

Abstract
Inflammation and lung remodeling are hallmarks of asbestos-induced fibrosis, but the molecular mechanisms that control these events are unclear. Using laser capture microdissection (LCM) of distal bronchioles in a murine asbestos inhalation model, we show that osteopontin (OPN) is up-regulated by bronchiolar epithelial cells after chrysotile asbestos exposures. In contrast to OPN wild-type mice (OPN+/+) inhaling asbestos, OPN null mice (OPN−/−) exposed to asbestos showed less eosinophilia in bronchoalveolar lavage fluids, diminished lung inflammation, and decreased mucin production. Bronchoalveolar lavage fluid concentrations of inflammatory cytokines (IL-1β, IL-4, IL-6, IL-12 subunit p40, MIP1α, MIP1β, and eotaxin) also were significantly less in asbestos-exposed OPN−/− mice. Microarrays performed on lung tissues from asbestos-exposed OPN+/+ and OPN−/− mice showed that OPN modulated the expression of a number of genes (Col1a2, Timp1, Tnc, Eln, and Col3a1) linked to fibrosis via initiation and cross talk between IL-1β and epidermal growth factor receptor-related signaling pathways. Novel targets of OPN identified include genes involved in cell signaling, immune system/defense, extracellular matrix remodeling, and cell cycle regulation. Although it is unclear whether the present findings are specific to chrysotile asbestos or would be observed after inhalation of other fibers in general, these results highlight new potential mechanisms and therapeutic targets for asbestosis and other diseases (asthma, smoking-related interstitial lung diseases) linked to OPN overexpression.
Asbestos constitutes a group of naturally occurring mineral fibers that are causally associated with the development of pleural and pulmonary fibrosis (asbestosis), malignant mesotheliomas, and lung carcinomas.1 Asbestosis, like idiopathic pulmonary fibrosis, is a chronic progressive disease characterized by lung inflammation and scarring, and is associated with a life expectancy averaging 3 years to 5 years after diagnosis.2 Although the prevalence of asbestosis has decreased in the aftermath of increased public awareness and regulated control of asbestos exposures in the workplace, asbestosis occurs in approximately 200,000 patients annually in the United States, and deaths are estimated at approximately 4000 per year.3 There is at present no cure for asbestos-related lung or pleural diseases, and treatment options are relatively ineffective.
The lung epithelial cell is a pivotal cell type initiating lung inflammation and adverse pathology via a number of cell signaling pathways induced by asbestos; however, understanding of the downstream molecular events critical to lung repair and/or disease development is limited. Inflammation and fibrogenesis are processes critical to lung remodeling, and the interplay between these events is under intense investigation in several laboratories. A number of profibrotic immunomodulatory proteins may be linked to the development and progression of airway and interstitial fibrosis including osteopontin (OPN), an acidic phosphoglycoprotein that functions primarily in modulating immune function and extracellular matrix (ECM) remodeling.4 Several studies have suggested that the presence of OPN in serum or bronchoalveolar lavage fluid (BALF) is a biomarker of exposure or early response, as well as a possible effector of disease in the development of idiopathic pulmonary fibrosis,5 lung cancers,6 mesothelioma,7–9 and asthma.10 Moreover, OPN gene or protein overexpression is induced in the lungs, BALF, or sera of humans and rodents after exposure to a variety of drugs and pathogenic agents, including bleomycin,11,12 cigarette smoke,13 titanium dioxide,14 and asbestos.9,15 A recent clinical study shows that OPN is overexpressed in BALF cells from smokers and is linked causally to smoking-related interstitial lung diseases.16 OPN has been detected in a number of cell types, including cells of the immune system, alveolar epithelial and carcinoma cells,5,11,17,18 and lung fibroblasts in vitro.19 However, the sources of up-regulation of OPN, how it enters BALF or serum, and its mechanistic roles in lung injury and remodeling after exposures to asbestos have not been explored.
Here we present a novel scenario for up-regulation and secretion of OPN by distal bronchiolar epithelial cells after inhalation of asbestos fibers. First, we show that Opn gene expression is up-regulated in lung homogenates of mice as well as in distal bronchiolar epithelium isolated by LCM. This observation is supported by IHC performed by others showing widespread association of OPN with luminal epithelial surfaces in human lung tissues.17 Asbestos inhalation is associated with epithelial injury, decreases in epithelial and endothelial barrier function, and protein extravasation into BALF.1 OPN secreted by bronchiolar epithelium might be disseminated to BALF or the bloodstream. Epithelial cell-derived OPN and contributions from other inflammatory cells may then elicit autocrine or paracrine effects on lung epithelial cells to produce OPN-dependent profibrotic gene expression, including up-regulation of procollagens, elastin interfacing protein, fibronectin, and matrix metalloproteinases (MMPs).
We further show, using a systems biology approach and comparing wild-type (OPN+/+) with OPN null (OPN−/−) C57BL/6 mice, that OPN mediates asbestos-associated lung injury and fibrogenesis by altering chemokine/cytokine levels, immune cell profiles in BALF and lung, and mucin production in distal bronchioles, sites of impaction of chrysotile asbestos fibers after inhalation. Combining these observations, gene ontological profiling and classification, and a functional network analysis, we report a complex interplay between several signaling pathways previously described in lung epithelial cells and other cell types after exposures to asbestos with novel signaling events and putative asbestos-associated genes leading to altered ECM remodeling and inflammation. These pathways are initiated by interaction of asbestos fibers with Areg, a ligand of the epidermal growth factor receptor (Egfr), and an inflammasome- or tumor necrosis factor α (TNF-α)-mediated IL-1β response. Moreover, they indicate reciprocal interactions between OPN and the transcription factor activator protein-1 (AP-1) to cause activation of cytokines and other transcription factors (NF-κB and Gata3), in part through receptors (Cd44 and integrins) that may be critical to asbestos-induced lung injury and inflammation. Based on these robust global analyses on data from functional analyses and gene profiling studies on lungs from OPN+/+ wild-type and OPN−/− null mice, we also identify a number of novel OPN up-regulated and down-regulated genes linked to ECM remodeling, muscle contraction, immune defense, cellular transport, and cell signaling/metabolism. The present study is novel for use of a functionally based global approach to discern cell signaling and transcriptome events in a physiological model of asbestos inhalation and fibrogenesis through characterization of OPN−/− mice exposed to inhaled mineral fibers.
Materials and Methods
Murine Inhalation Model of Asbestos Fibrogenesis
C57BL/6 male mice (The Jackson Laboratory, Bar Harbor, ME), 8 weeks to 12 weeks old, were maintained at the University of Vermont Association for Assessment and Accreditation of Laboratory Animal Care accredited Animal Inhalation Facility. Mice (n = 4/group to 5/group per time point) were placed in inhalation chambers and exposed to either clean air or chrysotile asbestos (8.5 mg/m3 air; National Institute of Environmental Health Sciences reference sample) for 3, 9, or 40 days (6 hours/day; 5 days/week). Animals were offered food and water ad libitum during the exposure period. After euthanasia of mice with sodium pentobarbital intraperitoneally, the lungs were perfused and inflated under pressure with PBS, and BALF was collected as described previously.20 The left lobes of the lung were sutured, excised, and placed in 4% paraformaldehyde for histology, and a portion was frozen and sectioned for LCM experiments.21 The right lobes were excised, minced, and placed in RNAlater solution (Ambion, Austin, TX) for isolation of RNA. In a separate experiment, age-matched male C57BL/6 (OPN+/+) and OPN−/− mice (The Jackson Laboratory, Bar Harbor, ME) were exposed identically to clean air or chrysotile asbestos for 9 days, and tissues and BALF were processed as described above. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Vermont.
Laser Capture Microdissection and mRNA Array Analysis on Epithelium from Distal Bronchioles
To perform LCM, frozen lung tissue sections were processed as previously described.21 We selectively captured epithelial cells in intact bronchioles (identified as structures ≤800 μm in perimeter at ×400 magnification without a smooth muscle peribronchiolar lining) using an Arcturus PixCell II laser capture microdissector (Arcturus Engineering, Mountain View, CA). The captured cells were extracted for 30 minutes at 42°C in an extraction buffer (Arcturus Engineering) and were stored at −80°C until further processing. A total of five slides were processed for each animal. Total RNA was isolated using phenol-chloroform extraction, followed by a clean-up protocol (Qiagen, Germantown, MD) as previously described.20 Relative mRNA expression levels were determined for 113 genes involved in cell adhesion and ECM homeostasis, using the GEArray (SA Biosciences, Frederick, MD), according to the manufacturer's protocol. Array images were digitized by densitometric scanning on a Fluor-S MultiImager (Bio-Rad Laboratories, Hercules, CA), and analyzed by using GEArray Expression Analysis Suite software version 2.0 (SA Biosciences). Values were normalized to the signal for the housekeeping gene Gapdh. Genes were considered to be differentially expressed if the ratio between control and treatment groups was >1.5.
Lactate Dehydrogenase, Total Protein, and Differential Cell Counts in BALF
BALF was collected, cells were centrifuged and isolated from supernatant fluid, and Cytospin slides were prepared as previously described.22 The amount of BALF supernatant retrieved from each mouse was recorded and analyzed for total protein and lactate dehydrogenase as a marker of lytic cell injury using standard methods.22 Cytospin slides were stained, and the numbers of macrophages, polymorphonuclear cells, eosinophils, and lymphocytes were quantified as described previously.20
Bio-Plex Analysis for Chemokines and Cytokines in BALF
To quantify cytokine and chemokine levels in BALF, a multiplex suspension protein array was performed using the Bio-Plex protein array system (Bio-Rad Laboratories), as described previously.20 Briefly, anti-cytokine/chemokine antibody-conjugated beads were adhered to individual wells of a 96-well filter plate. BALF was added for 30 minutes. The plate was then washed and incubated with multiplex detection antibody, followed by streptavidin-conjugated phycoerythrin. After a final wash, plates were analyzed using the Bio-Plex protein array system, and concentrations of each cytokine and chemokine were determined using Bio-Plex Manager version 3.0 software. Data are expressed as pg cytokine/mL BALF.
Scoring of Lung Inflammation and Mucin Production
Lung sections (two per mouse) were examined by a board-certified pathologist (K.J.B.) using a blinded coding system. The severity of inflammation was determined on lung sections stained with H&E using a scoring system previously described23, as follows: 0 = none, 1 = mild, 2 = moderate, 3 = severe. For grading of mucin production, sections were stained with Alcian Blue-PAS, and the epithelium of distal bronchioles (<800 μm perimeter at ×400 magnification) was scored semiquantitatively.20 The percentage of cells expressing Alcian Blue-PAS positivity per total percentage of epithelial cells in individual distal bronchioles was scored using a blinded coding system.
Gene Expression Profiling on Lung Tissue
Total RNA isolation and processing of microarrays (Affymetrix, Santa Clara, CA) was performed as previously described, using the mouse genome Plus 2.0 chips.20 For statistical analysis, we used the GCRMA (guanine cytosine robust multi-array) expression measure. Data was normalized24 using the online analysis tool ArrayQuest25 to implement the open-source Bioconductor GCRMA package (http://www.bioconductor.org/install/index.html, last accessed in 2009). Differentially expressed genes between control and other samples were then identified for each normalized data set, using fold-change ≥2 and P ≤ 0.05 as selection criteria. Validation of expression of select genes (Plunc and Areg) were performed by quantitative real-time PCR (qPCR) as previously described.20 Pathway analysis was performed with Pathway Studio Software version 7.0 (Ariadne Genomics, Rockville, MD).
Statistical Analysis
All experiments were performed with four to five animals per group per time point, except that in the microarrays three mouse lungs per group were used. For most experiments, data were analyzed using analysis of variance and the Student-Newman-Keuls procedure for adjustment of multiple pair-wise comparisons between treatment groups. Differences with P ≤ 0.05 were considered statistically significant. Statistical analysis for microarrays was as described in the previous section.
Results
Up-Regulation of OPN Is Induced by Asbestos in Small Airway Epithelial Cells
Inhalation of chrysotile asbestos by C57BL/6 mice results in subepithelial fibrosis in bronchioles at 40 days, preceded by epithelial cell proliferation detected at 3 days, with inflammation peaking at 9 days.20 There were significant (P ≤ 0.05) time-dependent increases in OPN mRNA expression, as determined by validation of mRNA from microarray analysis by qPCR in lung tissues of C57BL/6 mice before (day 0) and at 3, 9, and 40 days after inhalation of chrysotile asbestos (Figure 1). To test the hypothesis that bronchiolar epithelial cells produced increased mRNA levels of ECM-related genes, including genes encoding OPN and OPN-related receptors before the development of fibrosis, we performed LCM to selectively isolate distal bronchiolar epithelial cells from mice exposed to clean air or asbestos (Figure 1B). Isolated RNA was then analyzed by GEArray analysis version 2.0 (SABiosciences Qiagen, Frederick, MD). Out of 113 genes analyzed, 9 transcripts (OPN, cd44 antigen, procollagen IVα1, procollagen Vα1, elastin interface 1, fibronectin 1, Mmp14, Mmp20 and Timp1) were up-regulated (P ≤ 0.05) at 3 days and/or 9 days after inhalation of asbestos. Significant increases in Opn expression in epithelial cells occurred at 9 days after exposure to asbestos, compared with animals in clean air (Figure 1B). Additionally, expression levels of Cd44 antigen, a receptor for OPN, was significantly induced by asbestos at 3 days. Other previously documented receptors for OPN (including integrins αVβIII, αVβV, and αVβI) were not significantly altered at the mRNA level by asbestos, neither in whole lung TMA nor in LCM array studies (data not shown).
Figure 1.
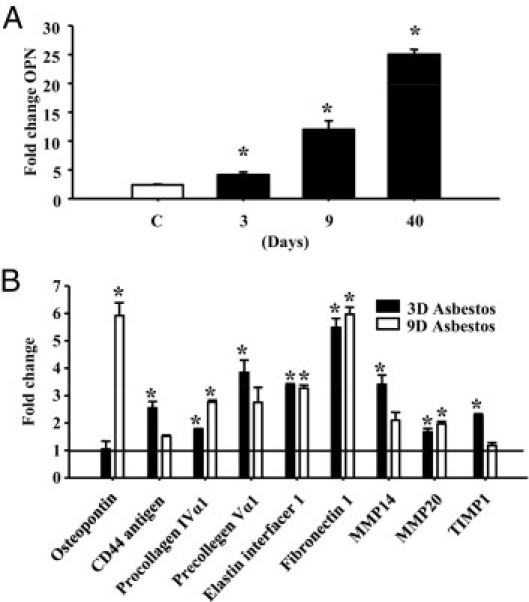
OPN mRNA expression is increased in lung epithelial cells exposed to asbestos. C57BL/6 mice were exposed to chrysotile asbestos (8 mg/m3 per day) for 3, 9, and 40 days. Lung tissue was collected and RNA was purified and analyzed for gene expression changes using microarrays and expression validated by qPCR. A: The transcriptional profile for OPN shows increased expression at all time points tested, compared with air-exposed animals (ie, clean air-exposed). B: Lungs were sectioned from mice exposed to asbestos for 3 days and 9 days, and LCM was performed on epithelial cells. Isolated RNA was examined for gene expression using GEArray analysis. Genes that were significantly altered by asbestos-exposed versus air-exposed animals are plotted. *P ≤ 0.05, air versus asbestos exposure.
Decreased Asbestos-Induced Injury and Inflammation Are Observed in OPN−/− Mice
To examine the functional roles of OPN in asbestos-induced lung remodeling, we exposed OPN+/+ and OPN−/− mice to clean air or chrysotile asbestos for 9 days, the peak time point of asbestos-induced inflammation and mucin production.20 Wild-type (OPN+/+) asbestos-exposed animals produced significantly elevated levels of lactate dehydrogenase, a marker of injury in BALF, whereby less overall injury was observed in asbestos-exposed OPN−/− mice (Figure 2A). Inhalation of asbestos caused an increase in total cell numbers in BALF in both OPN+/+ and OPN−/− mice, compared with the respective groups of mice in clean air, but total cell numbers were decreased (P ≤ 0.05) in asbestos-exposed OPN−/− versus OPN+/+ mice (Figure 2B). Although no differences in cell types were observed in mice in clean air, an altered differential cell profile was observed in BALF from asbestos-exposed mice, reflected in significant decreases in eosinophils in OPN−/− mice. These results correlated with histopathology: the severity of inflammation in lung tissue was less (P ≤ 0.05) in OPN−/− mice than in OPN+/+ mice after inhalation of asbestos (Figure 3A). A significant decrease in the numbers of distal bronchioles exhibiting mucin-positive epithelial cells was also observed with Alcian Blue-PAS staining in OPN−/− mice exposed to asbestos (Figure 3, B and C).
Figure 2.
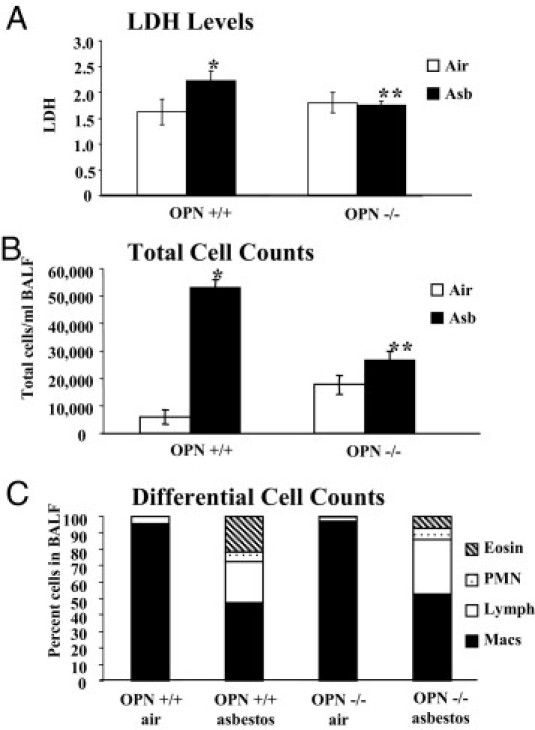
General asbestos-induced injury and immune cell profiles measured in BALF. OPN+/+ and OPN−/− mice were exposed to chrysotile asbestos (8 mg/m3 per day) for 9 days. The mice were lavaged and BALF was collected and processed for lactate dehydrogenase (A), total cell counts (B), and differential cell counts (C). *P ≤ 0.05, asbestos versus air exposure; **P ≤ 0.05, asbestos-exposed OPN+/+ versus OPN−/−. Cell types: eosinophils (Eosin), polymorphonuclear cells (PMN), lymphocytes (Lymph), and macrophages (Macs).
Figure 3.
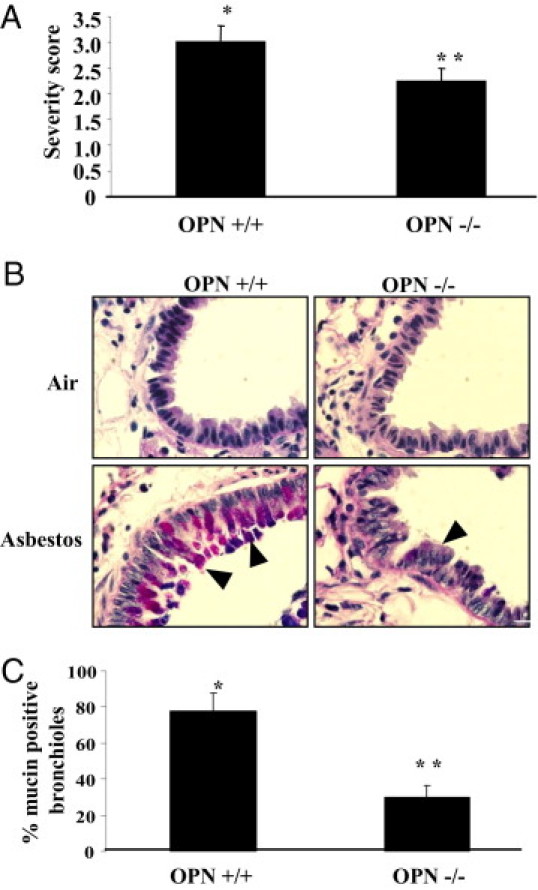
Decreased severity of asbestos-induced inflammation and mucin in OPN−/− mice. OPN+/+ and OPN−/− mice were exposed to chrysotile asbestos (8 mg/m3 per day) for 9 days. Lung tissue sections were stained with H&E or Alcian Blue-PAS and were scored for inflammation or mucin production, respectively. A: Severity of inflammation scored by assessing influx of macrophages and polymorphonuclear cells in H&E-stained tissue sections from asbestos-exposed mice. No detectable inflammation was observed in any air-exposed animals. B: A representative histological section of mucin staining with Alcian Blue-PAS for each treatment group. Arrowheads indicate examples of positive staining (pinkish-purple). C: Percentage of bronchioles affected with mucin in Alcian Blue-PAS stained tissue sections from asbestos-exposed mice. No detectable mucin was observed in any air-exposed animals. *P ≤ 0.05, asbestos versus air exposure; **P ≤ 0.05, asbestos-exposed OPN+/+ versus OPN−/−.
Changes in BALF Cytokine Profiles Are Observed in Asbestos-Exposed OPN Mice
No changes were seen in profiles of cytokines in OPN+/+ versus OPN−/− mice maintained in clean air (Figure 4). Out of 23 cytokines evaluated in Bio-Plex assays, 8 cytokines (IL-1β, IL-4, IL-5, IL-6, IL-12 subunit p40, MCP-1, MIP1α, and MIP1β) were significantly elevated in OPN+/+ animals exposed to asbestos, compared with air-exposed controls. IL-1β, IL-4, IL-6, IL-12 subunit p40, MIP1α, and MIP1β were significantly depressed in asbestos-exposed OPN−/− animals, compared with asbestos-exposed OPN+/+ animals (Figure 4). Eotaxin was undetectable in OPN−/− asbestos-exposed animals, compared with asbestos-exposed OPN+/+ animals. Although amounts of IL-5 and MCP-1 showed trends of repression in OPN−/− versus OPN+/+ mice after exposure to asbestos, these changes were not significant. Moreover, differences in amounts of other cytokines (IL-1α, IL-2, IL-3, IL-9, IL-10, IL-12(p70), IL-13, IL-17, INFγ, RANTES, and TNF-α) were not significantly altered between any groups (data not shown).
Figure 4.
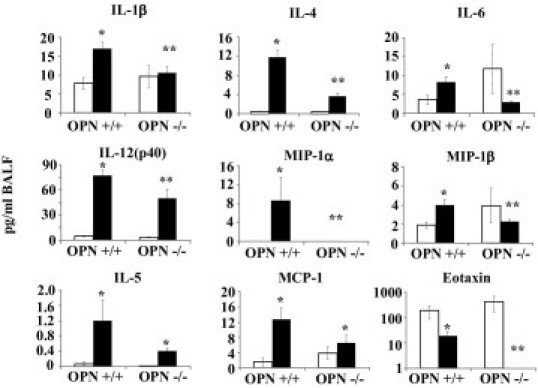
Inhibition of asbestos-induced production of immune-related proteins in BALF in OPN−/− mice. OPN+/+ and OPN−/− mice were exposed to chrysotile asbestos (8 mg/m3 per day) for 9 days. Bio-Plex analysis of BALF was performed. Chemokines and cytokines that were altered by asbestos in OPN+/+ and differentially produced in OPN−/− mice are shown. All values are presented as pg/mL BALF. Open bars: air-exposed animals; black bars: asbestos-exposed animals. *P ≤ 0.05, asbestos versus air exposure; **P ≤ 0.05, asbestos-exposed OPN+/+ versus OPN−/−.
Alterations in Gene Profiles Are Observed in Asbestos-Exposed OPN−/− Mice
The total numbers of genes significantly increased or decreased (≥2×) in expression in the lungs of OPN−/− clean-air mice and OPN+/+ or OPN−/− asbestos-exposed mice, compared with OPN+/+ mice in clean air, are shown in Figure 5A. Although some alterations were observed in mRNA levels between OPN−/− and OPN+/+ mice in clean air, gene expression changes (P ≤ 0.05) associated with asbestos were more robust. Expression of 52 genes was significantly altered in lungs of asbestos-exposed OPN+/+ versus OPN−/− mice.
Figure 5.
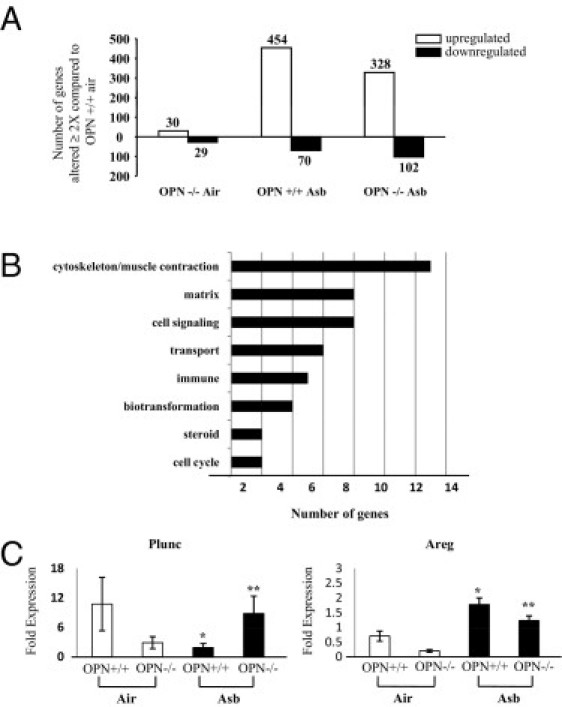
Gene profiling in OPN−/− mice exposed to asbestos. OPN+/+ and OPN−/− mice were exposed to chrysotile asbestos (8 mg/m3 per day) for 9 days. Total RNA was isolated from lung tissue and processed using microarrays. A: Total number of significant gene changes in lung tissue of OPN+/+ and OPN−/− asbestos-exposed mice and OPN−/− air-exposed mice, compared with OPN+/+ air-exposed mice. B: The number of genes that were significantly altered in lung tissue (P ≤ 0.05 and ≥2-fold compared with air-exposed OPN+/+ animals) and between asbestos-exposed OPN+/+ and OPN−/− mice, categorized by biological function. C: Validation of Plunc and Areg expression by qPCR in whole lung tissue from mice exposed to asbestos for 9 days. Data are reported as fold-change in expression relative to OPN+/+ air-exposed animals of each target, normalized to the expression of the housekeeping gene Hprt. Open bars: air-exposed animals; black bars: asbestos-exposed animals. *P ≤ 0.05, asbestos versus air exposure; **P ≤ 0.05, asbestos-exposed OPN+/+ versus OPN−/−.
The most prominent functional categories of these genes as determined by gene ontological analysis showed that changes in transcript levels reflected those linked to the cytoskeleton and muscle contraction, matrix, cell signaling, among others (Figure 5B). Validation of the microarray expression data was performed for two select targets (Plunc, Areg) by qPCR (Figure 5C). These patterns of expression are similar to those obtained by microarray analysis (Figure 6). Individual expression profiles were obtained for a subset of these transcripts involved in ECM remodeling (Figure 6A), cytoskeleton/muscle contraction (Figure 6B), cell signaling (Figure 6C), biotransformation enzymes (Figure 6D), cell cycle (Figure 6E), and immune system/defense (Figure 6F). Notably, a number of genes induced by asbestos in OPN+/+ mice showed significantly less expression in OPN−/− by comparison, including Adamts2, Areg, Ckap2, Nuf2, Col1a2, Col3a1, Cxcl10, Eln (elastin), Thbs1, Nrcam, Pbk, Pprc1, Stbd1, Timp1, Tnc, and Vcan. Additionally, for some targets, the opposite pattern was observed, wherein gene expression was increased (P ≤ 0.05) in asbestos-exposed OPN−/− mice, compared with OPN+/+ mice exposed to asbestos. These genes included Adipoq, Atp10, Ckmt2, Csrp3, Cyp2a4, Cytl1, Dbp, Fabp3, Fmo3, Gata3, Gp2, Hamp, Hs3st1, Hsd17b11, Ide, Marco, Mb, Sln, Smpx, Sult1d1, Tcap, Thrsp, Tnni3, Tnnt2, Myh6, Myl4, Myl7, Myoz1, Myrip, Pde4b, Pln, and Plunc.
Figure 6.
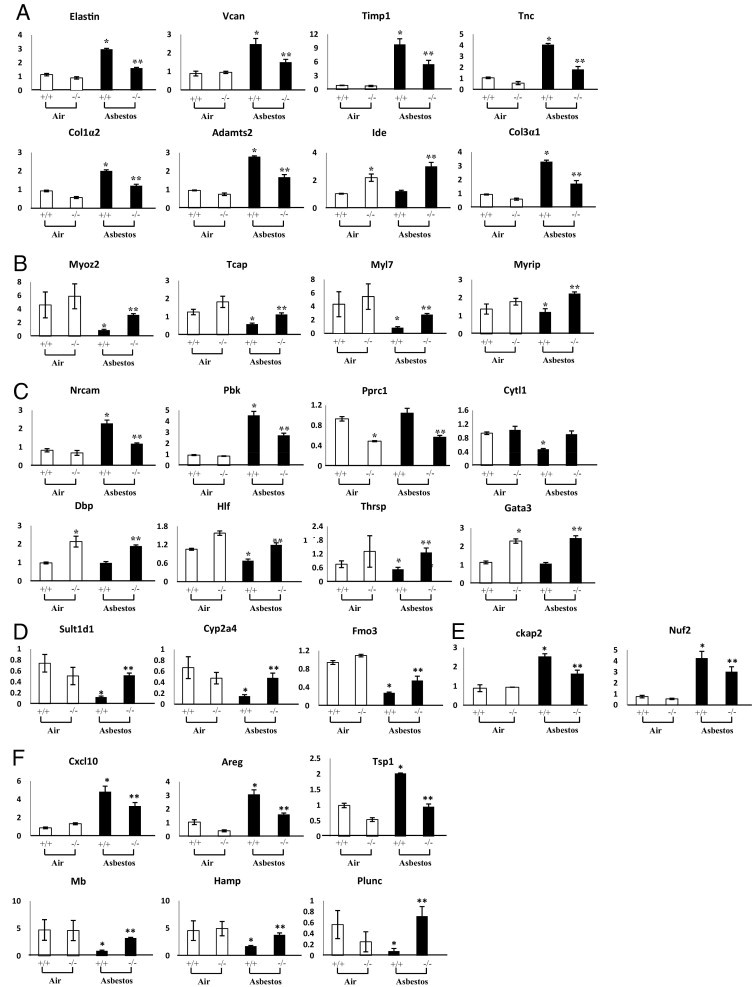
Expression profiles of select genes, by functional category. Data are reported as fold change relative to OPN+/+ air-exposed animals. A: Extracellular matrix. B: Cytoskeleton/muscle contraction. C: Cell signaling. D: Biotransformation. E: Cell cycle. F: Immune system/defense. *P ≤ 0.05, asbestos versus air exposure; **P ≤ 0.05, asbestos-exposed OPN+/+ versus OPN−/−.
Pathway Analyses of Asbestos and OPN-Related Gene Expression
Toward understanding the initiation of signaling cascades resulting in altered gene expression by asbestos, we generated a regulatory network based on relationships previously reported in scientific literature by our laboratory and others. This pathway was constructed using additional information from the present study on genes that were differentially up-regulated or down-regulated by asbestos in the lungs of OPN+/+ and OPN−/− mice. In addition, the pathway analysis incorporated information from Bio-Plex results and other key molecules (eg, EGFR, AP-1, NF-κB) previously shown to play an important role in asbestos-mediated signaling. A schematic of this function-based global network is presented in Figure 7. At the initiating pinnacle, asbestos fibers stimulate the production of the proinflammatory cytokine IL-1β through an inflammasome- and TNF-α-regulated pathway, and interact with Areg, a ligand for the EGFR. In turn, IL-1β via OPN and EGFR activates the AP-1 transcription factor. This activation of OPN likely occurs via Cd44 and integrin receptors. This convergence on AP-1 also occurs through MIP-1β. The downstream consequences of AP-1 activation involve the production of cytokines, including IL-6, IL-4, and IL-5, which then control the production of key proteins contributing to inflammation and ECM remodeling. OPN is also a downstream target of AP-1, which suggests the possibility of an autoregulatory mechanism.
Figure 7.
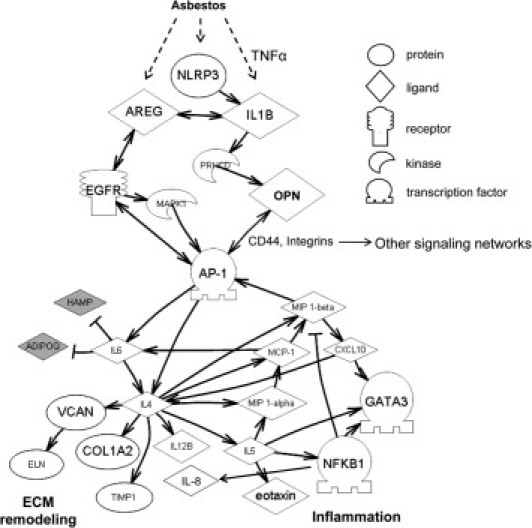
Schematic of plausible signaling networks affected by asbestos, and the role of OPN in regulating genes involved in inflammation and ECM remodeling. Solid arrows represent a positive relationship between two entities, through either expression or activation; double-headed arrows represent positive relationships that occur in both directions. Negative relationships are indicated by T-junction arrows. Shapes indicate protein or molecule type (as in legend). Filled shapes (gray) indicate likely down-regulation by OPN; open shapes (white) indicate up-regulation by OPN. Initial signaling by asbestos occurs through NLRP3-IL-1β-AREG, which activates EGFR and OPN, which then converge on AP-1. OPN acts via CD44 and integrin receptors to trigger the AP-1 transcription factor (and other pathways) that stimulate signaling pathways to regulate downstream genes or proteins related to extracellular matrix (ECM) remodeling and inflammation.
Discussion
To our knowledge, the present study is the first to characterize OPN−/− mice exposed to inhaled mineral fibers, and the results endorse OPN as a significant player in asbestos-induced injury in a model of chrysotile asbestos-induced fibrosis. We have previously shown that inhalation of asbestos for 3 days and 9 days results in epithelial cell proliferation, inflammation, and mucin production that precede the development of fibrotic lesions observed at 30 days to 40 days.20 Asbestos-associated inflammation is characterized by increased eosinophilia and inflammatory cytokines in BALF, including IL-1β, IL-4, IL-5, IL-6, MIP1β, and MCP-1.20,22 Moreover, neutrophilia and macrophage accumulation occurs in lungs of asbestos-exposed mice before the advent of fibrosis, as documented by histology and increased collagen content.26 Microarray analyses on lung tissues of mice exposed to chrysotile asbestos highlighted OPN as a potential target for further study, because it was significantly overexpressed in lungs of asbestos-exposed mice. Furthermore, previous results showed no alterations in OPN expression in mouse lung tissues after airborne exposure to a nonpathogenic particle control (titanium dioxide).20
These data suggest that the reaction of the lung to asbestos fibers is distinct from a generalized particle response, although in the present model we did not examine other types of asbestos or nonasbestos fibers. Increased OPN expression in lung was observed at 3, 9, and 40 days after exposure to asbestos, with levels increasing over time. We also observed the epithelial cell-specific expression of OPN in vivo, using LCM techniques to specifically dissect bronchiolar epithelial cells from mouse lung tissue section. The up-regulation of OPN observed in lung bronchiolar epithelial cells is in line with studies showing enhanced protein expression in lung epithelial cells after exposures to bleomycin or silica.11,27 Others have detected OPN in the respiratory epithelium of patients with idiopathic pulmonary fibrosis.5,11,28 The present results suggest that lung epithelial cells are a major contributor to increased transcription of OPN after inhalation of fibrogenic agents and likely contribute to amounts of OPN protein in BALF and plasma or serum.
In addition to up-regulation of OPN, using LCM we also observed up-regulation of Cd44 antigen specifically in bronchiolar epithelial cells in mice exposed to asbestos for 3 days. Cd44 antigen is a known receptor for OPN which we have also shown to play an important role in mesothelial cell transformation.29 Other receptors for OPN, including members of the integrin family (αVβIII, αVβV, and αVβI) were not significantly modulated by asbestos. From these data, we conclude that bronchiolar epithelial cells express OPN in response to asbestos and have the capacity to interact with secreted OPN protein through Cd44. Further studies are required to dissect the downstream consequences of OPN-Cd44 interactions specifically in bronchiolar epithelial cells and how they relate to cellular responses of inhaled asbestos in the lung.
Using OPN−/− mice to further investigate the functional role of OPN in asbestos injury, we showed that the loss of OPN reduces asbestos-induced cell injury and inflammation in the lung by reducing lactate dehydrogenase levels, eosinophilia, and inflammatory cytokines in BALF. These observations highlight a novel role of OPN in asbestos-induced pulmonary injury whereby recruitment of eosinophils, possibly mediated by cytokines such as IL-5 and eotaxin, are controlled in part by OPN and contribute to lung fibrogenesis. For example, in a bleomycin model, IL-5 overexpression increased eosinophilia and fibrosis, but this response was ameliorated in IL-5−/− mice.30 Another study showed that IL-4 recruits eosinophils via up-regulation of eotaxin.31 These results support trends observed here, in which the decreased IL-5 levels and significant reduction of IL-4 and eotaxin observed in OPN−/− mice were accompanied by decreased eosinophilia in BALF. We observed no significant alterations in the presence of other immune cells (including macrophages, lymphocytes, and neutrophils) in BALF between OPN+/+ and OPN−/− mice, suggesting that cytokines and other molecules modulated by OPN are central to recruitment of eosinophils in response to asbestos.
We also noted decreased production of mucin in distal bronchioles in OPN−/− mice exposed to asbestos. Other studies have shown the production of mucins is controlled in part by a number of cytokines, such as IL-1β32,33 and IL-4.34,35 The repression of these cytokines in OPN−/− mice exposed to asbestos likely contributes to decreases in mucin expression. It is still unclear precisely what role mucin plays in asbestos-induced fibrogenesis, and the interplay between the roles of mucin in fiber clearance and modulation of epithelial cell responses to asbestos requires further investigation.
A number of profibrotic cytokines and chemokines, including IL-1β, IL-4, IL-5, IL-6, MIP1β, and MCP-1, are increased in BALF after inhalation of asbestos.20,22 Although it has been shown that OPN modulates IL-5, IL-4, and IL-12 subunit p40 in other experimental models of fibrosis,30,36,37 we are unaware of published studies showing modulation of IL-1β, IL-6, MIP1α, MIP1β, eotaxin, or MCP-1 by OPN. These cytokines play a role in fibrosis,38–46 and our data suggest that their presence in BALF is in part controlled by OPN. Exactly how OPN modulates expression and elaboration of these cytokines after exposures to asbestos is unclear, but significant for the identification of new targets of inhaled fibrogenic agents.
In addition to altered immune profiles in BALF, gene expression signatures in lung tissues identified a number of targets affected by the loss of OPN in our inhalation model of fibrosis. A number of ECM-related genes induced by asbestos in OPN+/+ mice were depressed in response to asbestos in OPN−/− mice, including type I collagens, Eln, Timp1, Tnc, and Vcan, some of which were shown to be expressed specifically by epithelial cells in LCM-gene array studies (Figure 1B). Some of these genes (type I collagens and Timp1) are induced in the lung by other fibrotic agents, such as bleomycin, and are reduced after loss of OPN production.11 Nonetheless, the present findings suggest that Eln, Tnc, Vcan, and Adamts2 are novel OPN-regulated target genes in response to asbestos fibers.
Numerous genes related to the immune response also were repressed after asbestos exposures in OPN−/− mice, compared with the response in wild-type OPN+/+ mice. These included Cxcl10, Areg, and Tsp1. Decreased Cxcl10 expression is consistent with the dampened inflammatory responses observed in OPN−/− mice. Notably, Areg, a member of the Egf family, activates signaling via the EGFR. This is a major pathway affected by asbestos,47–49 and recent investigations suggest that it plays a role in fibroblast proliferation and fibrosis.50,51
We also noted changes in expression in whole lung tissues of other cytokines (Ccl3, Ccl6, Ccl7, Ccl8, Ccl9, Ccl11, Ccl12, Ccl17, Ccl22, Ccl24, Cxcl12, and Cxcl17) after asbestos exposure (data not shown). Of these, expression of Ccl24, Ccl3, and Ccl9 were significantly lower in asbestos-exposed OPN−/− mice, compared with OPN+/+ mice. These results suggest that OPN affects lung tissue levels of chemokines and cytokines at the transcriptional level. Identifying the source of these molecules and quantifying their protein levels was beyond the scope of the present work, but is relevant for future analysis. In our studies, asbestos exposures down-regulated Plunc expression in OPN+/+ mice but not in OPN−/− mice. The specific function of Plunc is unknown, but a recent study showed that this mRNA is overexpressed in patients with idiopathic pulmonary fibrosis, is localized to secretory/goblet bronchial columnar cells, and is secreted into mucus.52
We observed similar patterns of decreased expression of genes related to cytoskeletal and muscle homeostasis in OPN+/+ asbestos-exposed mice, including Smpx, Tnnt2, Tnni3, Myh6, Myl4, Tcap, Myoz2, Sln, Pln, and Myoz1. Their proteins are involved mainly in skeletal muscle hypertrophy and contraction, and could modulate the function of myofibroblasts in the lung associated with the development of pulmonary fibrosis. Of note, the repressed expression of these genes by asbestos was not observed in asbestos-exposed OPN−/− mice, a novel finding that indicates a regulatory connection between OPN and muscle-related genes in the lung. Further studies are required to assess the localization and functional roles of these gene products in lung repair and pulmonary fibrosis.
To gain an understanding of how OPN influences the expression of genes involved in asbestos-induced injury, inflammation, and subsequent fibrosis, we generated a regulatory network based on new results here combined with previously reported data describing cell interactions and signaling pathways stimulated or inhibited by asbestos. This global analysis revealed a complex interplay between several signaling pathways and asbestos-related responses (Figure 7), suggesting that OPN modulates inflammatory and ECM-related genes as a result of crosstalk between asbestos-fiber stimulated IL-1β-PKCδ and AREG-EGFR-MAPK1 pathways. These studies are in line with our recent work showing that asbestos fibers are sensed by the NALP3 inflammasome, which subsequently activates IL-1β and contributes to asbestos and silica-induced inflammatory responses.53 Although it has been reported that OPN expression is up-regulated by IL-1β in lung fibroblasts in vitro,19 we are unaware of any other prior studies linking IL-1β expression to regulation of OPN levels. It is also possible that stimulation of IL-1β is a result of upstream TNF-α production.
We also found convergence on the AP-1 transcription factor, by activation of IL-1β-OPN and EGFR-MAPK1 (Figure 7). Previous studies by our research group and by others have identified the significant involvement of AP-1 in many asbestos-induced responses, including proliferation, apoptosis, transformation, and cell differentiation.54 Activation of AP-1 by OPN likely occurs through activation of Cd44 and integrin receptors. Once AP-1 is activated, downstream genes involved in inflammation and ECM remodeling are modulated. Increased IL-4 is a key cytokine that controls the expression of many of the targets presented in our regulatory network (Figure 7), such as Col1a2, Timp1, Vcan, MIP1B, MCP-1, MIP1a, IL-8, and IL-5. These molecules are involved in general inflammation and eosinophil recruitment and ECM homeostasis. OPN activates AP-1 in melanoma cells,55 but it is also a downstream target of AP-1 in smooth muscle cells,55,56 suggesting that a positive feedback loop exists for regulation of gene expression by OPN. Of all of the relationships observed, only a few of the downstream targets (collagens, Timp1, and AP-1) in our regulatory network (Figure 7) have been reported previously to be modulated by OPN.11,54 We realize the complex nature of the signaling networks discussed, given that Cd44 and integrin receptors have been shown to activate other networks (notably, AKT); in our schematic (Figure 7), these pathways are represented as “other signaling networks.” We chose to concentrate our efforts on AP-1 pathways, because this has been a long-term focus of our asbestos research efforts over the years.57
In summary, we have presented novel findings on many functional effects of OPN after inhalation of asbestos fibers. This work shows that the lung epithelial cell is a major source of OPN up-regulation at the transcriptional level. Moreover, OPN plays a multifactorial role in immune cell recruitment and remodeling after exposure to fibrogenic asbestos fibers. Our novel, function-based genomic approach to identifying upstream signaling pathways and downstream events that occur after inhalation of asbestos by OPN wild-type versus OPN null mice revealed patterns of OPN-linked gene expression and key molecules that can be targeted in prevention and therapy of fibroproliferative lung diseases.
Acknowledgments
We thank Stacie Beuschel (University of Vermont), Jedd Hillegass, Ph.D. (University of Vermont), and David Hemenway, Ph.D. (University of Vermont), for technical assistance for the inhalation exposures. We also thank Pierre Revalier, Ph.D. (University of South Carolina), and USC INBRE staff for microarray analysis and Kevin Carnivale, Ph.D. (University of South Carolina School of Medicine), for helpful discussions regarding lung pathology.
Footnotes
Supported by NIH grants P01 HL67004 from the National Heart, Lung, and Blood Institute, T32 07122 (B.T.M.) and K22ES014742 (T.S.A.) from the National Institute of Environmental Health Sciences, and K01 CA104159 from the National Cancer Institute (M.E.R.).
References
- 1.Mossman B.T., Bignon J., Corn M., Seaton A., Gee J.B. Asbestos: scientific developments and implications for public policy. Science. 1990;247:294–301. doi: 10.1126/science.2153315. [DOI] [PubMed] [Google Scholar]
- 2.Mossman B.T., Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 3.O'Reilly K.M., McLaughlin A.M., Beckett W.S., Sime P.J. Asbestos-related lung disease. Am Fam Physician. 2007;75:683–688. [PubMed] [Google Scholar]
- 4.O'Regan A. The role of osteopontin in lung disease. Cytokine Growth Factor Rev. 2003;14:479–488. doi: 10.1016/s1359-6101(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 5.Pardo A., Gibson K., Cisneros J., Richards T.J., Yang Y., Becerril C., Yousem S., Herrera I., Ruiz V., Selman M., Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005;2:e251. doi: 10.1371/journal.pmed.0020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang Y.S., Kim H.J., Chang J., Ahn C.M., Kim S.K., Kim S.K. Elevated circulating level of osteopontin is associated with advanced disease state of non-small cell lung cancer. Lung Cancer. 2007;57:373–380. doi: 10.1016/j.lungcan.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Creaney J., Yeoman D., Demelker Y., Segal A., Musk A.W., Skates S.J., Robinson B.W. Comparison of osteopontin, megakaryocyte potentiating factor, and mesothelin proteins as markers in the serum of patients with malignant mesothelioma. J Thorac Oncol. 2008;3:851–857. doi: 10.1097/JTO.0b013e318180477b. [DOI] [PubMed] [Google Scholar]
- 8.Grigoriu B.D., Scherpereel A., Devos P., Chahine B., Letourneux M., Lebailly P., Grégoire M., Porte H., Copin M.C., Lassalle P. Utility of osteopontin and serum mesothelin in malignant pleural mesothelioma diagnosis and prognosis assessment. Clin Cancer Res. 2007;13:2928–2935. doi: 10.1158/1078-0432.CCR-06-2144. [DOI] [PubMed] [Google Scholar]
- 9.Pass H.I., Lott D., Lonardo F., Harbut M., Liu Z., Tang N., Carbone M., Webb C., Wali A. Asbestos exposure, pleural mesothelioma, and serum osteopontin levels. N Engl J Med. 2005;353:1564–1573. doi: 10.1056/NEJMoa051185. [DOI] [PubMed] [Google Scholar]
- 10.Hansel N.N., Cheadle C., Diette G.B., Wright J., Thompson K.M., Barnes K.C., Georas S.N. Analysis of CD4+ T-cell gene expression in allergic subjects using two different microarray platforms. Allergy. 2008;63:366–369. doi: 10.1111/j.1398-9995.2007.01540.x. [DOI] [PubMed] [Google Scholar]
- 11.Berman J.S., Serlin D., Li X., Whitley G., Hayes J., Rishikof D.C., Ricupero D.A., Liaw L., Goetschkes M., O'Regan A.W. Altered bleomycin-induced lung fibrosis in osteopontin-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1311–L1318. doi: 10.1152/ajplung.00394.2003. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi F., Takahashi K., Okazaki T., Maeda K., Ienaga H., Maeda M., Kon S., Uede T., Fukuchi Y. Role of osteopontin in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2001;24:264–271. doi: 10.1165/ajrcmb.24.3.4293. [DOI] [PubMed] [Google Scholar]
- 13.Woodruff P.G., Koth L.L., Yang Y.H., Rodriguez M.W., Favoreto S., Dolganov G.M., Paquet A.C., Erle D.J. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am J Respir Crit Care Med. 2005;172:1383–1392. doi: 10.1164/rccm.200505-686OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mangum J., Bermudez E., Sar M., Everitt J. Osteopontin expression in particle-induced lung disease. Exp Lung Res. 2004;30:585–598. doi: 10.1080/01902140490476346. [DOI] [PubMed] [Google Scholar]
- 15.Cristaudo A., Foddis R., Buselli R., Gattini V., Di Palma N., Guglielmi G. Medical surveillance of workers previously exposed to asbestos. Med Lav. 2006;97:475–481. [PubMed] [Google Scholar]
- 16.Prasse A., Stahl M., Schulz G., Kayser G., Wang L., Ask K., Yalcintepe J., Kirschbaum A., Bargagli E., Zissel G., Kolb M., Muller-Quernheim J., Weiss J.M., Renkl A.C. Essential role of osteopontin in smoking-related interstitial lung diseases. Am J Pathol. 2009;174:1683–1691. doi: 10.2353/ajpath.2009.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown L.F., Berse B., Van de Water L., Papadopoulos-Sergiou A., Perruzzi C.A., Manseau E.J., Dvorak H.F., Senger D.R. Expression and distribution of osteopontin in human tissues: widespread association with luminal epithelial surfaces. Mol Biol Cell. 1992;3:1169–1180. doi: 10.1091/mbc.3.10.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Z., Lin D., Yuan J., Xiao T., Zhang H., Sun W., Han N., Ma Y., Di X., Gao M., Ma J., Zhang J., Cheng S., Gao Y. Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin Cancer Res. 2005;11:4646–4652. doi: 10.1158/1078-0432.CCR-04-2013. [DOI] [PubMed] [Google Scholar]
- 19.Serlin D.M., Kuang P.P., Subramanian M., O'Regan A., Li X., Berman J.S., Goldstein R.H. Interleukin-1beta induces osteopontin expression in pulmonary fibroblasts. J Cell Biochem. 2006;97:519–529. doi: 10.1002/jcb.20661. [DOI] [PubMed] [Google Scholar]
- 20.Sabo-Attwood T., Ramos-Nino M., Bond J., Butnor K.J., Heintz N., Gruber A.D., Steele C., Taatjes D.J., Vacek P., Mossman B.T. Gene expression profiles reveal increased mClca3 (Gob5) expression and mucin production in a murine model of asbestos-induced fibrogenesis. Am J Pathol. 2005;167:1243–1256. doi: 10.1016/S0002-9440(10)61212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manning C.B., Mossman B.T., Taatjes D.J. Analysis of asbestos-induced gene expression changes in bronchiolar epithelial cells using laser capture microdissection and quantitative reverse transcriptase-polymerase chain reaction. Methods Mol Biol. 2006;319:231–236. doi: 10.1007/978-1-59259-993-6_11. [DOI] [PubMed] [Google Scholar]
- 22.Haegens A., Barrett T.F., Gell J., Shukla A., Macpherson M., Vacek P., Poynter M.E., Butnor K.J., Janssen-Heininger Y.M., Steele C., Mossman B.T. Airway epithelial NF-kappaB activation modulates asbestos-induced inflammation and mucin production in vivo. J Immunol. 2007;178:1800–1808. doi: 10.4049/jimmunol.178.3.1800. [DOI] [PubMed] [Google Scholar]
- 23.Fukagawa N.K., Li M., Sabo-Attwood T., Timblin C.R., Butnor K.J., Gagne J., Steele C., Taatjes D.J., Huber S., Mossman B.T. Inhaled asbestos exacerbates atherosclerosis in apolipoprotein E-deficient mice via CD4+ T cells. Environ Health Perspect. 2008;116:1218–1225. doi: 10.1289/ehp.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Z., Irizarry R.A., Gentleman R., Martinez-Murillo F., Spencer F.A. A model based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;468:909–917. [Google Scholar]
- 25.Argraves G.L., Jani S., Barth J.L., Argraves W.S. ArrayQuest: a Web resource for the analysis of DNA microarray data. BMC Bioinformatics. 2005;6:287. doi: 10.1186/1471-2105-6-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robledo R.F., Buder-Hoffmann S.A., Cummins A.B., Walsh E.S., Taatjes D.J., Mossman B.T. Increased phosphorylated extracellular signal-regulated kinase immunoreactivity associated with proliferative and morphologic lung alterations after chrysotile asbestos inhalation in mice. Am J Pathol. 2000;156:1307–1316. doi: 10.1016/S0002-9440(10)65001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohan M., Bader R., Puxeddu I., Levi-Schaffer F., Breuer R., Berkman N. Enhanced osteopontin expression in a murine model of allergen-induced airway remodelling. Clin Exp Allergy. 2007;37:1444–1454. doi: 10.1111/j.1365-2222.2007.02801.x. [DOI] [PubMed] [Google Scholar]
- 28.O'Regan A., Berman J.S. Osteopontin: a key cytokine in cell-mediated and granulomatous inflammation. Int J Exp Pathol. 2000;81:373–390. doi: 10.1046/j.1365-2613.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos-Nino M.E., Blumen S.R., Pass H., Mossman B.T. Fra-1 governs cell migration via modulation of CD44 expression in human mesotheliomas. Mol Cancer. 2007;6:81. doi: 10.1186/1476-4598-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huaux F., Liu T., McGarry B., Ullenbruch M., Xing Z., Phan S.H. Eosinophils and T lymphocytes possess distinct roles in bleomycin-induced lung injury and fibrosis. J Immunol. 2003;171:5470–5481. doi: 10.4049/jimmunol.171.10.5470. [DOI] [PubMed] [Google Scholar]
- 31.Mochizuki M., Bartels J., Mallet A.I., Christophers E., Schroder J.M. IL-4 induces eotaxin: a possible mechanism of selective eosinophil recruitment in helminth infection and atopy. J Immunol. 1998;160:60–68. [PubMed] [Google Scholar]
- 32.Gray T., Coakley R., Hirsh A., Thornton D., Kirkham S., Koo J.S., Burch L., Boucher R., Nettesheim P. Regulation of MUC5AC mucin secretion and airway surface liquid metabolism by IL-1beta in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L320–L330. doi: 10.1152/ajplung.00440.2002. [DOI] [PubMed] [Google Scholar]
- 33.Gray T., Nettesheim P., Loftin C., Koo J.S., Bonner J., Peddada S., Langenbach R. Interleukin-1beta-induced mucin production in human airway epithelium is mediated by cyclooxygenase-2, prostaglandin E2 receptors, and cyclic AMP-protein kinase A signaling. Mol Pharmacol. 2004;66:337–346. doi: 10.1124/mol.66.2.337. [DOI] [PubMed] [Google Scholar]
- 34.Cohn L., Homer R.J., Marinov A., Rankin J., Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. J Exp Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dabbagh K., Takeyama K., Lee H.M., Ueki I.F., Lausier J.A., Nadel J.A. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. 1999;162:6233–6237. [PubMed] [Google Scholar]
- 36.Huaux F., Arras M., Tomasi D., Barbarin V., Delos M., Coutelier J.P., Vink A., Phan S.H., Renauld J.C., Lison D. A profibrotic function of IL-12p40 in experimental pulmonary fibrosis. J Immunol. 2002;169:2653–2661. doi: 10.4049/jimmunol.169.5.2653. [DOI] [PubMed] [Google Scholar]
- 37.Huaux F., Liu T., McGarry B., Ullenbruch M., Phan S.H. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170:2083–2092. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 38.Emad A., Emad V. Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology. 2007;240:60–69. doi: 10.1016/j.tox.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 39.Gasse P., Mary C., Guenon I., Noulin N., Charron S., Schnyder-Candrian S., Schnyder B., Akira S., Quesniaux V.F., Lagente V., Ryffel B., Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalayarasan S., Sriram N., Sudhandiran G. Diallyl sulfide attenuates bleomycin-induced pulmonary fibrosis: critical role of iNOS, NF-kappaB, TNF-alpha and IL-1beta. Life Sci. 2008;82:1142–1153. doi: 10.1016/j.lfs.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Mastruzzo C., Crimi N., Vancheri C. Role of oxidative stress in pulmonary fibrosis. Monaldi Arch Chest Dis. 2002;57:173–176. [PubMed] [Google Scholar]
- 42.Saito F., Tasaka S., Inoue K., Miyamoto K., Nakano Y., Ogawa Y., Yamada W., Shiraishi Y., Hasegawa N., Fujishima S., Takano H., Ishizaka A. Role of interleukin-6 in bleomycin-induced lung inflammatory changes in mice. Am J Respir Cell Mol Biol. 2008;38:566–571. doi: 10.1165/rcmb.2007-0299OC. [DOI] [PubMed] [Google Scholar]
- 43.Wynn T.A. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang K., Gharaee-Kermani M., Jones M.L., Warren J.S., Phan S.H. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J Immunol. 1994;153:4733–4741. [PubMed] [Google Scholar]
- 45.Zhang K., Phan S.H. Cytokines and pulmonary fibrosis. Biol Signals. 1996;5:232–239. doi: 10.1159/000109195. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Z., Homer R.J., Wang Z., Chen Q., Geba G.P., Wang J., Zhang Y., Elias J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faux S.P., Houghton C.E., Hubbard A., Patrick G. Increased expression of epidermal growth factor receptor in rat pleural mesothelial cells correlates with carcinogenicity of mineral fibres. Carcinogenesis. 2000;21:2275–2280. doi: 10.1093/carcin/21.12.2275. [DOI] [PubMed] [Google Scholar]
- 48.Manning C.B., Cummins A.B., Jung M.W., Berlanger I., Timblin C.R., Palmer C., Taatjes D.J., Hemenway D., Vacek P., Mossman B.T. A mutant epidermal growth factor receptor targeted to lung epithelium inhibits asbestos-induced proliferation and proto-oncogene expression. Cancer Res. 2002;62:4169–4175. [PubMed] [Google Scholar]
- 49.Zanella C.L., Timblin C.R., Cummins A., Jung M., Goldberg J., Raabe R., Tritton T.R., Mossman B.T. Asbestos-induced phosphorylation of epidermal growth factor receptor is linked to c-fos and apoptosis. Am J Physiol. 1999;277:L684–L693. doi: 10.1152/ajplung.1999.277.4.L684. [DOI] [PubMed] [Google Scholar]
- 50.Hardie W.D., Davidson C., Ikegami M., Leikauf G.D., Le Cras T.D., Prestridge A., Whitsett J.A., Korfhagen T.R. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1217–L1225. doi: 10.1152/ajplung.00020.2008. [DOI] [PubMed] [Google Scholar]
- 51.Ishii Y., Fujimoto S., Fukuda T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am J Respir Crit Care Med. 2006;174:550–556. doi: 10.1164/rccm.200509-1534OC. [DOI] [PubMed] [Google Scholar]
- 52.Boon K., Bailey N.W., Yang J., Steel M.P., Groshong S., Kervitsky D., Brown K.K., Schwarz M.I., Schwartz D.A. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF) PLoS One. 2009;4:e5134. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dostert C., Petrilli V., Van Bruggen R., Steele C., Mossman B.T., Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramos-Nino M.E., Haegens A., Shukla A., Mossman B.T. Role of mitogen-activated protein kinases (MAPK) in cell injury and proliferation by environmental particulates. Mol Cell Biochem. 2002;234–235:111–118. [PubMed] [Google Scholar]
- 55.Rangaswami H., Kundu G.C. Osteopontin stimulates melanoma growth and lung metastasis through NIK/MEKK1-dependent MMP-9 activation pathways. Oncol Rep. 2007;18:909–915. [PubMed] [Google Scholar]
- 56.Bidder M., Shao J.S., Charlton-Kachigian N., Loewy A.P., Semenkovich C.F., Towler D.A. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J Biol Chem. 2002;277:44485–44496. doi: 10.1074/jbc.M206235200. [DOI] [PubMed] [Google Scholar]
- 57.Heintz N.H., Janssen-Heininger Y.M., Mossman B.T. Asbestos, lung cancers, and mesotheliomas: from molecular approaches to targeting tumor survival pathways. Am J Respir Cell Mol Biol. 2010;42:133–139. doi: 10.1165/rcmb.2009-0206TR. [DOI] [PMC free article] [PubMed] [Google Scholar]