

Abstract
Both the immune system and the epidermis likely have an important role in the pathogenesis of atopic dermatitis (AD). The objective of the present study was to develop a human skin equivalent model exhibiting morphologic and molecular characteristics of AD in a controlled manner. Skin equivalents generated from normal adult human keratinocytes were stimulated with type 2 T-helper cell (Th2) cytokines IL-4 and IL-13, and morphologic features and gene expression of the epidermis were studied. Th2 cytokines induced intercellular edema similar to spongiotic changes observed in lesional AD as assessed at histopathologic analysis and electron microscopy. Furthermore, genes known to be specifically expressed in epidermis of patients with AD such as CAII and NELL2 were induced. In contrast, expression of psoriasis-associated genes such as elafin and hBD2 was not changed. Th2 cytokines caused DNA fragmentation in the keratinocytes, which could be inhibited by the caspase inhibitor Z-VAD, which suggests that apoptosis was induced. In addition, up-regulation of the death receptor Fas was observed in keratinocytes after Th2 cytokine stimulation. IL-4 and IL-13 induced phosphorylation of the signaling molecule STAT6. It was concluded that the skin equivalent model described herein may be useful in investigation of the epidermal aspects of AD and for study of drugs that act at the level of keratinocyte biology.
Atopic dermatitis (AD) is a highly prevalent inflammatory skin disease caused by disturbed skin immune function.1 Although the pathologic features of AD were initially attributed to an imbalance in the adaptive immune system, several studies suggest important abnormalities of the innate immune system2,3 and skin barrier function.4 Recent studies have identified null mutations in the filaggrin gene as a major genetic risk factor for AD.5,6 Furthermore, dysregulation of several genes encoding proteases or antiproteases involved in the process of cornification, innate immune receptors, chemotactic factors, and antimicrobial proteins has been demonstrated in lesional AD skin.1,7 Imbalances in epidermal innate immunity may render AD skin more susceptible for bacterial and viral infections and may explain the higher rate of skin infections in AD skin compared with psoriasis.2,8
Depending on the stage, histologic examination of lesional AD skin demonstrated intraepidermal edema (spongiosis) and an inflammatory dermal infiltrate dominated by type 2 T-helper cells (Th2) producing high levels of cytokines such as IL-4 and IL-13.9 Gene expression profiling has identified increased expression of specific genes including carbonic anhydrase II (CAII) and neural epidermal growth factor–like 2 (NELL2)8,10 in lesional AD skin as compared with psoriasis. Subsequent confirmation of these results using quantitative real-time PCR (qPCR) demonstrated that gene expression of CAII and NELL2 was increased in AD compared with both psoriasis and healthy skin.11 CAII is a member of the family of metalloenzymes and is involved in maintenance of cellular pH, water transport, and ion homeostasis.12 NELL2 is primarily expressed in brain tissue,13 in which it promotes survival of hippocampal and cortical neurons.14 Expression of both CAII and NELL2 is induced in submerged cultured keratinocytes by the Th2 cytokines IL-4 and IL-13.15 The pathophysiologic role of both of these proteins in AD has not been established, although the role of CAII in water transport and pH regulation makes it a good candidate protein for mediation of spongiotic changes and pH abnormalities in AD.
A number of mechanistically distinct anti-AD therapies are available including UVB, corticosteroids, calcineurin inhibitors, and tar. Most of these drugs target the adaptive immune system rather than the epidermal keratinocytes or the interaction between the two. Whereas psoriasis therapy has advanced considerably as a result of development of biological agents, there is room for improvement in AD therapy insofar as efficacy and adverse effects. To study the pathologic features and pharmacologic intervention of AD, there is a need for skin models that mimic AD. Because use of in vivo AD models is limited by their low quality and the questionable translation of the disease process in mice to that in humans,16 in vitro three-dimensional tissue-engineered human skin equivalents may be useful for biological and pharmacologic studies. Development and validation of a human psoriatic skin equivalent model by addition of a selected set of proinflammatory cytokines to human skin equivalents has been recently demonstrated.17 A model for eczema was described by Engelhart et al,18 who added lymphocytes to tissue-engineered skin. The development of relevant high-content in vitro skin models that mimic AD would be of great value for pathologic studies and evaluation of novel therapeutic agents.
The present study describes a skin equivalent model for AD by addition of relevant proinflammatory cytokines to healthy human skin equivalents. This model enables controlled induction of AD-associated features such as structure and gene expression.
Materials and Methods
Generation of Human Skin Equivalents
Human adult abdominal keratinocytes were isolated and cultured as described previously.17 De-epidermized dermis was generated as reported previously,17 and 8-mm tissue samples were obtained using a biopsy punch. De-epidermized dermis was placed in transwells in a 24-well culture plate and seeded with 105 keratinocytes. After culturing the constructs submerged for 3 days in medium containing 5% serum, consisting of two parts Dulbecco's modified Eagle's medium and one part Ham's F12 medium (both from Life Technologies, Inc., Grand Island, NY) supplemented with 5% calf serum (Hyclone Laboratories, Inc., Logan, UT), 4 mmol/L l-glutamine and 50 IU/mL penicillin or streptomycin (both from Life Technologies, Inc.), 24.3 μg/mL adenine (Calbiochem, Los Angeles, CA), 1 μmol/L hydrocortisone (Merck KGaA, Darmstadt, Germany), and 50 μg/mL ascorbic acid, 0.2 μmol/L insulin, 1.36 ng/mL triiodothyronine, and 10−10 mmol/L cholera toxin (all from Sigma Aldrich, St. Louis, MO), the constructs were cultured at the air-liquid interface for 10 days in medium without serum, consisting of two parts Dulbecco's modified Eagle's medium and one part Ham's F12 medium (both from Life Technologies, Inc.) supplemented with 4 mmol/L l-glutamine and 50 IU/mL penicillin or streptomycin (both from Life Technologies, Inc.), 24.3 μg/mL adenine (Calbiochem), 1 mg/mL l-serine and 2 μg/mL l-carnitine (both from Sigma Aldrich), bovine serum albumin lipid mix (25 μmol/L palmitic acid, 7 μmol/L arachidonic acid, 15 μmol/L linoleic acid, and 0.4 μg/mL vitamin E; all from Sigma Aldrich), 1 μmol/L hydrocortisone (Merck KGaA), and 50 μg/mL ascorbic acid, 0.1 μmol/L insulin, 1.36 ng/mL triiodothyronine, 10−10 mmol/L cholera toxin, 5 ng/mL keratinocyte growth factor, and 2 ng/mL epidermal growth factor (all from Sigma Aldrich). AD skin equivalents were obtained by incubating healthy skin equivalents for the last 3 days of the air-liquid interface culture with indicated concentrations of IL-4 and IL-13 (PeproTech EC Ltd., London, England). For comparison, skin equivalents were stimulated with a mixture of 10 ng/mL IL-1α and 5 ng/mL tumor necrosis factor-α (TNF-α; both from PeproTech EC Ltd.), and 2 × 108 IU/mL IL-6 (Gentaur, Brussels, Belgium), with 30 ng/mL IL-17 or IL-22 (PeproTech EC Ltd.). To test the effect of anti-AD drugs, 10 μmol/L cyclosporine A (CsA), tacrolimus, dexamethasone, or ethoxzolamide was added to the cytokine mixtures for the last 3 days of the air-liquid interface cultures. Inhibition of apoptosis was studied using 40 μmol/L Z-VAD (Bachum AG, Bubendorf, Switzerland).
Histologic Analysis and IHC
Human skin equivalents were fixed in buffered 4% formalin for 4 hours and embedded in paraffin. Sections 6-μm thick were stained with H&E (Sigma Aldrich). Alternatively, sections were blocked for 15 minutes with PBS with 20% normal rabbit (for CAII and hBD2) or normal goat serum (for elafin), and subsequently incubated with anti-CAII (R&D Systems, Inc., Minneapolis, MN) in a 1:1000 dilution, anti-hBD2 (PeproTech EC Ltd.) in a 1:100 dilution, or anti-elafin (own laboratory) in a 1:500 dilution for 1 hour at room temperature. After washes in PBS, sections were incubated for 1 hour with a secondary antibody: biotinylated rabbit anti-sheep, biotinylated rabbit anti-goat, or biotinylated goat anti-rabbit in PBS containing 1% bovine serum albumin (Vector Laboratories, Inc., Burlingame, CA). After 30 minutes of incubation with avidin-biotin complex (Vector Laboratories, Inc.), sections were treated with 3-amino-9-ethylcarbazole (Calbiochem) for 10 minutes. Nuclei were stained with Mayer's hematoxylin solution (Sigma Aldrich), and coated using glycerol gelatin (Sigma Aldrich).
DNA fragmentation was detected using terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL). Sections 6 μm thick were incubated with 20 μg/mL proteinase K (Boehringer Mannheim GmbH, Mannheim, Germany) for 15 minutes, followed by incubation with a mixture of 200 to 400 U/mL terminal deoxynucleotidyl transferase enzyme (Pharmacia Biotech Inc., Piscataway, NJ) and 20 nmol/mL biotin-16-deoxyuridine triphosphate (Boehringer Mannheim GmbH) in terminal deoxynucleotidyl transferase buffer [0.5 mmol/L cacodylate (Fluka Chemie GmbH, Buchs, Switzerland)], 1 mmol/L cobalt(II) chloride, 0.5 mmol/L dithiothreitol, 0.05% Boseral Fraktion V, and 0.15 mmol/L NaCl). After incubation at 37°C for 1 hour, the reaction was stopped by rinsing in transformation buffer (300 mmol/L NaCl and 30 mmol/L 3-sodium citrate-2-hydrate). The sections were incubated for 30 minutes with the avidin-biotin complex, followed by 3-amino-9-ethylcarbazole at 37°C for 10 minutes . Nuclei were (weakly) stained with Mayer's hematoxylin solution, and coated using glycerol gelatin as described above.
Electron Microscopic Analysis
Samples were fixed at 4°C overnight with 2.5% glutaraldehyde in cacodylate buffer, and postfixed for 60 minutes at room temperature with 1% OsO4 and 1% K[Fe(CN)6]*H2O in Pallade buffer. After rinsing with cacodylate buffer and dehydration with several grades of ethanol, the samples were incubated with mixtures of ethanol absolute and increasing concentrations of Epon 812 (Agar Scientific Ltd., Stansted, Essex, England) until 100% Epon was reached. The constructs were removed from the transwell insert with a small diamond knife. Transverse sections approximately 1 mm thick were cut manually with a sharp razor blade and embedded in Epon. Semithin sections 1 μm thick were cut using a Leica Ultracut (Leica Microsystems GmbH, Wetzlar, Germany), stained with toluidine blue, and mounted on a glass slide. After examination and selection, ultrathin sections were prepared and mounted on copper grids, followed by staining with uranyl acetate and lead citrate. Photographs were obtained using an electron microscope (JEOL 1200 EX/II; JEOL, Tokyo, Japan) at 60 kV.
qPCR
Epidermis was separated from the skin equivalents via treatment with dispase (Roche Diagnostics GmbH, Mannheim, Germany) for 2 hours at 4°C, and total RNA was isolated from the epidermis using Trizol reagent (Life Technologies, Inc.). Generation of first-strand cDNA was performed as described previously.19 The reverse transcription reaction products were used for qPCR amplification, which was performed using the MyiQ Single-Color Real-Time Detection System for quantification with SYBR Green and melting curve analysis (Bio-Rad Laboratories, Inc., Hercules, CA). Primers were obtained from Biolegio BV (Nijmegen, the Netherlands), and sequences are given in Table 1. DNA was PCR-amplified using iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc.) under the following conditions: 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C, with data collected in the last 30 seconds. All primer concentrations were 300 nmol/L in a total reaction volume of 25 μL. The amount of each mRNA was normalized to the amount of mRNA of the human ribosomal phosphoprotein P0 (RPLP0) reference gene in the same sample. The RPLP0 gene is, at least for human skin, more suitable than commonly used genes such as actin or glyceraldehyde-3-phosphate dehydrogenase.20 Primers for qPCR were accepted only if their mean (SD) efficiency was 100% (10%) (Table 1). Relative mRNA expression levels of all examined genes were determined using the 2-ΔΔCT method.21
Table 1.
Primer Sequences of All Genes Examined Using qPCR
| Gene symbol | Description⁎ | Forward primer | Reverse primer | Primer efficiency, % |
|---|---|---|---|---|
| CA2 | Carbonic anhydrase II (CAII) | 5′-AACAATGGTCATGCTTTCAACG-3′ | 5′-TGTCCATCAAGTGAACCCCAG-3′ | 101 |
| NELL2 | Nellike2 (NELL2) | 5′-TAAGGGTATAATGCAAGATGTCCAATT-3′ | 5′-AGATCTGGGCACTGAGCAATAAA-3′ | 104 |
| DEFB4 | hBD-2, β-defensin-2 (hBD2) | 5′-GATGCCTCTTCCAGGTGTTTTT-3′ | 5′-GGATGACATATGGCTCCACTCTT-3′ | 99 |
| PI3 | SKALP, elafin (elafin) | 5′-CATGAGGGCCAGCAGCTT-3′ | 5′-TTTAACAGGAACTCCCGTGACA-3′ | 101 |
| IL8 | IL-8, CXCL8 (CXCL8) | 5′-CTTGGCAGCCTTCCTGATTT-3′ | 5′-TTCTTTAGCACTCCTTGGCAAAA-3′ | 105 |
| CXCL10 | IP-10, chemokine (C-X-C motif) ligand 10 (CXCL10) | 5′-TTCCTGCAAGCCAATTTTGTC-3′ | 5′-TCTTCTCACCCTTCTTTTTCATTGT-3′ | 100 |
| Fas | Fas, TNF receptor superfamily, member 6, CD95, APO-1 (Fas) | 5′-TGCAGAAGATGTAGATTGTGTGATGA-3′ | 5′-GGGTCCGGGTGCAGTTTATT-3′ | 104 |
| RPLP0 | Ribosomal phosphoprotein P0 (RPLP0) | 5′-CACCATTGAAATCCTGAGTGATGT-3′ | 5′-TGACCAGCCCAAAGGAGAAG-3′ | 100 |
Gene/protein name in parentheses is name used in text.
STAT6 Phosphorylation
First-passage keratinocytes were cultured to confluency in keratinocyte growth medium composed of keratinocyte basal medium (Clonetics Corp., San Diego, CA) supplemented with 5 μg/mL insulin, 0.1 mmol/L phosphoethanolamine, 0.1 mmol/L ethanolamine, and 10 ng/mL epidermal growth factor (all from Sigma Aldrich), 0.5 μg/mL hydrocortisone (BD Biosciences, Bedford, MA), and 0.4% bovine pituitary extract (Clonetics Corp.). Medium was refreshed 4 hours before stimulation with keratinocyte growth medium depleted with growth factors (bovine pituitary extract, epidermal growth factor, hydrocortisone, and insulin). After stimulation with 30 ng/mL IL-4 and IL-13 for 10 minutes in keratinocyte growth medium depleted with growth factors, cultures were washed with ice-cold PBS and lysed in ice-cold lysis buffer [10 mmol/L Tris (pH 7.8), 5 mmol/L EDTA, 50 mmol/L NaCl, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L sodium orthovanadate, 10 mmol/L pyrophosphate, and 50 mmol/L sodium fluoride] and 1x Roche protease inhibitor cocktail (Roche Diagnostics Nederland BV, Almere, the Netherlands) on ice. Insoluble material was removed via centrifugation, and Laemmli sample buffer was added. After heating for 5 minutes at 95°C, samples were subjected to SDS–polyacrylamide gel electrophoresis and Western blot analysis. Membranes were blocked with Tris-buffered saline solution [50 mmol/L Tris HCl (pH 7.5) and 150 mmol/L NaCl] supplemented with 0.2% Tween 20, 2% nonfat dry milk, and 2% bovine serum albumin. After blocking, membranes were incubated with one of the following antibodies: mouse monoclonal anti-pSTAT6 (pY641, 1:500; BD Biosciences Pharmigen, San Diego, CA) or rabbit polyclonal anti-STAT6 antibody (S-20, 1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After washing, the membranes were incubated with polyclonal goat anti-rabbit Alexa Fluor-680 (Molecular Probes, Eugene, OR) and goat anti-mouse IRDye800CW (LI-COR Biosciences, Lincoln, NE) as a secondary antibody, and analyzed using the LI-COR Odyssey Imaging system (LI-COR Biosciences).
Statistical Analysis
Statistical analysis was performed using a commercially available software package (STATISTICA; StatSoft, Inc., Tulsa, OK). qPCR data were analyzed using dCt values. For biopsy specimens, the average dCt values for healthy skin were compared with the average dCt values for lesional psoriatic and lesional AD skin. Paired Student's t-tests were performed on skin constructs, with comparison of the stimulated skin constructs with the unstimulated skin equivalents within the same experiment.
Results
IL-4 and IL-13 Induce Spongiosis in Human Skin Equivalents
Adult abdominal keratinocytes were seeded on de-epidermized dermis and cultured for 3 days submerged, followed by culturing for 10 days at the air-liquid interface. To induce an AD phenotype in the skin equivalents, the constructs were stimulated on the last 3 days of the air-liquid interface culture with 3, 10, or 30 ng/mL IL-4 and IL-13. Histologic examination of the skin equivalents demonstrated that stimulation of the constructs with IL-4 and IL-13 induced spongiosis-like intercellular spaces between the cells, which is a hallmark of lesional eczematous skin (Figure 1). In addition, the individual cytokines induced spongiosis-like intercellular spaces (data not shown). The greatest effect was accomplished at the highest concentration of IL-4 and IL-13, and this combination of cytokines was used throughout.
Figure 1.
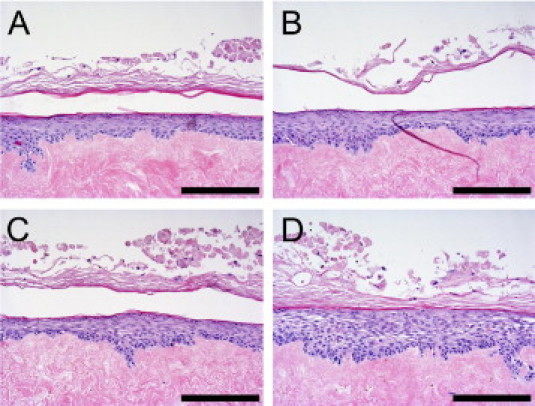
Cytokine-induced spongiosis in human skin equivalents. Skin equivalents were stimulated (B–D) or not (A) with 3 (B), 10 (C), or 30 (D) ng/mL IL-4 and IL-13. Skin equivalents were stained with H&E. Skin equivalents stimulated with IL-4 and IL-13 demonstrated increased intercellular spaces (spongiosis). Scale bar = 100 μm.
The induced spongiosis in human skin equivalents was confirmed at electron microscopy, which exhibited broadening of intercellular spaces in the epidermal compartment of the skin equivalents stimulated with 30 ng/mL IL-4 and IL-13 (Figure 2, A and B). Desmosomal contacts were clearly visible in both the unstimulated and the stimulated constructs, and morphologic features of the desmosomes were not affected by stimulation with IL-4 and IL-13 (Figure 2, A–C). While the number of desmosomes in the nonspongiotic areas of the epidermis was not affected by stimulation with Th2 cytokines, a slight reduction of desmosomal contacts was observed in spongiotic areas with relatively large intercellular spaces. Furthermore, E-cadherin protein levels were slightly decreased after stimulation with IL-4 and IL-13 (data not shown). While apoptotic cells were only sporadically present in the control constructs, the number of apoptotic cells was increased after stimulation with IL-4 and IL-13 (Figure 2D).
Figure 2.
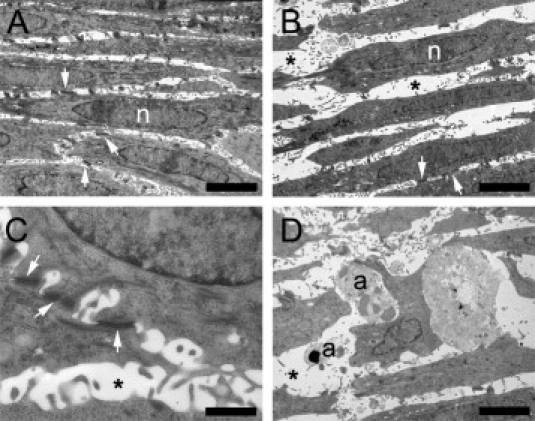
Effect of cytokine stimulation on morphologic features of human skin equivalents evaluated at electron microscopy. Skin equivalents were stimulated (B–D) or not (A) with 30 ng/mL IL-4 and IL-13. Desmosomal contacts (C) and apoptotic cells (D) were clearly visualized at electron microscopy. a, Apoptotic keratinocyte; n, nucleus; arrow, desmosome; asterisk, intercellular space (spongiosis). Scale bars: 8 μm (A and B); 1 μm (C); and 5 μm (D).
IL-4 and IL-13 Induce AD-Associated Genes in Keratinocytes of Skin Equivalents
The effect of cytokine stimulation on the skin equivalents was further investigated by studying genes known to be overexpressed in diseased epidermis using qPCR. These included the AD-related genes CAII and NELL2, the psoriasis-related genes hBD2 and elafin, and the chemokines CXCL8 and CXCL10. Skin equivalents were stimulated with IL-4 and IL-13, a mixture of the psoriasis-related proinflammatory cytokines IL-1α, TNF-α, and IL-6, or the Th17 cytokines IL-17 or IL-22. For comparison, gene expression was determined in epidermal sheets of skin biopsy specimens derived from healthy skin, lesional psoriatic skin, or lesional AD skin. Stimulation of the skin equivalents with IL-4 and IL-13 induced a strong increase in the AD-associated genes CAII (Figure 3A) and NELL2 (Figure 3B) compared with the control constructs. Stimulation with IL-4 or IL-13 alone also induced gene expression of CAII as demonstrated in submerged cultured keratinocytes (data not shown). In contrast to the effect on CAII and NELL2, IL-4 and IL-13 did not affect gene expression of the psoriasis-associated genes hBD2 (Figure 3C) and elafin (Figure 3D) or of the chemokines CXCL8 and CXCL10 (Figure 3E and 3F). Stimulation of the skin constructs with a mixture of the psoriasis-related cytokines IL-1α, TNF-α, and IL-6 or with the Th17 cytokine IL-17 increased expression of hBD2, elafin, and CXCL8, whereas expression of CAII, NELL2, and CXCL10 was not increased (Figure 3, A–F). IL-22 did not affect expression of the studied genes.
Figure 3.
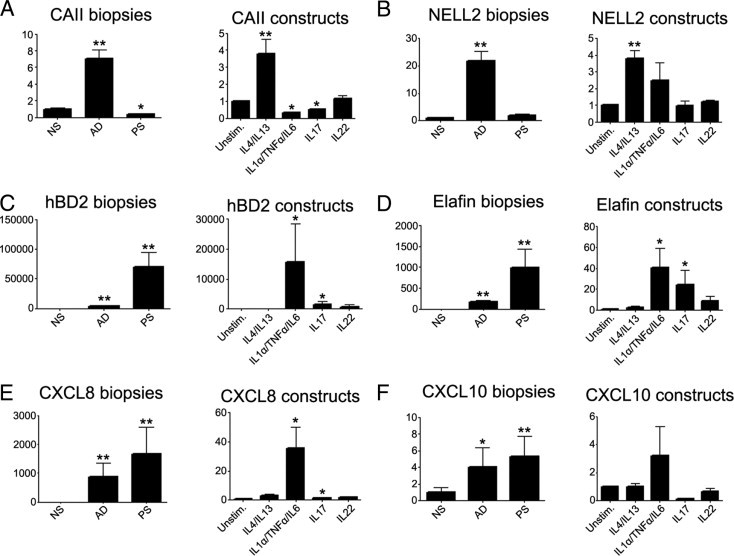
Effect of cytokine stimulation on gene expression in human skin equivalents. Skin equivalents were stimulated with 10 ng/mL IL-1α, 5 ng/mL TNF-α, and 5 ng/mL IL-6; 30 ng/mL IL-4 and IL-13; and 30 ng/mL IL-17 or 30 ng/mL IL-22. RNA was isolated. In addition, RNA was isolated from biopsy specimens from 20 healthy control individuals (NS), lesional skin of 11 patients with psoriasis (PS), and lesional skin of 10 patients with AD. Gene expression of CAII (A), NELL2 (B), hBD2 (C), elafin (D), CXCL8 (E), and CXCL10 (F) was determined using qPCR. The relative amount of mRNA in AD and psoriasis biopsy specimens was normalized to that of healthy skin, and the RNA amount of cytokine-stimulated constructs was normalized to their unstimulated skin equivalents. Error bars represent SEM. *P < 0.05 and **P < 0.01 (Student's t-test). The y axis represents the relative amount of mRNA of the indicated genes.
Induction of Apoptosis in Human Skin Equivalents by IL-4 and IL-13
Because increased keratinocyte apoptosis has been reported in acute forms of AD, the effect of IL-4 and IL-13 on DNA fragmentation in human skin equivalents was evaluated using the TUNEL assay (Figure 4). Stimulation with 30 ng/mL IL-4 and IL-13 increased the number of TUNEL-positive cells in the skin equivalents (Figure 4B), which suggests that these cytokines induce apoptosis in the keratinocytes. Because apoptotic cell death is characterized by activation of caspases, the effect of the caspase-inhibitor Z-VAD on DNA fragmentation induced by IL-4 and IL-13 was tested. Z-VAD inhibited IL-4– and IL-13–dependent DNA fragmentation (Figure 4C) for more than 50%, as calculated from the number of apoptotic cells (data not shown), which suggests that a caspase-dependent apoptotic process was induced in the skin equivalents. Th2 cytokine–induced spongiosis, however, was not inhibited by Z-VAD, which suggests that spongiosis and apoptosis are separate processes in this model. In contrast to stimulation with Th2 cytokines, stimulation of the constructs with a mixture of psoriasis-associated cytokines including IL-1α, TNF-α, and IL-6 did not result in an increase in the TUNEL-positive cell count (Figure 4D).
Figure 4.
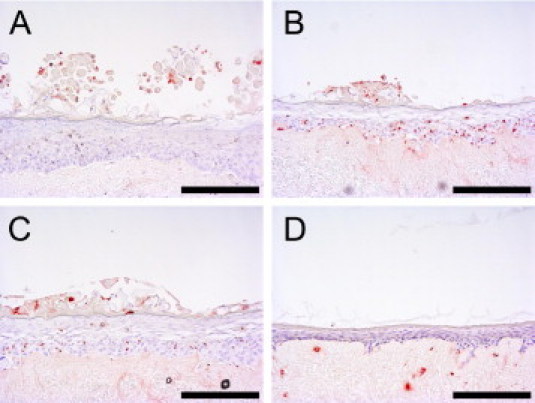
Cytokine-induced apoptosis in human skin equivalents. Skin equivalents were stimulated (B–D) or not (A) with 30 ng/mL IL-4 and IL-13 (B), 30 ng/mL IL-4 and IL-13 and 40 μmol/L Z-VAD (C), or a combination of 10 ng/mL IL-1α, 5 ng/mL TNF-α, and 5 ng/mL IL-6 (D). Induction of apoptosis was evaluated using the TUNEL assay, which results in red staining of cells. Scale bar = 100 μm.
Induction of apoptosis in AD was suggested to be mediated via Fas, also known as CD95,22 and increased protein expression of Fas was observed on keratinocytes of AD skin lesions. Therefore, Fas gene expression in skin equivalents was studied after stimulation with IL-4 and IL-13. Fas expression was increased by IL-4 and IL-13 (Figure 5).
Figure 5.
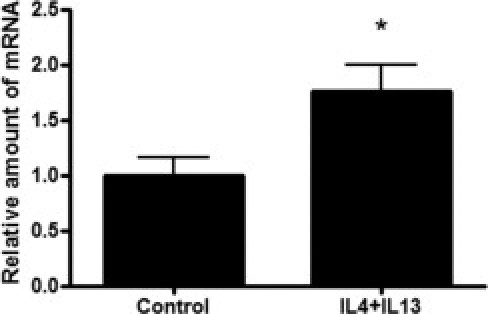
Increased gene expression of Fas in cytokine-stimulated human skin equivalents. Skin equivalents were stimulated with 30 ng/mL IL-4 and IL-13. Expression of Fas was determined using qPCR. The relative amount of mRNA (y axis) of cytokine-stimulated constructs was normalized to unstimulated skin equivalents. Error bars represent SEM. *P < 0.05 (Student's t-test).
STAT6 Phosphorylation by IL-4 and IL-13
Because STAT6 is involved in IL-4–mediated biological responses, activation of STAT6 by IL-4 and IL-13 was studied. First-passage keratinocytes were cultured submerged until confluency, and stimulated for 10 minutes with 30 ng/mL IL-4 and IL-13. Both phosphorylated and total STAT6 levels were determined using Western blot analysis, which revealed that stimulation with IL-4 and IL-13 induced STAT6 phosphorylation (Figure 6).
Figure 6.

Cytokine-induced STAT6 phosphorylation in human skin equivalents. First-passage submerged cultured human keratinocytes derived from three different donors (d1–3) were stimulated for 10 minutes with 30 ng/mL IL-4 and IL-13. Both phosphorylated STAT6 (pSTAT6) and total STAT6 (STAT6) levels in cellular lysates were determined using Western blot analysis.
Anti-AD Drugs Do Not Reverse the AD Phenotype in Skin Equivalents
Most of the known anti-AD drugs (calcineurin inhibitors, UVB, and corticosteroids) are presumed to target the immune system, although some (eg, corticosteroids) may also act at the level of the epidermal keratinocyte. The obvious prediction would be that the present model would not demonstrate an effect of T cell–directed drugs but could be suitable to screen drugs that interfere with keratinocyte activation. Three conventional drugs (CsA, tacrolimus, and dexamethasone) and an experimental drug (ethoxzolamide) were tested. Ethoxzolamide, a carbonic anhydrase inhibitor, was included because of the possible role of CAII in spongiotic changes after Th2 cytokine exposure. None of the drugs tested protected against Th2 cytokine–induced spongiosis. In addition, CsA, tacrolimus, and dexamethasone did not inhibit induction of CAII, NELL2, and Fas gene expression (data not shown).
Discussion
The objective of the present study was to develop a skin equivalent model that exhibited morphologic and molecular features of AD using a defined set of cytokines. The combination of cytokines IL-4 and IL-13 induced spongiotic changes in the epidermis of human skin equivalents, which is one of the characteristics of eczematous lesions. Furthermore, the cytokine mixture induced expression of the AD-associated genes CAII and NELL2, whereas expression of the psoriasis-associated genes hBD2 and elafin was not affected. In addition, IL-4 and IL-13 induced apoptosis in keratinocytes of human skin equivalents. The signaling molecule STAT6 was phosphorylated after IL-4 and IL-13 stimulation. The skin equivalent model, demonstrating histologic and molecular characteristics of AD, may be useful for study of the epidermal pathologic features of AD, and could enable discovery of anti-AD drugs that act at the level of the epidermal keratinocytes.
The Th2 cytokines IL-4 and IL-13 were used to induce an AD phenotype in normal human skin equivalents because these cytokines have an important role in AD.23,24 In the acute phase of the disease, the immune response is primarily coordinated by Th2 cells, and the Th2 cytokines IL-4, IL-5, and IL-13 predominate in lesional skin.25 During the chronic phase of AD, Th1 cytokines such as IL-12 and interferon-γ (IFN-γ) are increased in the skin.23,24 IL-4 and IL-13 promote immunoglobulin isotype switch to IgE and IgE production.26 In addition, IL-4 and IL-13 induce expression of vascular cell adhesion molecules such as VCAM1,26 which may suggest a role for these Th2 cytokines in regulation of eosinophilic infiltration in the skin. The present skin equivalent model consists of keratinocytes seeded on de-epidermized dermis in which no viable dermal cells were present. Therefore, the effects of IL-4 and IL-13 on epidermal morphologic features and gene expression are the result of interactions of IL-4 and IL-13 with the keratinocytes, which express receptors for IL-4 and IL-13.27,28
The combination of IL-4 and IL-13 induced increased intraepidermal intercellular edema, a clinical hallmark of acute and subacute eczema. A recent study suggested that induction of spongiosis by the cytokines IL-4, IL-13, and IFN-γ was mediated by decreased E-cadherin expression and simultaneously increased hyaluronan production by keratinocytes,29 resulting in osmotic movement of water into the intercellular compartment of the epidermis. Furthermore, T cell–induced Fas-mediated apoptosis of keratinocytes has been suggested as an important pathogenic event in the formation of spongiosis.22,30–32 Apoptosis induced by the death receptor Fas involves the binding of Fas ligand to its receptor and subsequent activation of caspase-8/10 and the effector caspase-3, resulting in DNA fragmentation. IFN-γ was suggested to have an important role in apoptosis induction by increasing the expression of Fas on keratinocytes.22 The present study exhibited an increased rate of DNA fragmentation in the epidermis of the skin equivalents after stimulation with IL-4 and IL-13, as demonstrated at TUNEL staining. Furthermore, the pan-caspase inhibitor Z-VAD-FMK inhibited cytokine-induced DNA fragmentation, which suggests that IL-4 and IL-13 induce apoptosis in the skin equivalents. In addition, mRNA expression of Fas was increased in the skin equivalents after stimulation with IL-4 and IL-13. Because no IFN-γ or IFN-γ–producing T cells were included in the model, it seems that IFN-γ is dispensable for increased Fas expression on keratinocytes. Several studies have reported increased apoptosis in lesional AD skin compared with healthy skin. Apoptosis is most apparent in biopsy specimens of acute AD, as described in studies by Ohtani et al29 and Trautmann et al.33 The present model may reflect acute rather than chronic AD, in which apoptosis is less prominent. Clearly, this model has limitations, and improvements and extensions would include addition of Th1 cytokines to mimic the chronic phase or even use of a defined activated T-cell population. This work is currently in progress.
To evaluate the induction of an AD phenotype in the skin equivalents by IL-4 and IL-13, expression of the AD-associated genes CAII and NELL2 was studied. Gene expression of both CAII and NELL2 was increased in lesional AD skin compared with healthy skin.11 However, IHC for CAII did not demonstrate increased staining in the Th2-stimulated skin equivalents (data not shown). This corresponds to the mild increase in CAII protein that was observed in AD lesions, in contrast with the elevated mRNA levels15 and the limited value of quantification at IHC. In the eye, CAII has a role in aqueous humor flow, and inhibition of this enzyme results in reduction of intraocular pressure in patients with glaucoma.34 Thus, it was tempting to speculate that CAII could be involved in epidermal abnormalities observed in AD such as spongiosis and increased surface pH. Inhibition of CAII by ethoxzolamide, however, did not prevent Th2 cytokine–induced spongiosis in the present model system. In addition to CAII, NELL2 expression was increased in AD.11 NELL2 is strongly expressed in adult and fetal brain,13 where it promotes survival of hippocampal and cortical neurons14 and increases differentiation of spinal cord progenitors.35 The function of NELL2 in AD has not been elucidated; however, it would be interesting to study whether there is a relation between increased NELL2 expression in AD and the increased amount of nerve fibers, which has been demonstrated in the epidermis of patients with AD.36 Corresponding to studies using submerged cultures of keratinocytes,15 the present study demonstrated that IL-4 and IL-13 increase gene expression of both CAII and NELL2 in the skin equivalents, demonstrating that the AD model exhibited molecular characteristics of AD skin.
In line with the known mechanism of action of CsA and tacrolimus, an effect of these compounds was not observed in the present model. Addition of dexamethasone did not demonstrate effects; however, the pan-caspase inhibitor Z-VAD inhibited apoptosis without affecting the spongiotic changes. This demonstrates that the model system is, in principle, suited to detect cytoprotective effects of drugs. Anti-AD drugs can be divided into compounds that act on the adaptive immune system and compounds that act on the keratinocytes. CsA and tacrolimus are anti-inflammatory drugs that act on lymphocyte activation,37 and because no lymphocytes are present in the skin equivalents, as expected, these drugs did not affect the IL-4– and IL-13–induced AD phenotype in the skin equivalents. While dexamethasone was suggested to affect lymphocytes and keratinocytes, no effect of dexamethasone on IL-4– and IL-13–induced morphologic and molecular changes in the skin equivalents was observed, which may be explained by the limited time in which dexamethasone was present in the culture medium; it may take more time for this drug to exert its effects. Furthermore, keratinocyte signaling pathways activated by T lymphocytes may be potential targets for dexamethasone, and these pathways were missing in the present model. In addition to the widely used drugs for AD, also tested was the effect of the carbonic anhydrase inhibitor ethoxzolamide38 on IL-4– and IL-13–induced spongiosis formation because a role for CAII in AD was suggested. However, ethoxzolamide did not affect spongiosis formation in the skin equivalent model.
hBD2 gene expression was increased in skin biopsy specimens from patients with AD compared with healthy volunteers. Previous studies, including that by de Jongh et al,8 also described higher expression levels of antimicrobial proteins in lesional AD skin compared with healthy skin.39–41 In contrast to these in vivo data, stimulation of the skin equivalents with IL-4 and IL-13 did not affect hBD2 gene expression. This may be explained in that the skin equivalents do not contain additional cell types such as lymphocytes or fibroblasts. IL-4 and IL-13 did not affect gene expression of the chemokines CXCL8 (IL-8) and CXCL10, both of which are increased in AD, psoriasis, and allergic contact dermatitis compared with healthy skin.11 In contrast, skin equivalents stimulated with either a mixture of psoriasis-related cytokines or IL-17 demonstrated increased gene expression of hBD2, elafin, and CXCL8, whereas CXCL10 gene expression was unaffected. The lack of effect of the Th1, Th2, and Th17 cytokines to induce CXCL10 may be explained by the lack of IFN-γ and IFN-γ–producing T cells in the model system because IFN-γ induces CXCL10 expression in vitro and in vivo.42,43 The present study demonstrated an increase in NELL2 after Th2 cytokine stimulation; however, the Th17-related cytokines IL-17 and IL-22 and the mixture of psoriasis-related cytokines IL-1α, TNFα, and IL-6 did not induce NELL2 in skin constructs. This is in accordance with the lack of NELL2 in biopsy specimens of lesional psoriatic skin because Th17 cytokines have an important role in psoriasis.44
In conclusion, it is suggested that the present model system can be used to study the epidermal pathologic features of AD, and may be useful for identification of drugs that could act at this level.
Acknowledgments
We thank Irene Otte-Holler (Radboud University Medical Centre), who performed the electron microscopy of skin equivalents.
Footnotes
Supported by grant bgt.6739 from the Dutch Program of Tissue Engineering (J.S.), AGIKO grant 920-03-355 from the Netherlands Organization for Health Research and Development (ZonMW) (M.K.), and grant 114000084 from the Alternatives to Animal Experiments program of ZonMW (J.S. and P.Z.).
Contributor Information
Joost Schalkwijk, Email: j.schalkwijk@derma.umcn.nl.
Geuranne S. Tjabringa, Email: G.S.Tjabringa@umcutrecht.nl.
References
- 1.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 2.Ong P.Y., Ohtake T., Brandt C., Strickland I., Boguniewicz M., Ganz T., Gallo R.L., Leung D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 3.Zeeuwen P.L., de Jongh G.J., Rodijk-Olthuis D., Kamsteeg M., Verhoosel R.M., van Rossum M.M., Hiemstra P.S., Schalkwijk J. Genetically programmed differences in epidermal host defense between psoriasis and atopic dermatitis patients. PLoS ONE. 2008;3:e2301. doi: 10.1371/journal.pone.0002301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elias P.M., Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol. 2009;9:437–446. doi: 10.1097/ACI.0b013e32832e7d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer C.N., Irvine A.D., Terron-Kwiatkowski A., Zhao Y., Liao H., Lee S.P., Goudie D.R., Sandilands A., Campbell L.E., Smith F.J., O'Regan G.M., Watson R.M., Cecil J.E., Bale S.J., Compton J.G., Digiovanna J.J., Fleckman P., Lewis-Jones S., Arseculeratne G., Sergeant A., Munro C.S., El Houate B., McElreavey K., Halkjaer L.B., Bisgaard H., Mukhopadhyay S., McLean W.H. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 6.Brown S.J., McLean W.H. Eczema genetics: current state of knowledge and future goals. J Invest Dermatol. 2009;129:543–552. doi: 10.1038/jid.2008.413. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu N., Saijoh K., Kuk C., Liu A.C., Khan S., Shirasaki F., Takehara K., Diamandis E.P. Human tissue kallikrein expression in the stratum corneum and serum of atopic dermatitis patients. Exp Dermatol. 2007;16:513–519. doi: 10.1111/j.1600-0625.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- 8.de Jongh G.J., Zeeuwen P.L., Kucharekova M., Pfundt R., van der Valk P.G., Blokx W., Dogan A., Hiemstra P.S., van de Kerkhof P.C., Schalkwijk J. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125:1163–1173. doi: 10.1111/j.0022-202X.2005.23935.x. [DOI] [PubMed] [Google Scholar]
- 9.Werfel T. The role of leukocytes, keratinocytes, and allergen-specific IgE in the development of atopic dermatitis. J Invest Dermatol. 2009;129:1878–1891. doi: 10.1038/jid.2009.71. [DOI] [PubMed] [Google Scholar]
- 10.Nomura I., Gao B., Boguniewicz M., Darst M.A., Travers J.B., Leung D.Y. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol. 2003;112:1195–1202. doi: 10.1016/j.jaci.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 11.Kamsteeg M., Jansen P.A., Vlijmen-Willems I.M., van Erp P.E., Rodijk-Olthuis D., van der Valk P.G., Feuth T., Zeeuwen P.L., Schalkwijk J. Molecular diagnostics of psoriasis, atopic dermatitis, allergic contact dermatitis and irritant contact dermatitis. Br J Dermatol. 2010;162:568–578. doi: 10.1111/j.1365-2133.2009.09547.x. [DOI] [PubMed] [Google Scholar]
- 12.Sly W.S., Hu P.Y. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe T.K., Katagiri T., Suzuki M., Shimizu F., Fujiwara T., Kanemoto N., Nakamura Y., Hirai Y., Maekawa H., Takahashi E. Cloning and characterization of two novel human cDNAs (NELL1 and NELL2) encoding proteins with six EGF-like repeats. Genomics. 1996;38:273–276. doi: 10.1006/geno.1996.0628. [DOI] [PubMed] [Google Scholar]
- 14.Aihara K., Kuroda S., Kanayama N., Matsuyama S., Tanizawa K., Horie M. A neuron-specific EGF family protein, NELL2, promotes survival of neurons through mitogen-activated protein kinases. Brain Res Mol Brain Res. 2003;116:86–93. doi: 10.1016/s0169-328x(03)00256-0. [DOI] [PubMed] [Google Scholar]
- 15.Kamsteeg M., Zeeuwen P.L., de Jongh G.J., Rodijk-Olthuis D., Zeeuwen-Franssen M.E., van Erp P.E., Schalkwijk J. Increased expression of carbonic anhydrase II (CAII) in lesional skin of atopic dermatitis: regulation by Th2 cytokines. J Invest Dermatol. 2007;127:1786–1789. doi: 10.1038/sj.jid.5700752. [DOI] [PubMed] [Google Scholar]
- 16.Jin H., He R., Oyoshi M., Geha R.S. Animal models of atopic dermatitis. J Invest Dermatol. 2009;129:31–40. doi: 10.1038/jid.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tjabringa G., Bergers M., van Rens D., de Boer R., Lamme E., Schalkwijk J. Development and validation of human psoriatic skin equivalents. Am J Pathol. 2008;173:815–823. doi: 10.2353/ajpath.2008.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelhart K., El Hindi T., Biesalski H.K., Pfitzner I. In vitro reproduction of clinical hallmarks of eczematous dermatitis in organotypic skin models. Arch Dermatol Res. 2005;297:1–9. doi: 10.1007/s00403-005-0575-7. [DOI] [PubMed] [Google Scholar]
- 19.Zeeuwen P.L., van Vlijmen-Willems I.M., Jansen B.J., Sotiropoulou G., Curfs J.H., Meis J.F., Janssen J.J., van Ruissen F., Schalkwijk J. Cystatin M/E expression is restricted to differentiated epidermal keratinocytes and sweat glands: a new skin-specific proteinase inhibitor that is a target for cross-linking by transglutaminase. J Invest Dermatol. 2001;116:693–701. doi: 10.1046/j.1523-1747.2001.01309.x. [DOI] [PubMed] [Google Scholar]
- 20.Minner F., Poumay Y. Candidate housekeeping genes require evaluation before their selection for studies of human epidermal keratinocytes. J Invest Dermatol. 2009;129:770–773. doi: 10.1038/jid.2008.247. [DOI] [PubMed] [Google Scholar]
- 21.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Trautmann A., Akdis M., Kleemann D., Altznauer F., Simon H.U., Graeve T., Noll M., Brocker E.B., Blaser K., Akdis C.A. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25–35. doi: 10.1172/JCI9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller K.M., Jaunin F., Masouye I., Saurat J.H., Hauser C. Th2 cells mediate IL-4–dependent local tissue inflammation. J Immunol. 1993;150:5576–5584. [PubMed] [Google Scholar]
- 24.Novak N., Bieber T. Allergic and nonallergic forms of atopic diseases. J Allergy Clin Immunol. 2003;112:252–262. doi: 10.1067/mai.2003.1595. [DOI] [PubMed] [Google Scholar]
- 25.Grewe M., Walther S., Gyufko K., Czech W., Schopf E., Krutmann J. Analysis of the cytokine pattern expressed in situ in inhalant allergen patch test reactions of atopic dermatitis patients. J Invest Dermatol. 1995;105:407–410. doi: 10.1111/1523-1747.ep12321078. [DOI] [PubMed] [Google Scholar]
- 26.Leung D.Y., Bieber T. Atopic dermatitis. Lancet. 2003;361:151–160. doi: 10.1016/S0140-6736(03)12193-9. [DOI] [PubMed] [Google Scholar]
- 27.Junghans V., Jung T., Neumann C. Human keratinocytes constitutively express IL-4 receptor molecules and respond to IL-4 with an increase in B7/BB1 expression. Exp Dermatol. 1996;5:316–324. doi: 10.1111/j.1600-0625.1996.tb00135.x. [DOI] [PubMed] [Google Scholar]
- 28.Akaiwa M., Yu B., Umeshita-Suyama R., Terada N., Suto H., Koga T., Arima K., Matsushita S., Saito H., Ogawa H., Furue M., Hamasaki N., Ohshima K., Izuhara K. Localization of human interleukin 13 receptor in non-haematopoietic cells. Cytokine. 2001;13:75–84. doi: 10.1006/cyto.2000.0814. [DOI] [PubMed] [Google Scholar]
- 29.Ohtani T., Memezawa A., Okuyama R., Sayo T., Sugiyama Y., Inoue S., Aiba S. Increased hyaluronan production and decreased E-cadherin expression by cytokine-stimulated keratinocytes lead to spongiosis formation. J Invest Dermatol. 2009;129:1412–1420. doi: 10.1038/jid.2008.394. [DOI] [PubMed] [Google Scholar]
- 30.Kerstan A., Leverkus M., Trautmann A. Effector pathways during eczematous dermatitis: where inflammation meets cell death. Exp Dermatol. 2009;18:893–899. doi: 10.1111/j.1600-0625.2009.00919.x. [DOI] [PubMed] [Google Scholar]
- 31.Trautmann A., Akdis M., Brocker E.B., Blaser K., Akdis C.A. New insights into the role of T cells in atopic dermatitis and allergic contact dermatitis. Trends Immunol. 2001;22:530–532. doi: 10.1016/s1471-4906(01)02004-x. [DOI] [PubMed] [Google Scholar]
- 32.Trautmann A., Akdis M., Blaser K., Akdis C.A. Role of dysregulated apoptosis in atopic dermatitis. Apoptosis. 2000;5:425–429. doi: 10.1023/a:1009620329213. [DOI] [PubMed] [Google Scholar]
- 33.Trautmann A., Akdis M., Schmid-Grendelmeier P., Disch R., Brocker E.B., Blaser K., Akdis C.A. Targeting keratinocyte apoptosis in the treatment of atopic dermatitis and allergic contact dermatitis. J Allergy Clin Immunol. 2001;108:839–846. doi: 10.1067/mai.2001.118796. [DOI] [PubMed] [Google Scholar]
- 34.McLaren J.W. Measurement of aqueous humor flow. Exp Eye Res. 2009;88:641–647. doi: 10.1016/j.exer.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Nelson B.R., Claes K., Todd V., Chaverra M., Lefcort F. NELL2 promotes motor and sensory neuron differentiation and stimulates mitogenesis in DRG in vivo. Dev Biol. 2004;270:322–335. doi: 10.1016/j.ydbio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Urashima R., Mihara M. Cutaneous nerves in atopic dermatitis: a histological, immunohistochemical and electron microscopic study. Virchows Arch. 1998;432:363–370. doi: 10.1007/s004280050179. [DOI] [PubMed] [Google Scholar]
- 37.Parsons W.H., Sigal N.H., Wyvratt M.J. FK-506: a novel immunosuppressant. Ann NY Acad Sci. 1993;685:22–36. doi: 10.1111/j.1749-6632.1993.tb35847.x. [DOI] [PubMed] [Google Scholar]
- 38.Supuran C.T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 39.Ballardini N., Johansson C., Lilja G., Lindh M., Linde Y., Scheynius A., Agerberth B. Enhanced expression of the antimicrobial peptide LL-37 in lesional skin of adults with atopic eczema. Br J Dermatol. 2009;161:40–47. doi: 10.1111/j.1365-2133.2009.09095.x. [DOI] [PubMed] [Google Scholar]
- 40.Gambichler T., Skrygan M., Tomi N.S., Othlinghaus N., Brockmeyer N.H., Altmeyer P., Kreuter A. Differential mRNA expression of antimicrobial peptides and proteins in atopic dermatitis as compared to psoriasis vulgaris and healthy skin. Int Arch Allergy Immunol. 2008;147:17–24. doi: 10.1159/000128582. [DOI] [PubMed] [Google Scholar]
- 41.Harder J., Dressel S., Wittersheim M., Cordes J., Meyer-Hoffert U., Mrowietz U., Folster-Holst R., Proksch E., Schroder J.M., Schwarz T., Glaser R. Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J Invest Dermatol. 2010;130:2138–2139. doi: 10.1038/jid.2009.432. [DOI] [PubMed] [Google Scholar]
- 42.Boorsma D.M., Flier J., Sampat S., Ottevanger C., de Haan P., Hooft L., Willemze R., Tensen C.P., Stoof T.J. Chemokine IP-10 expression in cultured human keratinocytes. Arch Dermatol Res. 1998;290:335–341. doi: 10.1007/s004030050314. [DOI] [PubMed] [Google Scholar]
- 43.Mori T., Kabashima K., Yoshiki R., Sugita K., Shiraishi N., Onoue A., Kuroda E., Kobayashi M., Yamashita U., Tokura Y. Cutaneous hypersensitivities to hapten are controlled by IFN-gamma–upregulated keratinocyte Th1 chemokines and IFN-gamma–downregulated Langerhans cell Th2 chemokines. J Invest Dermatol. 2008;128:1719–1727. doi: 10.1038/jid.2008.5. [DOI] [PubMed] [Google Scholar]
- 44.Lowes M.A., Kikuchi T., Fuentes-Duculan J., Cardinale I., Zaba L.C., Haider A.S., Bowman E.P., Krueger J.G. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]