitro and in vivo. The data suggest that NACA may be a novel treatment in rescuing retinal function and preventing vision loss secondary to retinal degenerative diseases, including age-related macular degeneration.
Retinal pigment epithelial (RPE) cells execute critical functions, including regulation of ion and metabolite transport, phagocytosis of shed photoreceptor outer segments (POSs), metabolism of retinol, formation of the outer blood-retinal barrier, and maintenance of the extracellular matrix.1 The RPE is subject to a particularly high level of oxidative stress from lipid peroxidation,2 intense exposure to light,3 extreme oxygen tension in the macular region,4 and its role in the phagocytosis of POSs.5 Light-induced peroxidation of shed POS polyunsaturated fatty acids leads to the formation of toxic reactive oxygen species (ROS) and lipofuscin. These by-products induce recurrent oxidative damage in and around the RPE, lead to RPE cell death, and seem to activate the complement system.6–8 The photoreceptors, which rely on the underlying RPE for nutritional and metabolic support,1 subsequently undergo secondary degeneration. As oxygen-using photoreceptors die, oxygen levels in the retina significantly increase due to lack of choroidal autoregulation.9,10 This results in additional oxidative damage to surrounding cells and further propagation of the vicious cycle.11,12 Secondary photoreceptor degeneration as a consequence of oxidative stress–induced RPE degeneration results in vision loss and is implicated in the pathogenesis of age-related macular degeneration (AMD).13–17
AMD is the leading cause of irreversible visual impairment in the elderly, affecting 30 million to 50 million people worldwide.18 Nonexudative or dry AMD accounts for 90% of disease prevalence. Drusen are deposits consisting of lipids with esterified and unesterified cholesterol found in Bruch's membrane early in the course of AMD. They are thought to be the result of RPE failure to process cellular debris associated with POS turnover, although recent evidence based on lipid composition may suggest another yet unknown vascular or metabolic source.19–21 Advanced nonexudative disease is characterized by diffuse RPE death, or geographic atrophy. Death of the RPE is accompanied by attenuation of overlying photoreceptors with concurrent visual impairment.22–24 Wet AMD accounts for 10% of disease. In wet AMD, new blood vessels develop underneath the retina and lead to exudation of fluid and hemorrhage. Ultimately, loss of visual acuity in both forms of AMD results from progressive degeneration of the RPE, photoreceptors, and choriocapillaris caused by factors such as oxidative stress, phototoxicity, inflammation, and hemorrhage.25
The only available treatment to slow the progression of visual loss in nonexudative or dry AMD includes vitamin and micronutrient supplementation, cessation of smoking, and possibly dietary modification.26 In a large prospective trial, use of a combination of zinc and antioxidants, including ascorbic acid, α-tocopherol, and β-carotene, was found to reduce the risk of vision loss in patients with AMD by 25% over 6 years.27 However, a recent in vitro evaluation of ascorbic acid demonstrated that it not only failed to protect RPE cells from oxidative stress–induced cell death but also potentiated cell damage at low concentrations, suggesting that a better treatment may be desirable.28
There are striking similarities between anatomical changes found in light-damaged rodent retinas and those found in advanced atrophic AMD.29 Ocular exposure to excessive light activates the visual transduction cascade, concurrently inducing RPE and photoreceptor cell death through phototoxic mechanisms. Apoptosis represents the primary mechanism of cell death in light-induced retinal degeneration in animal models and in a large variety of human retinal diseases, including AMD, providing a valuable model for investigating oxidative stress–induced degenerative photoreceptor loss.30–34
The need for more effective treatments to prevent RPE and photoreceptor cell death and the progression of retinal degenerations, including nonexudative AMD, is critical because the prevalence of these diseases is expected to double in the coming decades. In this study, we evaluated the effect of oxidative stress on the RPE and the role of a novel thiol antioxidant, N-acetylcysteine amide (NACA), in preventing RPE cell death and maintaining RPE cell homeostasis. NACA has been demonstrated to be effective in multiple oxidative stress–related diseases. It has been shown to be more effective than its parent compound, N-acetylcysteine (NAC), because the neutral carboxyl group of NACA increases its ability to permeate cell membranes and the blood-brain barrier. This allows NACA to be administered at a lower dose than NAC and prevents many adverse effects that are generally associated with NAC toxicity.35–39 We further evaluated the ability of NACA to prevent photoreceptor and RPE cell death in vivo using a light-induced toxicity model in 129/SvlmJ mice.
Materials and Methods
Materials
The RPE cell line ARPE-19 (American Type Culture Collection CRL-2302) was obtained from American Type Culture Collection (Manassas, VA). NACA was provided by Dr. Glenn Goldstein (David Pharmaceuticals, New York, NY) or was purchased from Daphne Atlas (Hebrew University of Jerusalem, Jerusalem, Israel). High-performance liquid chromatography (HPLC)–grade acetonitrile, glacial acetic acid, and o-phosphoric acid were obtained from Fisher Scientific (Pittsburgh, PA). Bradford reagent was obtained from BioRad (Hercules, CA). Cell culture reagents, such as Dulbecco's modified Eagle's medium:Ham's nutrient mixture F-12 (DMEM:F-12) media, trypsin-EDTA, penicillin-streptomycin, and fetal bovine serum (FBS), were purchased from American Type Culture Collection. Calcein AM was purchased from Biotium Inc. (Hayward, CA). All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Mice were purchased from The Jackson Laboratory, Bar Harbor, ME.
Cell Culture Conditions
ARPE-19 cells were grown in a DMEM:F-12 culture medium supplemented with 10% (v/v) FBS, to which 100 U/mL penicillin and 100 μg/mL streptomycin were added. Cells were maintained in a 37°C incubator and were supplied with 95% air and 5% CO2. All the experiments were performed on cells below passage number 20.
Cell Viability Determination
ARPE-19 cell viability was determined using propidium iodide and annexin V-fluorescein isothiocyanate (FITC) cell staining (Pharmingen, San Diego, CA) followed by fluorescence-activated cell sorting (FACS) (Beckman Coulter, Fullerton, CA), where media only and staurosporine were used as negative and positive controls, respectively. Briefly, cells were washed twice in cold PBS and then were resuspended in 1X binding buffer at a concentration of 1 × 106 cells/mL. A total of 100 μL of the solution (1 × 105 cells) was then transferred to a 5-mL culture tube, where 5 μL of annexin V-FITC and/or 5 μL of propidium iodide were added, depending on the sample. Cells were gently vortexed and incubated for 15 minutes at room temperature in the dark. Subsequently, 400 μL of 1X binding buffer was added to each tube, and the tubes were immediately analyzed by flow cytometry. The calcein AM assay was additionally used to determine cell viability as noted.40 ARPE-19 cells were seeded (12.5 × 103 cells per well) and were allowed to attach for 12 hours in DMEM:F-12 media containing 1% FBS. For tert-butyl hydroperoxide (tBHP) cytotoxicity studies, media was replaced with fresh media or media containing various concentrations of tBHP and was incubated for 4 hours. For NACA cytotoxicity studies, the cells were incubated with various concentrations of NACA for 24 hours. Protective effects of NACA were studied by first preincubating cells with 5 mmol/L NACA in media for 24 hours, followed by cell washing and complete removal of NACA, with subsequent exposure to 0.4 mmol/L tBHP for 4 hours. Cells were immediately stained using the specified annexin V-FITC or propidium iodide protocol, as described previously herein, and were analyzed using FACS.41 For the calcein assay, after the specified incubation times, the media was removed and the cells were incubated with 2 μmol/L calcein AM solution in PBS for 30 minutes. Fluorescence was measured at λexcitation = 485 nm and λemission = 530 nm.
ROS Measurement
Intracellular ROS accumulation was measured using the 2′,7′–dichlorofluorescein diacetate (Invitrogen, Carlsbad, CA) method.42 Briefly, 12.5 × 103 cells per well were seeded in a 96-well plate and were allowed to attach overnight (12 hours) in media containing 1% FBS. Media was then replaced with plain media in the control and tBHP-only groups or with 5 mmol/L NACA in the remaining groups. After 24 hours, cells were incubated with a solution of 20 μmol/L 2′,7′–dichlorofluorescein diacetate for 30 minutes. Then, cells were washed once with PBS containing 1 mmol/L CaCl2 and 1 mmol/L MgCl2. Immediately, media containing tBHP (0.1, 0.2, 0.4, and 0.6 mmol/L) was added to the cells. Fluorescence readings (λexcitation = 485 nm and λemission = 528 nm) were taken after incubating the cells with tBHP for 45 minutes.
Morphologic Assessment of ARPE-19 Cells
Changes in the overall cellular morphologic structure of cells in response to various treatments were observed and recorded using an inverted microscope (model CKX31SF; Olympus, Center Valley, PA).
Experimental Design for Oxidative Stress Parameters
Parameters, including reduced glutathione (GSH), malondialdehyde (MDA), and activities of glutathione peroxidase (GPx), were measured after the cells were treated as described later herein. Cells were seeded at a density of 1 × 106 cells per T-25 flask and were allowed to attach for 12 hours in 1% FBS media. The flasks were divided into various groups as follows: i) control, ii) NACA only, iii) tBHP only, and iv) NACA and tBHP. The control and tBHP-only groups were incubated in plain media, whereas the NACA-only and NACA and tBHP groups were incubated with 5 mmol/L NACA. After 24 hours, the media in the control and NACA-only groups was replaced with plain media, while the remaining two groups were exposed to media containing 0.4 mmol/L tBHP for 4 hours. At the end of the treatment period, cells were collected by trypsinization and were washed once with PBS (pH 7.4). The cell pellets thus obtained were further processed for appropriate assays.
GSH Measurement
GSH was determined by reverse-phase HPLC.43 The HPLC column was a 250 × 4.6-mm i.d Reliasil ODS-1 C18 column (5-μm packing material; Column Engineering, Ontario, CA). The mobile phase consisted of 70% acetonitrile, 30% water, 0.1% acetic acid, and 0.1% o-phosphoric acid. The cell pellet obtained was homogenized in serine borate buffer (100 mmol/L Tris buffer containing 10 mmol/L borate and 5 mmol/L serine with 1 mmol/L diethylenetriaminepentaacetic acid; pH 7.4). A total of 250 μL of homogenate was added to 750 μL of N-(1-pyrenyl)-maleimide (1 mmol/L in acetonitrile). The resulting solution was incubated at room temperature for 5 minutes, and the reaction was stopped by adding 10 μL of 2N HCl. The samples were then filtered through a 0.45-μm filter and were injected onto the HPLC system. The N-(1-pyrenyl)-maleimide derivatives of GSH were eluted from the column isocratically at a flow rate of 1 mL/min, and fluorescence was monitored at λexcitation = 330 nm and λemission = 376 nm.
Determination of MDA
MDA levels were determined according to the method described by Draper et al.44 Briefly, 550 μL of 5% tricholoroacetic acid and 100 μL of 500-ppm butylated hydroxytoluene in methanol were added to 350 μL of the cell homogenate. The mixture was then heated in a boiling water bath for 30 minutes. After cooling on ice, the mixture was centrifuged, and the supernatant fractions were mixed 1:1 with saturated thiobarbituric acid. The mixture was again heated in a boiling water bath for 30 minutes. After cooling on ice a second time, 500 μL of the mixture was extracted with 1 mL of n-butanol and was centrifuged to facilitate the separation of phases. The resulting organic layers were first filtered through 0.45-μm filters and then were injected onto the HPLC system. The column used was a 100 × 4.6-mm i.d C18 column (3-μm packing material; Astec, Bellefonte, PA). The mobile phase used contained 69.4% sodium phosphate buffer, 30% acetonitrile, and 0.6% tetrahydrofuran. The fluorescent product was monitored at λexcitation = 515 nm and λemission = 550 nm. MDA bis (dimethyl acetal), which gives MDA on acid treatment, was used as the standard.
GPx Activity
GPx activity was measured using the GPx colorimetric assay kit from OxisResearch (Foster City, CA). The cell pellets were homogenized in 50 mmol/L phosphate buffer (pH 7.4) containing 1 mmol/L EDTA. The homogenates were centrifuged at 8500 × g for 10 minutes, and the supernatant thus obtained was used for the assay. In brief, the assay buffer, supernatant, and NADPH reagent (containing glutathione reductase, GSH, and NADPH) were taken in a cuvette, and the reaction was initiated by the addition of tBHP. The decrease in absorbance at 340 nm was recorded for 2 minutes, and the change in absorbance at 340 nm per minute from the initial linear portion of the curve was used to calculate the GPx enzyme activity. GPx activity was calculated using the extinction coefficient of NADPH (6220 M−1 cm−1) and is expressed as units per milligram of protein.
Transepithelial Electrical Resistance Measurement
ARPE-19 cells were seeded on transwell inserts and were allowed to grow until confluence. Media in inserts was replaced with either plain media or 5 mmol/L NACA, and cells were incubated for 24 hours. The bottom plate wells always contained plain media. After 24 hours, the media in inserts was replaced with plain media or 0.4 mmol/L tBHP and was incubated for 24 hours. At the end of the incubation period, transepithelial electrical resistance (TEER) was measured using an EVOM voltohmmeter and STX2 electrode (World Precision Instruments, Sarasota, FL). Blank resistance (insert without cells) was subtracted from the resistance reading across cell monolayers to obtain the true resistance.
Determination of Protein
The protein levels of the cell samples were measured by the Bradford method.45 Bovine serum albumin was used as the protein standard.
Animal Research
All the animal experiments were approved by the Animal Studies Committee at Washington University School of Medicine, and mice were treated in accordance with the recommendations of the Association for Research in Vision and Ophthalmology. Litters of 8-week-old 129/SvlmJ mice (The Jackson Laboratory) were given i.p. injections of NACA (250 mg/kg in PBS) versus PBS alone 1 day before and 1 day after light exposure. The mice were subsequently dark-adapted overnight. The following day, the mice were dilated with 0.5% cyclogyl and 5 minutes later with 2.5% phenylephrine hydrochloride and then were placed back in the dark for 30 minutes. They were then exposed to 13,000 lux of light for 4 hours, with redilation at 2 hours. The mice were placed in the dark for 24 hours after light exposure, followed by a normal 12-hour light to dark cycle. Six days after light exposure, the mice underwent full-field electroretinography (ERG) using a UTAS-E 3000 Visual Electrodiagnostic System (LKC Technologies, Gaithersburg, MD).46,47 Mice were anesthetized by i.p. injection of a mixture of 86.9 mg/kg ketamine and 13.4 mg/kg xylazine. The recording electrode was a platinum loop placed in a drop of methylcellulose on the surface of the cornea; a reference electrode was placed subdermally at the vertex of the skull and a ground electrode under the skin of the back or tail. Stimuli were brief white flashes delivered via a Ganzfeld integrating sphere, and signals were recorded with bandpass settings of 0.3 Hz to 500 Hz. After a 10-minute stabilization period, a scotopic intensity series was recorded that included rod-specific and scotopic bright flash responses. After a 10-minute light adaptation period on a steady white background, a photopic intensity series was recorded. Scotopic and photopic b-wave amplitudes and scotopic a-wave amplitudes were recorded for all flash intensities. After the recordings were completed, the mice were euthanized, a mark was placed at 12:00 at the corneal limbus, and the eyes were removed and embedded and fixed in 4% paraformaldehyde. Sections were cut parallel to 12:00 meridian through the optic nerve and were stained with H&E. With the observer masked with respect to treatment group, outer nuclear layer (ONL) thickness was measured at eight defined points along the vertical meridian. The optic nerve head was used as a reference point, and in the superior and inferior hemispheres, the distance between each point was 100 μm.
Statistics
All values are reported as mean ± SE of multiduplicates (run in at least triplicate). Statistical significance was calculated using an unpaired two-tailed t-test. Results with P < 0.05 were considered statistically significant.
Results
tBHP Increases ROS, Leading to Oxidative Stress and Cell Death in ARPE-19
To investigate RPE cell viability in response to tBHP treatment, we used FACS with annexin V-FITC and propidium iodide–labeled ARPE-19 cells. A dose-dependent decrease in cell viability was observed to occur with RPE cell exposure to tBHP (P < 0.0001; Figure 1A), which was confirmed using a calcein AM assay. Based on the dose-response relationship, incubation of cells in 0.4 mmol/L tBHP for 4 hours was determined to be optimal in evaluating potential cell rescue with NACA. To further substantiate the role of tBHP in RPE cell death through oxidative stress, ROS levels were measured. An increase in the production of ROS in ARPE-19 cells was seen with exposure to tBHP (P < 0.0001; Figure 1B).
Figure 1.
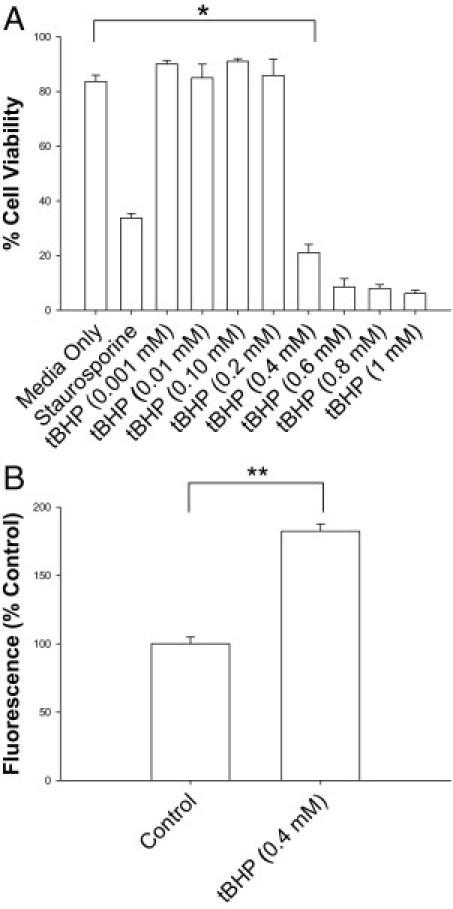
A: Cytotoxicity of tBHP in ARPE-19 cells. Cells were incubated with various concentrations of tBHP, media, or staurosporine; stained with annexin V-FITC/propidium iodide; and counted using FACS. B: Effects of tBHP on ROS levels in ARPE-19 cells. Cells loaded with 2′,7′–dichlorofluorescein diacetate were treated with tBHP, and fluorescence levels were measured. Fluorescence levels are directly correlated with ROS levels. tBHP exposure of ARPE-19 led to significant cell death and increased ROS. *P < 0.0001; **P < 0.0001. Error bars represent SE.
NACA Is Nontoxic to ARPE-19 Cells
To evaluate whether measured changes in cell response were due to an induction of stress by NACA, cells were incubated in NACA at various concentrations over a 24-hour period, after which cell viability was assessed using FACS. There were no statistically significant differences measured between the media-only group and any of the NACA concentrations after 24-hour incubation. ROS levels were measured in ARPE-19 cells incubated in NACA for 24 hours. There was no statistically significant difference in ROS levels between cells preincubated in NACA versus controls (data not shown).
Maintenance of Cellular Homeostasis and Prevention of Oxidative Stress–Induced Cell Damage and Death
To evaluate whether NACA could protect against the severe oxidative stress and cell death induced by tBHP, sequential exposure of ARPE-19 cells was tested. NACA preincubation was shown to rescue RPE cells from tBHP-induced oxidative stress and cell death as demonstrated by a 322% increase in cell survival found in NACA pretreated cells compared with cells exposed to tBHP without NACA preincubation (P < 0.00001; Figure 2A). In addition, no significant difference was found between tBHP-exposed cells preincubated in NACA and negative controls. The overall cellular homeostasis and integrity of the tBHP-exposed RPE cells were then evaluated by cell morphologic analysis, as seen by conventional microscopy. Figure 2B demonstrates that NACA preincubation allows gross maintenance of cellular morphologic features and cell attachment even after toxic exposure to tBHP. To further evaluate the cellular integrity of ARPE-19 cells, TEER was assessed. Reductions in TEER represent an early expression of cell damage. We found that ARPE-19 exposure to tBHP significantly decreased TEER compared with controls, whereas pretreatment with NACA prevented any tBHP-induced decrease in TEER (P < 0.0001 for both; Figure 3).
Figure 2.
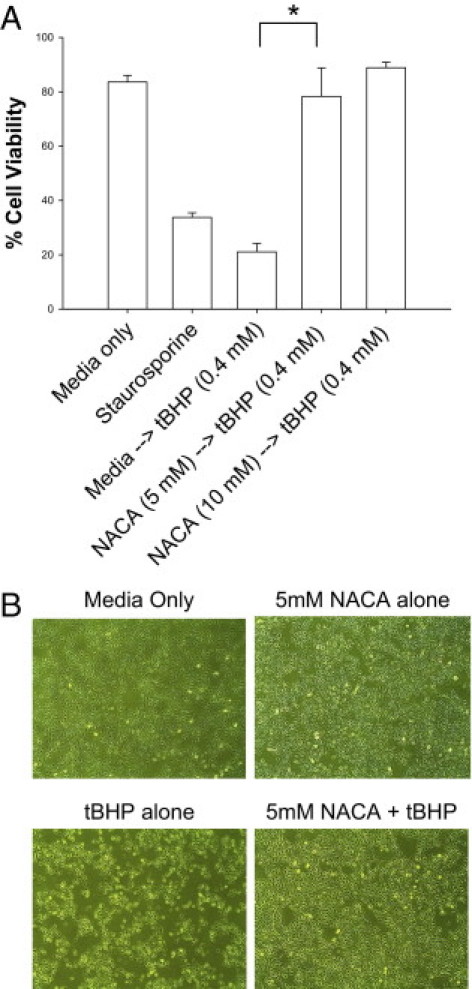
Prevention of tBHP-induced cell death in ARPE-19 with NACA. A: Cells were pretreated with NACA for 24 hours before treatment with 0.4 mmol/L tBHP for 4 hours. The staurosporine group is included as a positive control. *P < 0.00001. B: ARPE-19 cell photographs taken during treatment with labeled compounds. Note the significant cell death with tBHP exposure significantly reversed by NACA preincubation. Error bars represent SE.
Figure 3.
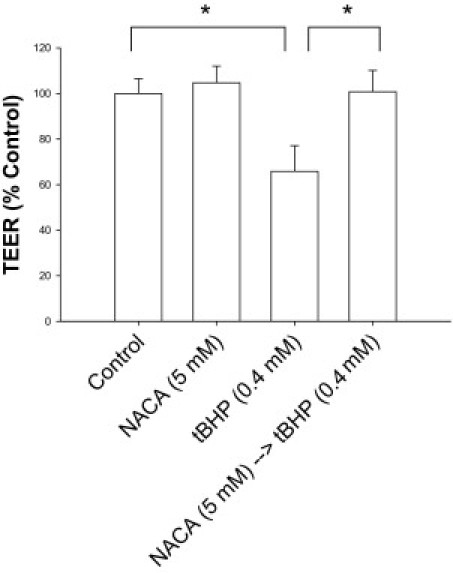
Maintenance of tBHP-exposed ARPE-19 cellular homeostasis, as measured by TEER, with NACA. ARPE-19 cells were grown on transwell inserts, and TEER was measured using an EVOM voltohmmeter. *P < 0.0001. Error bars represent SE.
Preservation of RPE Cellular Homeostasis and Reversal of Lipid Peroxidation
To evaluate the ability of NACA to prevent tBHP-induced ROS proliferation, ARPE-19 cells were incubated in NACA, after which a reliable ROS-detecting reagent was loaded into the cells before exposure to tBHP.42 Levels of ROS production in tBHP-exposed cells pretreated with NACA were significantly reduced compared with those in the tBHP-treated group without NACA preincubation (P < 0.01; Figure 4A). To investigate whether NACA affects lipid peroxidation in RPE cells, we measured MDA levels in ARPE-19 cells. Figure 4B depicts MDA levels in cells treated with tBHP with and without pretreatment using NACA. Cells treated with tBHP have significantly higher levels of MDA compared with NACA pretreated cells exposed to tBHP (P < 0.01). Pretreatment of cells with NACA before tBHP exposure prevented any increase in MDA levels above control.
Figure 4.
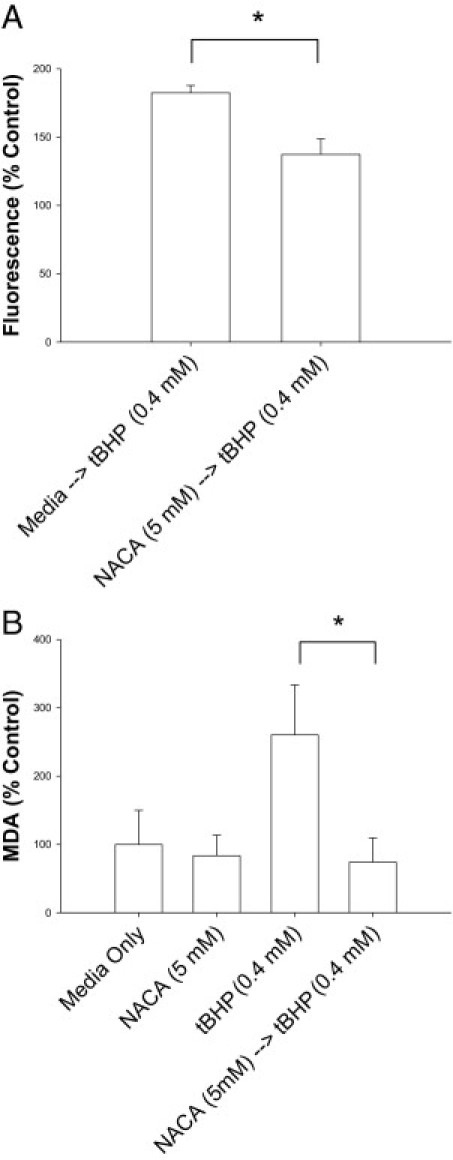
Protective effect of NACA against ROS production and lipid peroxidation in ARPE-19 cells. A: Cells were pretreated with media or NACA for 24 hours followed by exposure to 0.4 mmol/L tBHP. Fluorescence measured is directly correlated to ROS present in the cell. B: MDA levels in tBHP-exposed cells. Cells were pretreated with 5 mmol/L NACA for 24 hours, followed by exposure to tBHP. The control media group was incubated in media for 28 hours, The NACA group was exposed to NACA for 24 hours, followed by media alone for 4 hours. Cells were collected, and MDA levels, a biomarker that measures the level of oxidative stress in a cell due to lipid peroxidation, were measured. *P < 0.01. Error bars represent SE.
NACA Increases Levels of Cellular GSH and Induces GPx
The effects of NACA on cellular GSH and the critical phase II antioxidant enzyme GPx were investigated using reverse-phase HPLC43 and a GPx colorimetric assay kit. Cell exposure to 0.4 mmol/L tBHP for 4 hours resulted in a 75% decrease in GSH levels compared with controls (P < 0.001; Figure 5A). NACA-preincubated cells exposed to tBHP demonstrated cellular GSH levels greater than those of controls and significantly greater than with tBHP exposure alone (P < 0.001; Figure 5A). Cells pretreated with NACA (without exposure to tBHP) also showed significantly increased levels of GSH compared with controls, a trend previously seen with NACA exposure in other cells (P < 0.001; Figure 5A).36 tBHP was further shown to cause a dramatic decline in GPx activity (P < 0.01; Figure 5B). NACA preincubation before tBHP exposure caused a significant improvement in GPx activity in cells to near baseline levels (P < 0.001; Figure 5B).
Figure 5.
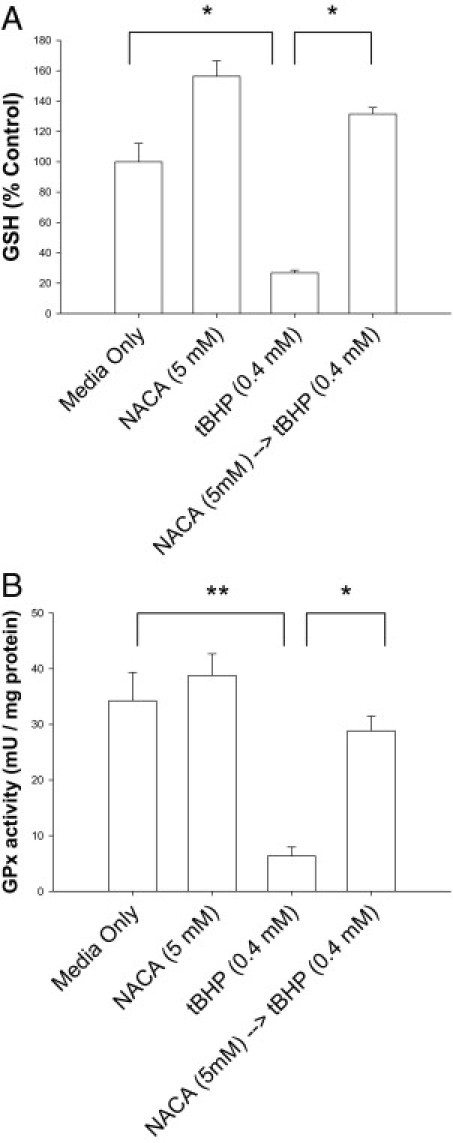
Maintenance of cellular GSH and induction of GPx with NACA. A: GSH level was quantified in cells treated for 24 hours with media, NACA, or tBHP and in cells pretreated with NACA and exposed to tBHP. B: GPx activity was measured in cells treated as described previously herein. *P < 0.001; **P < 0.01. Error bars represent SE.
Prevention of Oxidative Damage, Photoreceptor Cell Death, and RPE Cell Damage in Vivo
A model of light-induced toxicity with 129/SvlmJ mice was used to investigate the ability of NACA to prevent photoreceptor death in vivo. The mice were treated with an i.p. injection of vehicle only or vehicle containing NACA (250 mg/kg) 24 hours before and 24 hours after light exposure. Mice were then euthanized 6 days after light exposure, and retinal sections were evaluated by a masked observer. Specific attention was given to the photoreceptor cell body density in the ONL at eight locations. Results at all eight measurement locations demonstrate significant preservation of photoreceptor cell density in the ONL of mice treated with NACA compared with controls (P < 0.01 for each location; Figure 6). Further evaluation of the RPE by a masked observer using a ×630 confocal microscope demonstrated significant preservation of RPE integrity in NACA-pretreated light-exposed mice compared with vehicle-treated light-exposed mice (Figure 7).
Figure 6.
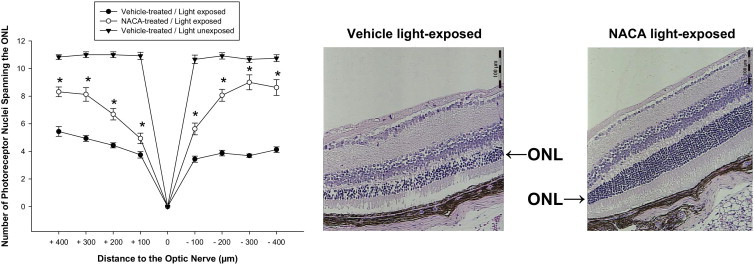
Suppression of light-induced ONL thinning with NACA. Six days after light exposure, retinal ONL thickness was measured. Reduction in ONL thickness induced by light exposure was significantly reversed with NACA pretreatment compared with vehicle alone. Representative images of H&E staining for retinal sections at the same location 400 μm superior to the optic nerve head demonstrating prevention of ONL thinning with NACA (right) compared with vehicle alone (left). Scale bars = 100 μm. n = 6. *P < 0.01. Error bars represent SE.
Figure 7.
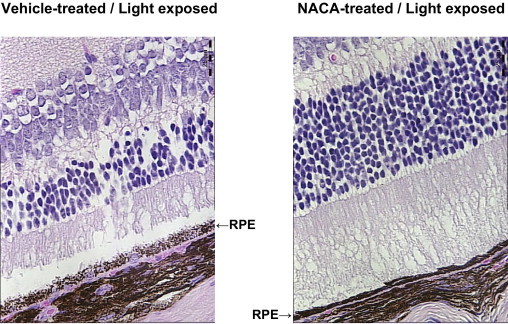
Maintenance of RPE cell integrity in vivo with NACA. Confocal microscopy (×630) obtained 6 days after light exposure. RPE cell disruption as induced by light exposure was significantly prevented with NACA pretreatment compared with vehicle alone. Representative images of H&E staining for retinal sections at the same location 400 μm superior to the optic nerve head demonstrating prevention of RPE disruption with NACA (right) compared with vehicle alone (left). Scale bars = 25 μm.
Preservation of Visual Potential and Photoreceptor Function
To investigate the effect of NACA on light-induced retinal dysfunction, ERG evaluation was performed on light-unexposed mice and 6 days after light exposure in NACA and vehicle-treated mice. Light exposure of vehicle-treated mice led to a significant reduction in the mean ± SE peak amplitudes of scotopic a-wave (127.8 ± 29.3 μV), scotopic b-wave (522.9 ± 135.88 μV), and photopic b-wave (175.4 ± 14.4 μV) compared with those of vehicle-treated mice receiving no light exposure [scotopic a-wave, 252.4 ± 39.6 μV (P < 0.01), scotopic b-wave, 889.7 ± 73.6 μV (P < 0.01), and photopic b-wave, 296.0 ± 79.9 μV (P = 0.02)] (Figure 8). Systemic administration of NACA to light-exposed mice significantly prevented the reduction in mean ± SE peak amplitude (P < 0.05 for all) of the scotopic a-wave (271.2 ± 48.5 μV), scotopic b-wave (1015.7 ± 128.4 μV), and photopic b-wave (284.9 ± 49.2 μV) compared with vehicle treatment to light-exposed animals (scotopic a-wave, 127.8 ± 29.3 μV; scotopic b-wave, 522.9 ± 135.88 μV; and photopic b-wave, 175.4 ± 14.4 μV).
Figure 8.
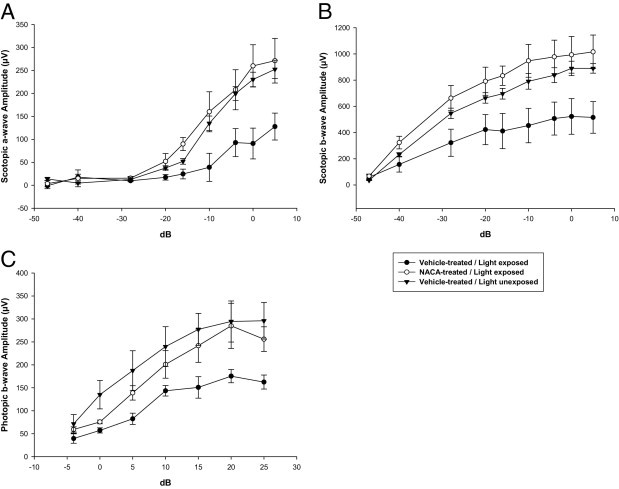
Prevention of light-induced retinal dysfunction with NACA as demonstrated by ERG. Data obtained 6 days after light exposure. Mean ERG wave responses for scotopic a-wave (A), scotopic b-wave (B), and photopic b-wave (C) demonstrating significant reductions in ERG responses in all waveforms comparing vehicle-treated unexposed mice with vehicle-treated light-exposed mice (P < 0.01 for all). Pretreatment with NACA prevented significant reductions in all ERG waveform peak amplitudes compared with vehicle-treated light-exposed mice and further prevented significant reduction in all ERG waveform peak amplitudes compared with unexposed mice (P < 0.05 for all). n = 6. (Standard flash 0 dB = 3.0 cds/m2.) Error bars represent SE.
Discussion
Oxidative stress is believed to play a fundamental role in the pathogenesis of AMD and other retinal degenerations.6,48,49 Patients with late AMD have been shown to have significantly lower antioxidant enzyme levels and higher lipid peroxidation by-product levels compared with patients with early AMD (P < 0.001 for each).50 The role of oxidative stress in AMD has been further supported by several epidemiologic studies.26,27,51,52
Although studies have demonstrated protection of RPE cells from oxidative stress–induced cell death in vitro, none have shown maintenance of RPE cell homeostasis. In addition, to our knowledge, no compound shown to be successful in RPE cell rescue against oxidative stress in vitro has also demonstrated protection against photoreceptor and RPE degeneration and loss of visual potential in vivo. We hypothesized that a more robust antioxidant, such as NACA, might prevent RPE and photoreceptor cell death in retinal degenerations, including AMD. We evaluated the potential of the novel thiol antioxidant, NACA, to prevent oxidative stress–related damage in vitro and in vivo. NACA was generated as an antioxidant with greater potential to cross the blood-brain barrier and cell membranes due to the substitution of an amide group in place of the carboxyl group in its parent compound, NAC. Studies using NACA have confirmed these properties, demonstrating its ability to penetrate the cell membrane significantly better than other antioxidants, including NAC, vitamin E, and carotenoids.39,53 It scavenges free radicals directly and indirectly by providing GSH, substitutes itself for depleted GSH, and chelates Cu2+, ultimately preventing catalyzation of ROS formation.54
In this study, we demonstrate that NACA protects RPE cells against oxidative stress–induced cell damage in vitro. We used the ARPE-19 cell line, a spontaneously arising human RPE cell line with normal karyologic features that has structural and functional properties characteristic of RPE cells in vivo.55 tBHP was used as the oxidant, causing oxidative stress in cells by inducing free radicals, decreasing GSH, and promoting lipid peroxidation,56–58 all thought to be essential in the pathogenesis of retinal degenerations, including AMD.6 tBHP triggers RPE cell death by induction of the apoptotic signaling cascade through severe oxidative stress.59 tBHP increased ROS in a dose-dependent manner, leading to severe oxidative stress, and, ultimately, cell death in ARPE-19 cells. NACA was nontoxic to the RPE cells even at high concentrations. NACA preincubation robustly prevented ARPE-19 oxidative stress–induced cell death from tBHP. No significant differences were found between NACA-preincubated cells exposed to tBHP and those exposed only to media throughout the experiment, an unprecedented finding in ARPE-19 antioxidant cell rescue experiments.
To further establish the ability of NACA to protect RPE health from oxidative stress, cellular integrity after tBHP exposure was measured using TEER. TEER provides a measurement of movement of ions across the paracellular pathway. TEER assessment has proved to be a successful model in predicting how oxidative stress damage to a monolayer affects the presence and stability of tight junctions and barrier function and how particular compounds are able to prevent this damage.60 A vital function of the RPE in vivo is the formation of the outer blood-retinal barrier, essential to retinal health. A decline in RPE monolayer cellular homeostasis and tight junction integrity places the RPE at risk and leads to barrier impairment. This exposes the retina to detrimental entities and can cause retinal degeneration and damage and become a nidus for neovascularization. NACA prevented a tBHP-induced decrease in TEER, verifying its ability to protect cellular homeostasis and outer blood-retinal barrier integrity under severe oxidative stress conditions.
In RPE cells, damage to molecules in the POSs occurs as a result of free radical chain reactions initiated by oxygen or light metabolism. Lysosomal enzymes in the RPE fail to digest lipid peroxidation–modified POSs, preventing molecular degradation.61 The result is an accumulation of lipofuscin in the RPE that reduces cytoplasmic space and cell volume available to organelles, further decreasing the cell's potential to phagocytose additional POSs. Each RPE cell must continue to engulf spent POSs for life, with undigested residual bodies added to an exponentially growing stockpile of lipofuscin.62 Accumulation of lipofuscin is a hallmark of aging in metabolically active postmitotic RPE cells and induces their degeneration.63–65 Deposition of excess lipofuscin and its peroxidized lipids in the surrounding Bruch's membrane manifests as drusen, a classic early clinical finding in AMD.66 NACA prevented ROS propagation and completely reversed lipid peroxidation as measured by MDA, a biomarker that measures the level of oxidative stress in a cell due to lipid peroxidation.67 These results are even more critical in light of recent literature demonstrating significant increases in human plasma and erythrocyte MDA levels in patients with AMD that directly correlate with disease severity.50
Accumulation of ROS-laden lipofuscin granules, combined with elevated local oxygen tension, high polyunsaturated lipid content, focused light exposure, and secondary photosensitizing agents that accumulate with aging, creates a severe oxidative stress environment for the RPE. This leads to a significant reduction in levels of GSH, the most critical antioxidant and redox buffer of cells.68,69 The decline in GSH seen with oxidative stress creates a redox imbalance that enhances cell susceptibility to injury and ROS-induced apoptosis. Altering the redox state to a more reduced level by adding GSH has been shown to decrease sensitivity of RPE cells to apoptosis from ROS.70 NACA prevented oxidative stress and cell damage in RPE cells by increasing levels of cellular GSH and inducing the critical phase II detoxification enzyme GPx, a selenoenzyme that reduces each peroxide molecule to two waters and oxidatively couples two GSH molecules. NACA has been shown to reduce apoptosis by inhibiting the p38 mitogen-activated protein kinase/inducible nitric oxide synthase pathway, likely leading to induction of the nuclear factor erythroid 2–related factor.71 NACA-induced nuclear factor erythroid 2–related factor activation would yield increased GSH and ultimate induction of the GPx catalytic cycle, as demonstrated in the results.72 This is vital in light of evidence using in vivo models of oxidative damage–induced retinal degeneration that independent increased expression of GPx provides powerful protection of retinal structure and function.73 Human evidence further demonstrates that GPx activity is significantly lower in the red blood cells and plasma of patients with AMD maculopathy than in control subjects.50
Protecting the RPE from oxidative damage is critical in the treatment of retinal degeneration, but the ultimate goal is the preservation of retinal photoreceptors. A shared feature between RPE and photoreceptor cell damage as well as apoptosis seems to be light-induced ROS production, generated by rhodopsin bleaching or from toxic compounds, such as A2E.74 Photoreceptors have demonstrated significantly increased vulnerability to death during aging, likely related to this ROS production and resultant oxidative stress.75–77 The 129/SvlmJ light-induced toxicity mouse model of retinal degeneration enabled investigation of the potential for NACA to slow photoreceptor degeneration and rescue visual potential in vivo. NACA demonstrated the ability to prevent oxidative damage and slow photoreceptor cell death as measured by ONL cell density and RPE cell integrity. Although histologic analysis is generally limited to small regions in the retina, ERG can measure a complete retinal response of surviving cells. NACA demonstrated preservation of visual potential and photoreceptor function in the 129/SvlmJ mouse as measured by scotopic a-wave, scotopic b-wave, and photopic b-wave ERG. Furthermore, there was no difference in ERG waveforms comparing light-unexposed mice and NACA-pretreated light-exposed mice. The results, therefore, illustrate that NACA can prevent photoreceptor degeneration and loss of visual potential in vivo.
Overall, NACA shows remarkable protection of RPE and photoreceptors against oxidative cell damage and death, in vitro and in vivo. In conjunction with the considerable evidence supporting the role of oxidative stress in the pathogenesis of multiple significant blinding retinal disease processes, including AMD, and NACA's superior chemical properties and bioavailability, this compound may be a critical agent in the delay or prevention of retinal degenerative diseases.
With the prevalence of AMD and other retinal degenerations expected to double in the coming decades, the need for more effective therapy to prevent its progression is imperative. Additional work is needed to find an optimized regimen of NACA dosing and to elucidate further mechanisms through which NACA can act to prevent oxidative stress and cell death in the RPE and photoreceptors. Because NACA has been proved successful as an oral medication in previous mouse studies, the potential for a readily available oral medication to prevent or even slow the progression of AMD is promising.
Footnotes
Supported by NIH grants K08EY016139 and R01EY019287 (R.S.A.), NIH Vision core grant P30 EY 02687, a Carl Marshall Reeves and Mildred Almen Reeves Foundation Inc. Award (R.S.A.), a Research to Prevent Blindness Inc. Career Development Award (R.S.A.), the International Retina Research Foundation (R.S.A.), an American Federation for Aging Research grant (R.S.A.), the American Retina Foundation (R.S.A.), an International Retinal Research Foundation Callahan Award (D.S.D.), a Lacey Foundation Research Award (A.S.), NIH and National Institute on Drug Abuse awardR15DA023409 (N.E.), and a Research to Prevent Blindness Inc. unrestricted grant to Washington University.
A.M.S. and L.A. contributed equally to this work.
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Marmorstein A.D., Finnemann S.C., Bonilha V.L., Rodriguez-Boulan E. Morphogenesis of the retinal pigment epithelium: toward understanding retinal degenerative diseases. Ann N Y Acad Sci. 1998;857:1–12. doi: 10.1111/j.1749-6632.1998.tb10102.x. [DOI] [PubMed] [Google Scholar]
- 2.Tate D.J., Jr., Miceli M.V., Newsome D.A. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:1271–1279. [PubMed] [Google Scholar]
- 3.Dorey C.K., Delori F.C., Akeo K. Growth of cultured RPE and endothelial cells is inhibited by blue light but not green or red light. Curr Eye Res. 1990;9:549–559. doi: 10.3109/02713689008999595. [DOI] [PubMed] [Google Scholar]
- 4.Alder V.A., Cringle S.J. The effect of the retinal circulation on vitreal oxygen tension. Curr Eye Res. 1985;4:121–129. doi: 10.3109/02713688508999977. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy C.J., Rakoczy P.E., Constable I.J. Lipofuscin of the retinal pigment epithelium: a review. Eye. 1995;9(pt 6):763–771. doi: 10.1038/eye.1995.192. [DOI] [PubMed] [Google Scholar]
- 6.Cai J., Nelson K.C., Wu M., Sternberg P., Jr., Jones D.P. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 7.Bressler N.M., Munoz B., Maguire M.G., Vitale S.E., Schein O.D., Taylor H.R., West S.K. Five-year incidence and disappearance of drusen and retinal pigment epithelial abnormalities: Waterman study. Arch Ophthalmol. 1995;113:301–308. doi: 10.1001/archopht.1995.01100030055022. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J., Jang Y.P., Kim S.R., Sparrow J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu D.Y., Cringle S.J., Su E.N., Yu P.K. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci. 2000;41:3999–4006. [PubMed] [Google Scholar]
- 10.Yu D.Y., Cringle S., Valter K., Walsh N., Lee D., Stone J. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Invest Ophthalmol Vis Sci. 2004;45:2013–2019. doi: 10.1167/iovs.03-0845. [DOI] [PubMed] [Google Scholar]
- 11.Shen J., Yang X., Dong A., Petters R.M., Peng Y.W., Wong F., Campochiaro P.A. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol. 2005;203:457–464. doi: 10.1002/jcp.20346. [DOI] [PubMed] [Google Scholar]
- 12.Komeima K., Rogers B.S., Lu L., Campochiaro P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2006;103:11300–11305. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green W.R., Key S.N., III Senile macular degeneration: a histopathologic study. Trans Am Ophthalmol Soc. 1977;75:180–254. [PMC free article] [PubMed] [Google Scholar]
- 14.Green W.R., McDonnell P.J., Yeo J.H. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- 15.Spraul C.W., Lang G.E., Grossniklaus H.E. Morphometric analysis of the choroid, Bruch's membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:2724–2735. [PubMed] [Google Scholar]
- 16.Zarbin M.A. Age-related macular degeneration: review of pathogenesis. Eur J Ophthalmol. 1998;8:199–206. doi: 10.1177/112067219800800401. [DOI] [PubMed] [Google Scholar]
- 17.Lu L., Oveson B.C., Jo Y.J., Lauer T.W., Usui S., Komeima K., Xie B., Campochiaro P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid Redox Signal. 2009;11:715–724. doi: 10.1089/ars.2008.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Resnikoff S., Pascolini D., Etya'ale D., Kocur I., Pararajasegaram R., Pokharel G.P., Mariotti S.P. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L., Li C.M., Rudolf M., Belyaeva O.V., Chung B.H., Messinger J.D., Kedishvili N.Y., Curcio C.A. Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest Ophthalmol Vis Sci. 2009;50:870–877. doi: 10.1167/iovs.08-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J.D., Curcio C.A., Johnson M. Morphometric analysis of lipoprotein-like particle accumulation in aging human macular Bruch's membrane. Invest Ophthalmol Vis Sci. 2008;49:2721–2727. doi: 10.1167/iovs.07-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jager R.D., Mieler W.F., Miller J.W. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- 22.Pauleikhoff D., Sheraidah G., Marshall J., Bird A.C., Wessing A. Biochemical and histochemical analysis of age related lipid deposits in Bruch's membrane [in German] Ophthalmologe. 1994;91:730–734. [PubMed] [Google Scholar]
- 23.Pauleikhoff D., Harper C.A., Marshall J., Bird A.C. Aging changes in Bruch's membrane: a histochemical and morphologic study. Ophthalmology. 1990;97:171–178. [PubMed] [Google Scholar]
- 24.Yanoff M., Duker J.S., Augsburger J.J. Mosby; St. Louis MO: 2004. Ophthalmology. [Google Scholar]
- 25.Klein R., Peto T., Bird A., Vannewkirk M.R. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004;137:486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 26.Coleman H., Chew E. Nutritional supplementation in age-related macular degeneration. Curr Opin Ophthalmol. 2007;18:220–223. doi: 10.1097/ICU.0b013e32814a586b. [DOI] [PubMed] [Google Scholar]
- 27.Age-Related Eye Disease Study Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeitz O., Schlichting L., Richard G., Strauss O. Lack of antioxidative properties of vitamin C and pyruvate in cultured retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2007;245:276–281. doi: 10.1007/s00417-006-0384-5. [DOI] [PubMed] [Google Scholar]
- 29.Marc R.E., Jones B.W., Watt C.B., Vazquez-Chona F., Vaughan D.K., Organisciak D.T. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–806. [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Z., Rogers B., Kachi S., Hackett S.F., Sick A., Campochiaro P.A. Reduction of p66Shc suppresses oxidative damage in retinal pigmented epithelial cells and retina. J Cell Physiol. 2006;209:996–1005. doi: 10.1002/jcp.20819. [DOI] [PubMed] [Google Scholar]
- 31.Wenzel A., Grimm C., Marti A., Kueng-Hitz N., Hafezi F., Niemeyer G., Reme C.E. c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J Neurosci. 2000;20:81–88. doi: 10.1523/JNEUROSCI.20-01-00081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abler A.S., Chang C.J., Ful J., Tso M.O., Lam T.T. Photic injury triggers apoptosis of photoreceptor cells. Res Commun Mol Pathol Pharmacol. 1996;92:177–189. [PubMed] [Google Scholar]
- 33.Hafezi F., Marti A., Munz K., Reme C.E. Light-induced apoptosis: differential timing in the retina and pigment epithelium. Exp Eye Res. 1997;64:963–970. doi: 10.1006/exer.1997.0288. [DOI] [PubMed] [Google Scholar]
- 34.Reme C.E., Grimm C., Hafezi F., Marti A., Wenzel A. Apoptotic cell death in retinal degenerations. Prog Retin Eye Res. 1998;17:443–464. doi: 10.1016/s1350-9462(98)00009-3. [DOI] [PubMed] [Google Scholar]
- 35.Penugonda S., Mare S., Goldstein G., Banks W.A., Ercal N. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC12. Brain Res. 2005;1056:132–138. doi: 10.1016/j.brainres.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 36.Wu W., Abraham L., Ogony J., Matthews R., Goldstein G., Ercal N. Effects of N-acetylcysteine amide (NACA), a thiol antioxidant on radiation-induced cytotoxicity in Chinese hamster ovary cells. Life Sci. 2008;82:1122–1130. doi: 10.1016/j.lfs.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 37.Price T.O., Uras F., Banks W.A., Ercal N. A novel antioxidant N-acetylcysteine amide prevents gp120- and Tat-induced oxidative stress in brain endothelial cells. Exp Neurol. 2006;201:193–202. doi: 10.1016/j.expneurol.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 38.Am J., Atlas D., Fibach E. N-acetylcysteine amide (AD4) attenuates oxidative stress in β-thalassemia blood cells. Biochim Biophys Acta. 2008;1780:249–255. doi: 10.1016/j.bbagen.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Grinberg L., Fibach E., Am J., Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005;38:136–145. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 40.Wang X.M., Terasaki P.I., Rankin G.W., Jr., Chia D., Zhong H.P., Hardy S. A new microcellular cytotoxicity test based on calcein AM release. Hum Immunol. 1993;37:264–270. doi: 10.1016/0198-8859(93)90510-8. [DOI] [PubMed] [Google Scholar]
- 41.Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 42.Wang H., Joseph J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- 43.Winters R.A., Zukowski J., Ercal N., Matthews R.H., Spitz D.R. Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide. Anal Biochem. 1995;227:14–21. doi: 10.1006/abio.1995.1246. [DOI] [PubMed] [Google Scholar]
- 44.Draper H.H., Squires E.J., Mahmoodi H., Wu J., Agarwal S., Hadley M. A comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials. Free Radic Biol Med. 1993;15:353–363. doi: 10.1016/0891-5849(93)90035-s. [DOI] [PubMed] [Google Scholar]
- 45.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 46.Peachey N.S., Ball S.L. Electrophysiological analysis of visual function in mutant mice. Doc Ophthalmol. 2003;107:13–36. doi: 10.1023/a:1024448314608. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Y., Zhang Y., Ojwang B.A., Brantley M.A., Jr., Gidday J.M. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-1α and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
- 48.Beatty S., Koh H., Phil M., Henson D., Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 49.Seddon J.M., Hennekens C.H. Vitamins, minerals, and macular degeneration: promising but unproven hypotheses. Arch Ophthalmol. 1994;112:176–179. doi: 10.1001/archopht.1994.01090140052021. [DOI] [PubMed] [Google Scholar]
- 50.Evereklioglu C., Er H., Doganay S., Cekmen M., Turkoz Y., Otlu B., Ozerol E. Nitric oxide and lipid peroxidation are increased and associated with decreased antioxidant enzyme activities in patients with age-related macular degeneration. Doc Ophthalmol. 2003;106:129–136. doi: 10.1023/a:1022512402811. [DOI] [PubMed] [Google Scholar]
- 51.Seddon J.M., George S., Rosner B. Cigarette smoking, fish consumption, omega-3 fatty acid intake, and associations with age-related macular degeneration: the US Twin Study of Age-Related Macular Degeneration. Arch Ophthalmol. 2006;124:995–1001. doi: 10.1001/archopht.124.7.995. [DOI] [PubMed] [Google Scholar]
- 52.Evans J.R. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–253. doi: 10.1016/s1350-9462(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 53.Offen D., Gilgun-Sherki Y., Barhum Y., Benhar M., Grinberg L., Reich R., Melamed E., Atlas D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J Neurochem. 2004;89:1241–1251. doi: 10.1111/j.1471-4159.2004.02428.x. [DOI] [PubMed] [Google Scholar]
- 54.Penugonda S., Mare S., Lutz P., Banks W.A., Ercal N. Potentiation of lead-induced cell death in PC12 cells by glutamate: protection by N-acetylcysteine amide (NACA), a novel thiol antioxidant. Toxicol Appl Pharmacol. 2006;216:197–205. doi: 10.1016/j.taap.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 55.Dunn K.C., Aotaki-Keen A.E., Putkey F.R., Hjelmeland L.M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- 56.Thornalley P.J., Trotta R.J., Stern A. Free radical involvement in the oxidative phenomena induced by tert-butyl hydroperoxide in erythrocytes. Biochim Biophys Acta. 1983;759:16–22. doi: 10.1016/0304-4165(83)90183-6. [DOI] [PubMed] [Google Scholar]
- 57.Masaki N., Kyle M.E., Farber J.L. Tert-butyl hydroperoxide kills cultured hepatocytes by peroxidizing membrane lipids. Arch Biochem Biophys. 1989;269:390–399. doi: 10.1016/0003-9861(89)90122-7. [DOI] [PubMed] [Google Scholar]
- 58.Comporti M. Glutathione depleting agents and lipid peroxidation. Chem Phys Lipids. 1987;45:143–169. doi: 10.1016/0009-3084(87)90064-8. [DOI] [PubMed] [Google Scholar]
- 59.Cai J., Wu M., Nelson K.C., Sternberg P., Jr., Jones D.P. Oxidant-induced apoptosis in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40:959–966. [PubMed] [Google Scholar]
- 60.Omatsu T., Naito Y., Handa O., Hayashi N., Mizushima K., Qin Y., Hirata I., Adachi S., Okayama T., Kishimoto E., Takagi T., Kokura S., Ichikawa H., Yoshikawa T. Involvement of reactive oxygen species in indomethacin-induced apoptosis of small intestinal epithelial cells. J Gastroenterol. 2009;44(Suppl 19):30–34. doi: 10.1007/s00535-008-2293-3. [DOI] [PubMed] [Google Scholar]
- 61.Young R.W. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]
- 62.Feeney-Burns L., Berman E.R., Rothman H. Lipofuscin of human retinal pigment epithelium. Am J Ophthalmol. 1980;90:783–791. doi: 10.1016/s0002-9394(14)75193-1. [DOI] [PubMed] [Google Scholar]
- 63.Terman A., Brunk U.T. Oxidative stress, accumulation of biological “garbage,” and aging. Antioxid Redox Signal. 2006;8:197–204. doi: 10.1089/ars.2006.8.197. [DOI] [PubMed] [Google Scholar]
- 64.Terman A., Gustafsson B., Brunk U.T. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Aspects Med. 2006;27:471–482. doi: 10.1016/j.mam.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Sparrow J.R., Fishkin N., Zhou J., Cai B., Jang Y.P., Krane S., Itagaki Y., Nakanishi K. A2E, a byproduct of the visual cycle. Vision Res. 2003;43:2983–2990. doi: 10.1016/s0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]
- 66.Spaide R.F., Ho-Spaide W.C., Browne R.W., Armstrong D. Characterization of peroxidized lipids in Bruch's membrane. Retina. 1999;19:141–147. doi: 10.1097/00006982-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 67.Moore K., Roberts L.J., II Measurement of lipid peroxidation. Free Radic Res. 1998;28:659–671. doi: 10.3109/10715769809065821. [DOI] [PubMed] [Google Scholar]
- 68.Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 69.Sternberg P., Jr., Davidson P.C., Jones D.P., Hagen T.M., Reed R.L., Drews-Botsch C. Protection of retinal pigment epithelium from oxidative injury by glutathione and precursors. Invest Ophthalmol Vis Sci. 1993;34:3661–3668. [PubMed] [Google Scholar]
- 70.Nelson K.C., Armstrong J.S., Moriarty S., Cai J., Wu M.W., Sternberg P., Jr., Jones D.P. Protection of retinal pigment epithelial cells from oxidative damage by oltipraz, a cancer chemopreventive agent. Invest Ophthalmol Vis Sci. 2002;43:3550–3554. [PubMed] [Google Scholar]
- 71.Gong X, Celsi G, Carlsson K, Norgren S, Chen M: N-acetylcysteine amide protects renal proximal tubular epithelial cells against iohexol-induced apoptosis by blocking p38 MAPK and iNOS signaling. Am J Nephrol 31:178–188 [DOI] [PubMed]
- 72.Heverly-Coulson G.S., Boyd R.J. Reduction of hydrogen peroxide by glutathione peroxidase mimics: reaction mechanism and energetics. J Phys Chem A. 2010;114:1996–2000. doi: 10.1021/jp910368u. [DOI] [PubMed] [Google Scholar]
- 73.Lu L., Oveson B.C., Jo Y.J., Lauer T., Usui S., Komeima K., Xie B., Campochiaro P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid Redox Signal. 2009;11:715–724. doi: 10.1089/ars.2008.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Organisciak D.T., Vaughan D.K. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134. doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao H., Hollyfield J.G. Aging of the human retina: differential loss of neurons and retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1–17. [PubMed] [Google Scholar]
- 76.Curcio C.A., Millican C.L., Allen K.A., Kalina R.E. Aging of the human photoreceptor mosaic: evidence for selective vulnerability of rods in central retina. Invest Ophthalmol Vis Sci. 1993;34:3278–3296. [PubMed] [Google Scholar]
- 77.Dorey C.K., Wu G., Ebenstein D., Garsd A., Weiter J.J. Cell loss in the aging retina: relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–1699. [PubMed] [Google Scholar]